

Abstract
Background/Aims
Canonical transient receptor potential (TRPC) channels modulate membrane potential and intracellular Ca2+. We examined the role of TRPC1 and TRPC3 channels in vasocontraction and relaxation in mouse aorta.
Methods
Vasocontraction and relaxation of aorta from wild type (WT), TRPC1 KO, and TRPC3 KO mice were measured for phenylephrine (Phe) and carbachol (CCh). Intracellular Ca2+ was measured in primary aorta endothelial cells (EC) and whole cell K+ current in freshly isolated smooth muscle cells (SMC).
Results and Conclusion
TRPC1 KO aorta showed increased vasocontraction to Phe compared to WT and TRPC3 KO due to diminished role of BKCa channels. BKCa mRNA (aorta) and whole cell current (SMC) was reduced versus WT. Contraction in WT aorta was increased to TRPC1 KO level by BKCa channel inhibition. Relaxation to CCh was reduced in TRPC1 KO and TRPC3 KO aortas with concomitant reduction in EC Ca2+ response. Pyr3 (TRPC3 blocker) reduced the Ca2+ response to CCh in EC from WT, but not TRPC3 KO mice. In summary, TRPC1 attenuates receptor-mediated contraction through activation and/or expression of SMC BKCa channels while TRPC3 does not contribute to receptor-mediated constriction. Both TRPC1 and TRPC3 participate in EC Ca2+ influx and vasorelaxation of aorta.
Keywords: TRPC1, TRPC3, endothelium, smooth muscle, vascular, calcium, vasorelaxation, vasoconstriction, knockout mouse
Introduction
The canonical transient receptor potential (TRPC) channels comprise a family of non-selective cation channels (TRPC1–7) that have been shown to be expressed throughout the vasculature in both smooth muscle (SMC) and endothelial cells (EC) [1]. These channels are non-selective cation channels which primarily conduct Ca2+, Na+, and K+ along their respective electrochemical gradients [2]. As such, TRPC channels have been considered candidates for regulating intracellular calcium levels and membrane potential in the vasculature.
Despite several years of study regarding the possible role of TRPC channels as store-operated Ca2+ channels (SOC) and receptor-operated Ca2+ channels (ROC), surprisingly little information exists regarding their role in the mechanisms of vasoconstriction and vasorelaxation in intact arteries. The paucity of these studies may be a result of the general absence of selective pharmacological tools for these channels. For this reason, the use of gene knockout mice offers a good option for studying specific roles of these channels in the regulation of vascular tone.
In mouse aorta, a comparison of TRPC mRNA expression levels demonstrated greatest expression for TRPC1 and TRPC3 channels [3]. However, the few studies that have examined the functional role of these channels in the control of vascular tone have led to divergent conclusions and left numerous unanswered questions. Only one study has thus far examined the role of TRPC1 in agonist-mediated vasoconstriction and vasorelaxation in TRPC1 KO mice [4] and no study to date has examined vascular responsiveness in TRPC3 KO mice.
The goal of the present study was to determine the role of TRPC1 and TRPC3 in the mechanism of vasocontraction and vasorelaxation in mouse aorta from TRPC1 KO and TRPC3 KO mice. The aorta is the largest compliance artery in the body and contributes to peak blood pressure as well as pulse pressure. Reduced compliance of this artery through greater active tone or reduced active relaxation promotes greater pulse wave velocity and can contribute to altered coupling between the heart and aorta and subsequent cardiovascular pathology [5–7]. We sought to determine 1) if absence of TRPC1 or TRPC3 results in reduced agonist-mediated vasoconstriction and 2) if loss of TRPC1 or TRPC3 results in loss of endothelial-mediated vasorelaxation.
Methods
Animal models
All animal experiments were performed in accordance with Baylor College of Medicine (BCM) Institutional Animal Care and Use Committee guidelines (August 31 2010 revision). Generation of TRPC1 KO and TRPC3 KO mice has been described previously [3,8]. TRPC1 KOand TRPC3 KO mice were provided by Dr. Lutz Birnbaumer. All experiments were performed with male mice.
Aorta EC primary culture
EC were obtained from mouse aorta explants as described previously [9]. In brief, 35 mm culture dishes were coated for 1–2 hr at room temperature with BD Matrigel™ Basement Membrane Matrix (BD Biosciences, CA, USA), diluted 1:1 with DMEM (Gibco, CA, USA) containing glucose and L-glutamine. The Aorta was dissected out, cleaned of fat and thoroughly perfused with PBS to remove blood. The vessel was then cut open and 2 mm fragments were placed intima-down onto matrigel-coated dishes, containing DMEM supplemented with 10 % FBS (Gibco), 100 ug/ml Endothelial Cell Growth Supplement (BD Biosciences), 10 U/ml Heparin (Sigma, MO, USA), 100 U/ml Penicillin/Streptomycin (Invitrogen, CA, USA) and 2% Minimal Essential Amino Acid mixture (Sigma). EC that migrated from the explant were allowed to grow for 4–12 weeks at 37 C in 5% CO2. We confirmed the purity of the resulting cells in preliminary experiments using Tie2-GFP transgenic mice [10] (Supplemental Figure 1). For experiments, EC were harvested from the gel matrix using 1 hr incubation with neutral protease (7 mg/ml; Worthington Biochemical, NJ, USA) in PBS, washed twice with DMEM and plated onto Petri dishes or 12 mm glass cover-slips coated with Fibronectin (BD Biosciences), 50 ug/ml for 2 hr. Cells were supplemented with the same media and maintained at 37 C, 5% CO2.
Patch clamp and Ca2+ imaging experiments
All patch clamp and Ca2+ imaging measurements were conducted at room temperature with continuous bath perfusion. Standard physiological bath solution consisted of (in mM) 140 NaCl, 5.6 KCl, 1.6 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES (pH 7.3). For experiments in Ca2+-free extracellular solution, bath saline contained no Ca2+ and 4 mM BAPTA.
For patch clamp studies, freshly dispersed aortic smooth muscle cells (SMC) were obtained from WT and TRPC1 KO mice by enzymatic dissociation (30 min at 37 C) using papain (80 U/ml) with 5 mg/ml dithiothreitol (DTT) in the following digestion buffer (in mM): 138 NaCl, 5 KCl, 1.5 MgCl2, 0.42 Na2HPO4, 0.44 NaH2PO4, 0.1 CaCl2, 10 Hepes, 4.2 NaHCO3 and 0.5 % BSA. This was followed by an 8 min incubation with collagenase type 2 (565 U/ml) and hyaluronidase 2000 U/ml in the same digestion buffer. Dispersed SMC were placed in the recording chamber and used within 6 hours of isolation.
Whole cell currents of SMC were recorded in the voltage clamp mode using the perforated patch configuration [11,12]. All data was recorded with a Multiclamp 700B patch-clamp amplifier, DigiData 1440A computer interface, and Clampex 10.0 acquisition and analysis software (Molecular Devices, Sunnyvale, CA). The bath was grounded with Ag-AgCl pellet via a 1 M KCl, 2% agarose bridge. The pipette solution contained (in mM) 40 KCl, 100 K gluconate, 1 MgCl2, 10 NaCl, 0.1 EGTA, 10 NaCl, 10 HEPES, 0.1 EGTA (pH 7.2, adjusted with KOH). Amphotericin B (0.5 mg/ml) was added to the pipette solution immediately prior to the recording and acquisition was started when access resistance decreased below 35 MΩ. 70 % of series resistance was compensated by the amplifier circuit. Whole cell currents were evoked by 500 ms voltage ramps from −100 to +60 mV and the resulting signal was filtered at 1 kHz using builtin amplifier filter and sampled at 500 μs rate.
Ca2+ imaging experiments were performed with cultured EC grown on glass cover-slips when a nearly confluent monolayer was observed, typically between 3 and 10 days after passage. Cells were washed with standard physiological solution to remove the culture media and loaded with Fura-2 AM Ca2+ sensitive dye [13] (TefLabs, TX, USA), 1 ug/ml for 1 hr at room temperature. Fura-2 loaded EC were imaged by acquiring 340/380 nm excitation – 510 nm emission fluorescent ratios (R340/380) every 3.9 s, using an Olympus IX81 fluorescent microscope equipped with Uplan S-Apo 20× 0.75 NA lens (Olympus, USA), Lambda LS Xenon Arc lamp, Lambda 10-2 filter wheel shutter controller (both from Sutter Instruments, CA, USA), RET-EXi-F-M-12-C CCD-camera (QImaging, Canada) and controlled with Slidebook 4.2 Imaging software (Olympus).
Isometric Tension Measurements in Aorta
The thoracic aorta from just below the decending portion of the arch to the diaphragm was flushed with Hanks buffer in situ to remove blood, then dissected out and cut into 2.5 mm segments. Each segment was mounted in a myograph (ChuelTech, Houston, TX) to measure isometric tension changes. The ring segments were bathed in Krebs buffer consisting of (in mM): (119 NaCl, 4.7 KCl, 1 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 11.1 Glucose, and 2.5 CaCl2) maintained at 37°C and bubbled with a gas mixture of 5% CO2/20% O2/75% N2 (pH 7.4) [14]. Changes in tension were measured using Model 2 Research Grade isometric force transducer (Harvard Apparatus, Holliston, MA). The outputs from the transducers were digitized by Powerlab/8sp (AD Instruments, Colorado Springs, CO) and captured at 1 KHz by LabChart v4.2.4 (AD Instruments). Aortic ring segments were equilibrated in the organ chamber for one hour during which resting tension was gradually increased to 0.8 g. This amount of resting tension was determined based on length-tension measurements from a separate group of aortas from WT and KO mice. Once the arteries were at resting tension, they were contracted with 40 mM KCl 3 or 4 times to ensure that the arteries were capable of producing reproducible contraction. Aortas from WT, TRPC1 KO, TRPC3 KO mice were evaluated for contractile responses using phenylephrine (Phe; 3 μM) followed by endothelial-mediated relaxations to carbachol (CCh; 10 μM). Carbachol was used in lieu of acetylcholine as a muscarinic agonist due to its resistance to hydrolysis by acetylcholinesterase. Contraction to phenylephrine was normalized to the final KCl contraction and expressed as a percent of the KCl response. Relaxation to carbachol was normalized to the contraction produced by Phe where relaxation to pre-Phe tension corresponds to a 100% relaxation.
Real Time PCR
RNA was extracted from EC grown on fibronectin coated Petri dishes at day 7–9 after the 1st passage using the TRIzol Isolation protocol described in the Pure Link RNA Micro Kit (Invitrogen). RNA was quantified and evaluated for purity by UV Photospectrometer at the 260nm and 280nm wavelengths. The RNA was treated with DNAse I (Invitrogen) and equal amounts of RNA were used to generate cDNA by Super Script III First Strand Synthesis System (Invitrogen). Real-Time PCR was performed with a Mastercycler ep realplex thermal cycler (Eppendorf, Hamburg, Germany) using QuantiTect SYBR Green (Qiagen, USA). Expression was evaluated relative to Beta Actin. The PCR primers are listed in the supplemental data (Supplemental Table).
Data analysis
SigmaPlot 11.0 (Systat Software, Chicago, IL) was used for statistical analyses. Data are shown as mean ± s.e.m. Fura-2 R340/380 time lapse data acquired from individual cells were baseline subtracted and agonist-induced ΔR340/380 were averaged for each coverslip (12–59 cells/coverslip). In some cases, the integral of Ca2+ increase was measured as an area under the curve. Data were compared using ANOVA with Holm-Sidak test for multiple group comparison when appropriate and compared with the one-way ANOVA or two-way RM-ANOVA when appropriate, using the Holm-Sidak test for pair-wise multiple comparison procedures. Real time PCR data was analyzed by Kruskal-Wallis one-way ANOVA on ranks.
Results
The mean age of mice in this study were 11.3±0.3, 11.9±0.2, and 11.3±0.2 weeks for WT, TRPC1 KO, and TRPC3 KO and were not significantly different between groups. Given that TRPC channel expression plasticity has been shown in some TRPC KO systems [15], we used qRT-PCR to measure mRNA expression in EC for the TRPC channel family. These studies did not demonstrate statistically significant changes in expression levels compared to WT except for the respective knockouts (Supplemental Fig 2).
Role of TRPC1 and TRPC3 in vasocontraction
Contraction of aorta segments was evaluated from wild-type (WT), TRPC1 KO, and TRPC3 KO mice. We evaluated vascular responsiveness to Phe, an alpha adrenergic receptor agonist, at a sub-maximal concentration (3 μM) (Fig 1A&B). Mean Phe-induced contraction for the three groups was 92.9±6.2, 129.7±7.0, and 103.3±14.6% of KCl constriction for WT, TRPC1, and TRPC3 respectively. Aortas from the TRPC1 KO group had significantly greater contraction compared to the WT and TRPC3 KO groups (P<0.001); the WT and TRPC3 KO groups were not statistically different.
Fig 1.
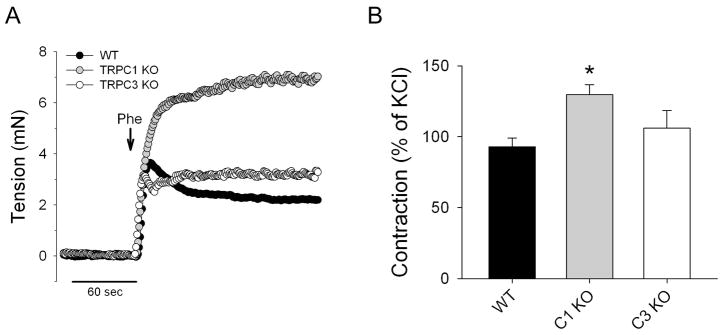
The effect of TRPC1 and TRPC3 gene KO on isometric contraction of mouse aorta: (A) Representative traces demonstrating changes in aorta tension after application of 3 μM phenylephrine (Phe) and (B) Average tension developed to Phe in aorta from WT (n = 23), TRPC1 KO (n = 17) or TRPC3 KO (n = 12) mice. * - p<0.05.
Mechanism of greater contractility in aorta of TRPC1 KO mice
We next sought to determine the mechanism by which loss of TRPC1 channels leads to enhanced vasocontraction. We first tested the possibility that the greater contraction of aorta from TRPC1 mice was due to reduced basal or Phe-stimulated NO production in this group. If this were the case, we would expect the contractions in the presence of a NOS inhibitor to be equalized in both groups. However, contractions to Phe in the presence of L-NAME (300 μM) were still significantly greater in the TRPC1 KO group (19.1±0. 5 vs. 22.1±0. 8 mN; P=0.01).
TRPC1 was recently shown to co-localize with BKCa channels and contribute to activation of this channel in rat [16]. In this model, TRPC1 channels are thought to provide or trigger the local elevation of Ca2+ which promotes activation of BKCa channels. This K+ channel activation produces SMC membrane hyperpolarization which then tempers the contraction of the artery. If this model is valid in our system, we would predict that the absence of TRPC1 would lead to less BKCa channel opening, greater smooth muscle depolarization and greater vasocontraction. Additionally, we would predict that pharmacological inhibition of BKCa channels in aorta of WT mice should produce greater contraction to Phe similar to that in the TRPC1 KO group. Figure 2A shows the result of Phe contraction in the absence and presence of a BKCa channel blocker, penitrem A (200 nM), for aorta of WT and TRPC1 KO mice. Note that BKCa channel inhibition in the WT group led to a significant increase in developed tension (P<0.05) that was comparable to the TRPC1 KO value without penitrem A. Addition of penitrem A to the TRPC1 KO group led to a smaller increase in tension that was not statistically different from TRPC1 KO control (P=0.18).
Fig 2.

The role of large conductance Ca activated potassium channels (BK) in modulation of aorta contractile response in WT and TRPC1 KO mice. (A) Average contraction to phenylephrine (3 μM) in aorta pretreated with penitrem A (0.2 μM, pen) or vehicle (0.2 % DMSO, con) for 5 min (n=6 for each WT and KO groups). (B, left panel) Membrane current in aorta smooth muscle cells activated by command voltage changes shown on the upper panel. Scatter plots – baseline current in untreated cells from WT (black circles) and TRPC1 KO (gray circles) mice. Line plots – representative traces acquired after application of phenyephrine (25 uM) to WT (black) or TRPC1 KO (gray) cells. (B, right panel) Average density of phenylephrine activated outward current at 60 mV in WT (n=5) and TRPC1 KO (n=6) smooth muscle cells. (C) Expression of BKCa channel alpha subunit mRNA by real time PCR in aorta from WT and TRPC1 KO mice (n=6 each). Expression is reported relative to β-actin.
Whole cell patch clamp was used to determine the effect of TRPC1 expression on Phe-stimulated BKCa current in freshly isolated aortic SMC. Voltage clamp recordings demonstrated small baseline currents at rest that were similar between WT and TRPC1 KO SMC (Fig 2B, left panel). Subsequent application of 25 μM Phe led to activation of large outward currents in both groups typical of BKCa channels. The activated current could be completely blocked by penitrem A, confirming that it was the result of BKCa channel opening (Supplemental Figure 3). Mean and peak current density (pA/pF) of Phe-stimulated cells at 60 mV was significantly reduced in the TRPC1 KO group (Fig 2B, right panel and Supplemental Figure 3) (P<0.01). From the above studies, it was clear that BKCa channel activation attenuates receptor-mediated vasocontraction in this artery and that this moderation of contraction was notably less in SMC from TRPC1 KO mice.
While the preceding experiments support the model wherein TRPC1 channels facilitate BKCa channel activation, an alternative possibility is that BKCa channel expression is reduced in SMC from TRPC1 KO mice. We therefore examined BKCa channel alpha subunit mRNA expression by real time PCR (Fig 2C). Interestingly, BKCa mRNA expression was reduced by 42% in aorta isolated from TRPC1 KO mice compared with WT (n=6, P=0.047). These data correlate with the observation that the peak Phe-activated BKCa current is decreased in TRPC1 KO SMC (Supplemental Fig 3). Thus, an additional mechanism by which vascular contractility is augmented in TRPC1 KO mice may be through BKCa channel expression. It should be noted that the above two mechanisms are not mutually exclusive.
Role of TRPC1 and TRPC3 in endothelium-dependent relaxation of a compliance artery
Relaxation to a cholinergic agonist, carbachol (CCh), was performed in Phe pre-contracted aorta from WT, TRPC1 KO, and TRPC3 KO mice (Fig 3). Application of CCh produced sustained relaxations in all groups studied, however, relaxations were significantly reduced in both TRPC1 KO and TRPC3 KO groups compared with WT (n=12–20 mice, P<0.02). Mean relaxations were 81±2, 71±3, and 71±3% for WT, TRPC1 KO, and TRPC3 KO respectively.
Fig 3.
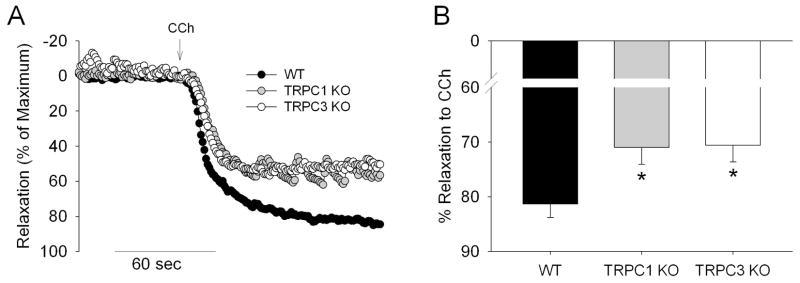
The effect of TRPC1 and TRPC3 gene KO on isometric relaxation of mouse aorta. (A) Representative relaxation recordings of mouse aorta in response to carbachol (CCh, 10 μM) from WT(black), TRPC1 KO (gray) and TRPC3 KO (white circles) mice. (B) Average relaxation to CCh normalized to the maximal constriction to phenylephrine (3 μM). * - p < 0.05 in TRPC1 KO (n = 16) or TRPC3 KO (n = 12) vs. WT group (n = 20).
Role of TRPC1 and TRPC3 channels in the Ca2+ response to CCh in aorta EC
Application of CCh to EC from WT mice demonstrated a dose-dependent increase in intracellular Ca2+ concentration (Supplemental Figure 4) which was largely dependent on Ca2+ influx and completely dependent on PLC activation and IP3 receptor activation. The Ca2+ response to CCh was significantly reduced in the presence of Ca2+-free/EGTA bath solution and also demonstrated Ca2+ influx by the Mn2+ quenching technique (Figure 4A&B). Application of a PLC inhibitor (U73122, 4 μM) or an IP3 receptor blocker (Xestospongin C, 50 μM) resulted in profound inhibition of the Ca2+ response to CCh.
Fig 4.

Dependence of EC CCh-induced Ca response on extracellular Ca and PLC-IP3 signaling pathway. (A) EC were superfused with the regular bath saline (1.6 mM Ca, control) or “0 Ca” saline (Ca-free, 4 mM BAPTA). CCh (100 uM) was applied at the point indicated by the arrow. (B) CCh induced increase in Fura-2 quenching. Fura-2 fluorescence at the F360 was recorded in the presence of 1 mM Mn2+ for 4.5 min before application of CCh to obtain baseline quenching rate. Then CCh (100 μM) was applied (arrow) in the same Mn-containing saline. Dashed line reflects an extension of the basal quenching rate before CCh. (C) Cells were pretreated with 0.1 % DMSO or PLC inhibitor U73122 for 5 min before application of CCh (100 μM). (D) Cells were preincubated with 50 μM XeC of vehicle (5 % DMSO) for 30 min before application of CCh (100 μM).
We next evaluated the specific role of EC TRPC1 and TRPC3 channels in the CCh-mediated Ca2+ signaling pathway. The Ca2+ responses to CCh were significantly decreased in EC from both TRPC1 KO and TRPC3 KO mice (Fig 5A&B). We additionally examined the contribution of TRPC3 to the Ca2+ response using a recently described TRPC3 channel blocker, Ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate (Pyr3) [17]. Treatment with 10 μM Pyr3 resulted in significant inhibition of the CCh response in EC of WT mice. Pyr3 produced a slight inhibition of the peak response in EC of TRPC1 KO mice but no effect on the subsequent sustained response (Fig 5A&B). Similar treatment of EC from TRPC3 KO mice had no effect on the Ca2+ response, demonstrating the specificity of Pyr3 in these experimental conditions (Fig 5C). The Pyr3-mediated inhibitions represented as a percentage of the respective control response (area under the curve) were 51.3±2.1 % (WT), 19.9±4.3 % (TRPC1), and 4.3±5.6 % (TRPC3). Together, these data demonstrate a functional role of TRPC1 and TRPC3 channels in CCh-mediated Ca2+ regulation in aorta EC by gene knockout (TRPC1 and TRPC3) and by selective pharmacological blocker (TRPC3).
Fig 5.
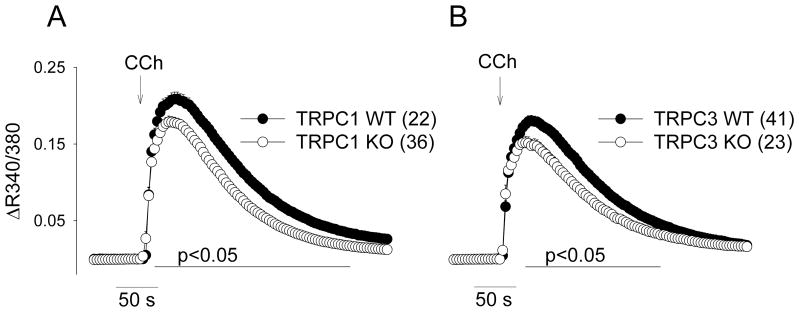
Intracellular Ca2+ responses to CCh in EC from TRPC1 (left) and TRPC3 KO mice (right). Average Ca response to CCh (100 μM) was determined from 22–41 coverslips derived from EC of 3 to 17 mice per group. All cells per a given coverslip were averaged to obtain a single Ca response. All coverslip responses were then averaged and presented as mean±SE. The region where statistical difference between individual points exists between groups (Holm-Sidak test) is indicated by the horizontal line.
Discussion
In the present study we systematically examined the role of TRPC1 and TRPC3 channels in the mechanisms of vasocontraction and vasorelaxation in a major compliance artery using knockout mice. An increased state of active tone of central conduit arteries, such as the aorta, contribute to an increase in systolic blood pressure and pulse wave velocity (PWV) (reviewed in Schiffrin 2004[6]). This greater PWV results in an earlier wave reflection from distal resistance arteries that summates with the anterorgrade wave thus producing an increased peak pressure and in pulse pressure. Reduced compliance of the aorta (through reduced active relaxation mechanisms or exaggerated contractile mechanisms) can therefore contribute to cardiovascular pathologies such as isolated systolic hypertension [6]. In this regard, mechanisms of active relaxation and contraction of the aorta must be considered in the mechanisms of development and progression of hypertension as well as in possible therapeutic strategies in reducing arterial and ventricular stiffening [5].
In the present studies, we report the following novel findings: 1) Vasocontraction is augmented in aorta from TRPC1 mice, 2) Greater aorta contractility from TRPC1 KO mice results from attenuated BKCa channel current, 3) Vasorelaxation is reduced in aorta from both TRPC1 KO and TRPC3 KO mice, and 4) TRPC1 and TRPC3 channels contribute to receptor-mediated Ca2+ influx in EC as determined by gene knockout and a selective TRPC3-specific blocker. These findings are discussed in greater detail below.
Role of TRPC1 and TRPC3 in Vasocontraction
Given the general lack of selective pharmacological tools for studying the TRPC channels, investigators have had to rely on alternate methods to elucidate the role of these channels such as channel blocking antibodies, RNAi knockdown, and gene knockout mice. In the case of TRPC1, many of the studies demonstrating a role for this channel in the vasculature have used overnight incubation with the TRPC1-blocking antibody, T1E3 [18]. Investigations with T1E3 incubation have demonstrated involvement of TRPC1 channels in store-operated Ca2+ entry in SMC [18] as well as isolated caudal artery contraction to ET-1 [19]. The role in ET-1-mediated contraction is not universal though, as the blocking antibody had no effect on the contraction in basilar artery from the same study. RNAi methods of knockdown applied to isolated SMC have also indicated a role for the TRPC1 channel in Ca2+ entry [20]. While still quite limited in number, the aforementioned studies implicate the TRPC1 channel in SMC Ca2+ regulation and receptor-mediated vascular contractility.
In TRPC1 KO mice, the role for TRPC1 channels has been less apparent. While TRPC1 is the most abundantly expressed TRPC mRNA in aorta and cerebral artery, there is only one previous study to date which examined agonist-mediated constriction. In this study, Schmidt et al found that KCl and Phe-mediated constriction of pressurized carotid artery was not altered by TRPC1 gene deletion [4]. In contrast to these findings in carotid artery, we found significantly greater vascular contractility to both agents in aorta. One possible reason for the difference between these two studies may involve the relative role of TRPC1 channels in the activation of BKCa channels between artery types. Kwan et al. recently described a mechanism in which TRPC1 channels contribute to BKCa channel activation in vascular SMC during stimulation by contractile agonists [16]. In this mechanism, BKCa channel activation provides a SMC hyperpolarizing current that attenuates the contractile response. The authors found that SMC depolarization and artery contraction were greater when treated with BKCa channel blockers or the TRPC1 blocking antibody, T1E3. Thus, if TRPC1 similarly contributes to BKCa activation in our study, we would expect to see, as we did, greater contraction in aorta from TRPC1 mice. In addition, we demonstrated that contractility of aorta from WT mice could be increased to the level of TRPC1 KO mice by inhibition of BKCa channels. Furthermore, Phe-stimulated currents, both mean and peak, were significantly reduced in SMC from TRPC1 KO mice. If this model of TRPC1 regulation of contractility through BKCa activation is correct, it would explain how loss of a channel involved in Ca2+ influx (or release) can seemingly paradoxically lead to greater contraction. Schmidt et al. also evaluated the potential for TRPC1 to regulate activity of BKCa channels in freshly isolated cerebral artery SMC. They determined that frequency and amplitude of Ca2+ sparks and spontaneous transient outward currents (STOCs) were not different in resting SMC isolated from TRPC1 KO mice and thus concluded that BKCa channel activity was not affected by absence of TRPC1 [4]. However, it should be noted that we did not find a difference in BKCa current in resting conditions either; agonist stimulation was required to demonstrate a difference between aorta SMC from WT and TRPC1 KO mice.
While our results are consistent with the above model proposed by Kwan et al. [16], we also found evidence for an alternative mechanism in which BKCa channel expression is reduced in aorta SMC of TRPC1 KO mice. We are unaware of any prior report of TRPC1 channels and BKCa channel expression. However, there is recent evidence for another TRPC family channel (TRPC6) in trafficking BKCa channels to the plasma membrane [21]. This finding raises the intriguing, though speculative, possibility that TRPC1 may also play a role in BKCa channel trafficking or regulation of expression. It should be pointed out that BKCa trafficking or expression would not appear to explain the findings in the study by Kwan et al., as they obtained functional results with an acute (1 hour) incubation with a pore blocking TRPC1 antibody. In our study, either or both mechanisms may contribute to TRPC1 regulation of vascular contractility.
Lastly, while BKCa channel expression (mRNA) and function (patch clamp and isolated vessels) are reduced in the smooth muscle of TRPC1 KO mice, these differences alone cannot completely account for the difference in contractility between groups. It is likely that additional differences must exist such as voltage-operated Ca2+ channel expression/function, calcium sensitivity, etc.
Role of TRPC1 and TRPC3 in EC-mediated Vasorelaxation and Ca2+ Regulation
TRPC channels are functionally expressed in EC and have been demonstrated to contribute to EC Ca2+ regulation [22,23]. These channels are believed to promote vasorelaxation through increases in endothelial calcium, with subsequent activation of nitric oxide (NO), prostacyclin, or endothelium-dependent hyperpolarization (EDH). In the earliest study of TRPC channels in vascular function, Freichel et al. demonstrated a critical role for TRPC4 channels in store-operated Ca2+ entry in mouse EC [24]. They found that Ca2+ entry following store depletion was significantly diminished in EC of TRPC4 KO mice. Furthermore, the authors demonstrated that knockout of TRPC4 resulted in approximately 50% reduction in ACh-mediated vasorelaxation of isolated aorta. While this seminal study demonstrated that the bulk of store-operated Ca2+ entry occurs via TRPC4 channels, it did leave the question of whether other TRPC channels participate in the mechanism of agonist-stimulated Ca2+ entry.
Our present study demonstrates a role for TRPC3 channels in CCh-mediated vasorelaxation. To our knowledge, this study provides the first evidence of TRPC3 channels in the mechanism of EC-dependent vasorelaxation using a knockout model. We subsequently demonstrated that TRPC3 contributes to EC Ca2+ regulation using TRPC3 KO mice as well as a newly described TRPC3 channel blocker, Pyr3. Interestingly, the effect of Pyr3 on EC from WT mice was greater than the effect of TRPC3 gene deletion. The greater inhibition with Pyr3 was not due to non-specific actions of the blocker, as it had no effect in EC from TRPC3 KO mice. One possible explanation for the greater effect of Pyr3 could involve blocking TRPC3 in the context of a heteromeric channel. TRPC3 has the potential to form functional channels with several other TRPC family members including TRPC1, TRPC6, and TRPC7 [25][11,26,27]. Application of Pyr3 might thus block TRPC3-containing heteromeric channels in addition to any TRPC3 homomeric channels. In contrast, gene deletion would eliminate the TRPC3 protein but would not necessarily prevent a binding partner from forming a homomeric channel or a heteromeric channel with another TRPC protein.
Our data similarly showed a role for TRPC1 channels in CCh-mediated vasorelaxation and EC Ca2+ regulation. Few prior studies have examined the role of TRPC1 in vasorelaxation. In one recent study, TRPC1 was shown to be involved in endothelial-mediated flow-induced dilation, likely as a part of a heteromeric channel with TRPV4. In this study, Ma et al. additionally demonstrated that induction of luminal flow was associated with an increase in EC Ca2+ that was diminished following incubation with a TRPC1 blocking antibody [28]. In contrast to the positive role in flow-induced dilation, one recent study using TRPC1 KO mice found an inhibitory role for TRPC1 in agonist-mediated vasorelaxation. Schmidt et al. found that EDH-mediated dilation to ACh was actually enhanced in pressurized carotid artery and cremaster microcirculation of TRPC1 KO mice [4]. Although the mechanism by which dilation was enhanced was not determined, the authors ascribed a role for TRPC1 as a negative regulator specifically of the EDH-mediated pathway. In this same study, the NO-mediated dilation response (by NO donor or ACh stimulation) was unaffected by the loss of TRPC1 channels. While it is not presently clear how to account for the difference between our results and those of Schmidt et al., one potential explanation is that loss of TRPC1 produces different results in arteries that utilize EDH-dependent vasodilation versus those that do not. EDH-mediated vasodilation depends on activation of small and intermediate conductance KCa (SK/IKCa) channels to produce endothelial hyperpolarization that is then transferred to the surrounding smooth muscle layers [29]. A reduction in the magnitude of EC hyperpolarization would result in less smooth muscle hyperpolarization and thus less vasodilation [30]. Schmidt et al. report that depolarizing current is reduced in EC of TRPC1 KO mice, which permits greater hyperpolarization and thus greater EDH-mediated vasodilation [4]. The aorta, however, does not utilize EDH-mediated relaxation and rather relies completely on NO-mediated relaxation [31]. The NO-mediated mechanism depends principally on EC Ca2+ which is unaffected by membrane potential in intact endothelium [32][33]. Thus, larger arteries that do not employ the EDH-dependent mechanism, such as aorta, would not be adversely affected by TRPC1-mediated depolarization of the EC and therefore would not be expected to have greater relaxation upon loss of TRPC1.
In summary, we show that TRPC1 but not TRPC3 contributes to vasocontraction in mouse aorta. Our data support the role of SMC TRPC1 as an attenuator of agonist-mediated contraction through expression or activation of membrane BKCa channels. Thus, loss of TRPC1 channels results in greater aorta contraction. In contrast to TRPC1, TRPC3 does not play a role in constriction, as knockout of TRPC3 failed to alter either agonist-mediated vasocontraction to Phe or non-specific depolarization with KCl. In the mechanism of vasorelaxation, both TRPC1 and TRPC3 contribute to the CCh-mediated response in aorta through contributions in EC calcium regulation. In light of the known plasticity of TRPC channel expression, these conclusions regarding calcium regulation will need to be followed up in native endothelial cells. In total, these findings support a role for TRPC1 in attenuating smooth muscle contraction and a role for TRPC1 and TRPC3 in EC calcium regulation and vasorelaxation.
Supplementary Material
Fig 6.
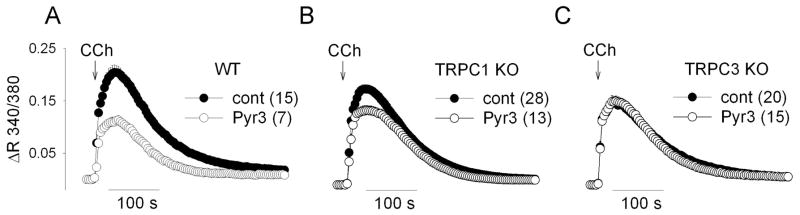
The effect of a TRPC3 channel blocker (Pyr3) on the CCh-induced Ca response in EC from WT, TRPC1 KO, and TRPC3 KO mice. Cells were pretreated with Pyr3 for 10 min before the recording after which Pyr3 was continuously present in the bath. DMSO (0.1 %) was used as a vehicle control. CCh (100 μM) was applied at the time indicated by arrow.
Acknowledgments
Sources of Funding: NIH R01 HL088435 (Sean Marrelli).
Footnotes
Disclosures: None.
References
- 1.Earley S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca(2+)-activated K(+) channels, J Cardiovasc Pharmacol. 2011;57:148–153. doi: 10.1097/FJC.0b013e3181f580d9. [DOI] [PubMed] [Google Scholar]
- 2.Clapham DE, Julius D, Montell C, Schultz G International Union of Pharmacology. XLIX Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich A, Kalwa H, Storch U, Mederos y Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt K, Dubrovska G, Nielsen G, Fesus G, Uhrenholt TR, Hansen PB, Gudermann T, Dietrich A, Gollasch M, de Wit C, Kohler R. Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br J Pharmacol. 2010;161:1722–1733. doi: 10.1111/j.1476-5381.2010.00985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kass DA. Ventricular arterial stiffening: integrating the pathophysiology. Hypertension. 2005;46:185–193. doi: 10.1161/01.HYP.0000168053.34306.d4. [DOI] [PubMed] [Google Scholar]
- 6.Schiffrin EL. Vascular stiffening and arterial compliance. Implications for systolic blood pressure. Am J Hypertens. 2004;17:39S–48S. doi: 10.1016/j.amjhyper.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 7.Cavalcante JL, Lima JA, Redheuil A, Al-Mallah MH. Aortic stiffness: current understanding and future directions. J Am Coll Cardiol. 2011;57:1511–1522. doi: 10.1016/j.jacc.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suh SH, Vennekens R, Manolopoulos VG, Freichel M, Schweig U, Prenen J, Flockerzi V, Droogmans G, Nilius B. Characterisation of explanted endothelial cells from mouse aorta: electrophysiology and Ca2+ signalling. Pflugers Arch. 1999;438:612–620. doi: 10.1007/s004249900085. [DOI] [PubMed] [Google Scholar]
- 10.Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, Richardson CD, Kawate T, Kuno J, Weinstein BM, Stainier DY, Sato TN. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 11.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 12.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 13.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 14.Chen J, Crossland RF, Noorani MM, Marrelli SP. Inhibition of TRPC1/TRPC3 by PKG contributes to NO-mediated vasorelaxation. Am J Physiol Heart Circ Physiol. 2009;297:H417–24. doi: 10.1152/ajpheart.01130.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich A, Mederos Y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwan HY, Shen B, Ma X, Kwok YC, Huang Y, Man YB, Yu S, Yao X. TRPC1 associates with BK(Ca) channel to form a signal complex in vascular smooth muscle cells. Circ Res. 2009;104:670–678. doi: 10.1161/CIRCRESAHA.108.188748. [DOI] [PubMed] [Google Scholar]
- 17.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circ Res. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- 19.Bergdahl A, Gomez MF, Wihlborg AK, Erlinge D, Eyjolfson A, Xu SZ, Beech DJ, Dreja K, Hellstrand P. Plasticity of TRPC expression in arterial smooth muscle: correlation with store-operated Ca2+ entry. Am J Physiol Cell Physiol. 2005;288:C872–C880. doi: 10.1152/ajpcell.00334.2004. [DOI] [PubMed] [Google Scholar]
- 20.Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur J Pharmacol. 2008;583:135–147. doi: 10.1016/j.ejphar.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol. 2009;75:466–477. doi: 10.1124/mol.108.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang DX, Gutterman DD. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011;57:133–139. doi: 10.1097/FJC.0b013e3181fd35d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietrich A, Gudermann T. TRP channels in the cardiopulmonary vasculature. Adv Exp Med Biol. 2011;704:781–810. doi: 10.1007/978-94-007-0265-3_41. [DOI] [PubMed] [Google Scholar]
- 24.Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol. 2001;3:121–127. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 25.Zagranichnaya TK, Wu X, Villereal ML. Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in HEK-293 cells. J Biol Chem. 2005;280:29559–29569. doi: 10.1074/jbc.M505842200. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Bandyopadhyay BC, Singh BB, Groschner K, Ambudkar IS. Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel. Heteromeric assembly of TRPC1-TRPC3. J Biol Chem. 2005;280:21600–21606. doi: 10.1074/jbc.C400492200. [DOI] [PubMed] [Google Scholar]
- 27.Goel M, Sinkins WG, Schilling WP. Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem. 2002;277:48303–48310. doi: 10.1074/jbc.M207882200. [DOI] [PubMed] [Google Scholar]
- 28.Ma X, Qiu S, Luo J, Ma Y, Ngai CY, Shen B, Wong CO, Huang Y, Yao X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler Thromb Vasc Biol. 2010;30:851–858. doi: 10.1161/ATVBAHA.109.196584. [DOI] [PubMed] [Google Scholar]
- 29.Bryan RM, Jr, You J, Golding EM, Marrelli SP. Endothelium-derived hyperpolarizing factor: a cousin to nitric oxide and prostacyclin. Anesthesiology. 2005;102:1261–1277. doi: 10.1097/00000542-200506000-00028. [DOI] [PubMed] [Google Scholar]
- 30.Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res. 2000;87:474–479. doi: 10.1161/01.res.87.6.474. [DOI] [PubMed] [Google Scholar]
- 31.Beny JL, Nguyen MN, Marino M, Matsui M. Muscarinic receptor knockout mice confirm involvement of M3 receptor in endothelium-dependent vasodilatation in mouse arteries. J Cardiovasc Pharmacol. 2008;51:505–512. doi: 10.1097/FJC.0b013e31816d5f2f. [DOI] [PubMed] [Google Scholar]
- 32.Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol Heart Circ Physiol. 2003;285:H1590–H1599. doi: 10.1152/ajpheart.00376.2003. [DOI] [PubMed] [Google Scholar]
- 33.Cohen KD, Jackson WF. Membrane hyperpolarization is not required for sustained muscarinic agonist-induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation. 2005;12:169–182. doi: 10.1080/10739680590904973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.