

Abstract
Metabolic syndrome has been widely associated with an increased risk for acute cardiovascular events. Emerging evidence supports metabolic syndrome as a condition favoring an adverse cardiac remodeling, which might evolve towards heart dysfunction and failure. This pathological remodeling has been described to result from the cardiac adaptive response to clinical mechanical conditions (such as hypertension, dyslipidemia, and hyperglycemia), soluble inflammatory molecules (such as cytokines and chemokines), as well as hormones (such as insulin), characterizing the pathophysiology of metabolic syndrome. Moreover, these cardiac processes (resulting in cardiac hypertrophy and fibrosis) are also associated with the modulation of intracellular signalling pathways within cardiomyocytes. Amongst the different intracellular kinases, mitogen-activated protein kinases (MAPKs) were shown to be involved in heart damage in metabolic syndrome. However, their role remains controversial. In this paper, we will discuss and update evidence on MAPK-mediated mechanisms underlying cardiac adverse remodeling associated with metabolic syndrome.
1. Introduction
The prevalence of metabolic syndrome is rapidly increasing in the western world [1]. Metabolic syndrome has been defined as a cluster of multiple disorders including insulin resistance, abdominal obesity, dyslipidemia, increased blood pressure, hypercholesterolemia, and proinflammatory state [2]. Several definitions have been historically proposed during the last decades, also including oxidative stress, leptin resistances and endothelial dysfunction as key pathophysiological mechanisms contributing to the increase of cardiovascular risk that affect metabolic syndrome patients [2, 3]. Considering these paradigms, it is clear that the metabolic syndrome is a fully heterogeneous construction, raising important scientific limitations for meta-analyses of clinical investigations. Although some common components (such as dyslipidemia, hypertension, and hyperglycemia) are recurrent in the different definitions [4], these other disorders might represent an important variable in the analysis of different cohorts. Several conditions included in the metabolic syndrome have been shown as strongly associated with an acceleration of atherogenesis and an increased incidence of acute ischemic events [1]. In addition, some processes (mainly systemic insulin resistance and inflammation) have been proposed to contribute to physiological organ remodelling and pathological damage in metabolic syndrome. In particular, different chronic adverse heart remodeling and the development of liver steatosis have been widely described [5, 6]. In this paper, we will focus on the pathophysiology of heart remodelling and damage during metabolic syndrome. Metabolism syndrome patients affected by diabetes are associated with altered myocardial substrate metabolism, which has emerged as an important contributor to the development of cardiomyopathy [7]. In diabetes and concomitant metabolic syndrome, an increased cardiac fatty acid metabolism and reduced glucose metabolism have been reported [7]. Although initially profitable, the rate of fatty acid uptake reaches a point where it exceeds the rate of fatty acid oxidation, thereby promoting the accumulation of lipids, resulting in lipotoxicity and associated cardiac dysfunction [8]. This leads to some complications, such as cardiac hypertrophy, which is identified as a cardiac pathological remodeling. However, cardiac remodeling does not necessarily refer to pathological adaptation of the myocardium. Indeed, short-term compensatory mechanisms are beneficial for the heart because it adapts cardiac output to physiological or pathological loading conditions such as exercise, hypertension, or aortic stenosis. In contrast, sustained overload leads to maladaptive and detrimental remodeling, as reported in detail by Buckberg and coworkers [9]. Altogether, these studies suggest that cardiac remodeling in metabolic syndrome depends on the different component disorders and might not be a disease per se but rather an adaptive response.
Several intracellular signaling pathways, continuously sensing the extracellular stimuli and modulating the different intracellular responses, have been investigated to characterize their role in cardiomyocyte modifications and potential injury associated with metabolic syndrome. Mitogen-activated protein kinases (MAPKs) are cytosolic signaling proteins that become activated after specific phosphorylation [10]. In response to wide extracellular stimuli, MAPKs have been shown to modulate various cellular processes, such as cell growth and cell size regulation [11]. Although not specifically performed in models of metabolic syndrome, in vitro studies using isolated cardiomyocytes have shown that MAPKs might be involved in cardiac hypertrophy via three traditional phases: (i) the activation of specific transmembrane proteins; (ii) intracellular signal transduction; (iii) the activation of cytosolic and nuclear events [12]. Since cardiac hypertrophy is characterized by increased cell size, it has been suggested that MAPKs might play a critical role in cardiac remodeling in hypertensive patients with metabolic syndrome. This appears to be achieved by modulating the activity of numerous transcription factors that target specific genes involved in structural response of the myocardium. In this paper, we will provide an overview on the role of MAPKs in the adverse cardiac remodeling that is associated with metabolic syndrome.
2. Different Structural Adverse Cardiac Remodeling in Metabolic Syndrome
Diagnostic criteria of metabolic syndrome (as indicated by ATP III classification [24]) have been reported to independently predict the development of diastolic dysfunction and cardiac hypertrophy [13, 19, 20] (Table 1). Cardiac hypertrophy is commonly defined as an increase in heart size or more particularly as an increase in ventricular size with or without increased wall thickness relative to body size [25]. This cardiac modification was shown to be a common alteration in subjects with different stages of obesity, which is one of the central features in metabolic syndrome [15, 16, 26–28]. These studies have used noninvasive methods such as echocardiography and magnetic resonance imaging (MRI) to assess cardiac adaptations in obese patients. The results indicate that obesity is associated with a high prevalence of cardiac hypertrophy, characterized by an enhancement of left ventricular cavity size as well as wall thickness. Moreover, it has been observed that wall thickness was increased to greater extent than left ventricular cavity size, revealing a concentric instead of eccentric cardiac hypertrophy. Indeed, computed tomography and MRI demonstrated that fat tissue deposits are well detectable within the heart of obese subjects and commonly accumulate anterior to the right ventricle [29–31]. Although noninvasive methods are useful to characterize cardiac size, structure, and function, they have limitations; in that they do not allow analyzing the biochemical composition of the hypertrophic heart in metabolic syndrome. Interestingly, postmortem studies confirmed the presence of cardiac hypertrophy and cardiac fat tissue deposits in obese patients [17, 18]. The amount of epicardial fat has been reported to be correlated with both visceral fat and the severity of ventricular hypertrophy [14]. Several studies have been conducted in animal models to better understand the cardiac adaptation in diabetes [32, 33]. Diabetes was associated with an increase in left ventricular internal dimension during diastole (LVIDD) and systole (LVIDS) in rats [34]. This adverse cardiac remodeling was independent of hypertension [34]. These results are consistent with a previous study, demonstrating that diabetic rats develop an eccentric left ventricular hypertrophy associated with a decreased cardiac systolic function, and related to impaired collagen turnover [35].
Table 1.
Different disorders in metabolic syndrome are associated with cardiac structural and functional changes.
| Metabolic syndrome characteristic | Adverse cardiac remodelling | Reference | Method of assessment |
|---|---|---|---|
| Obese women have higher end-diastolic septal and posterior wall thickness, left ventricle mass, and relative wall thickness than nonobese | [13] | Echocardiography and tissue Doppler imaging | |
| Obesity | Uncomplicated severe obesity is associated with adapted and appropriate changes in cardiac structure and function | [14] | Echocardiography |
| Reduced left ventricle systolic and diastolic function and increased myocardial reflectivity characterize obese patients as compared to referents | [15] | Transthoracic echocardiography, myocardial Doppler-derived systolic and early diastolic velocity, strain and strain rate imaging, and tissue characterization with cyclic variation and calibrated integrated backscatter | |
|
| |||
| Diabetes, fasting glucose, and fasting insulin levels are associated with left ventricular hypertrophy | [16] | Echocardiography and laboratory testing | |
| Diabetes | Increased heart size in obese men | [17] | Autopsy |
| Postmortem analysis of obese patients that died from gastric bypass complication revealed cardiac hypertrophy | [18] | Autopsy | |
|
| |||
| Hypertension | Left ventricle mass is positively associated with the number of metabolic risk factors in normotensive and hypertensive participants | [19] | Echocardiography |
|
| |||
| Increased left ventricular mass and reduce left ventricular relaxation | [20] | Echocardiography was used to assess pulse-wave Doppler and tissue Doppler imaging | |
|
Metabolic syndrome (defined as a cluster of all previously cited disorders [2]) |
Ventricular diastolic dysfunction, mean left ventricular mass, and left ventricular diameter significantly increase with the number of features of the metabolic syndrome | [21] | Structured clinical interview with a physician, ECG and a transthoracic M-mode, and 2D echocardiogram |
| High levels of IL-6 that could be observed in metabolic syndrome induce cardiac fibrosis | [22] | Blood-perfused isolated heart | |
| Cardiotrophin-1 treatment, mimicking the upregulated level found in metabolic syndrome, induces cardiac fibrosis | [23] | Echocardiography, Doppler, and echo tracking device and ex vivo approach | |
The Prospective Cardiovascular Münster (PROCAM) study, recruiting a cohort of 2754 males aged 40–65 years over a four-year period, showed that patients, who had either hypertension or diabetes, had a 2.5-fold increased risk of cardiovascular morbidity. However, when developing both, patients had an eightfold increase in cardiovascular risk. This multiplicative relevance was confirmed by a twentyfold increase of cardiovascular risk in patients with concomitant diabetes mellitus, hypertension, and abnormal lipid profile [36]. Since the metabolic syndrome might include all these disorders by definition, it is easy to understand that the alterations of an atherosclerotic targeted organ, such as the myocardium, might result from both acute and chronic ischemia. However, also in the absence of traditional atherosclerotic complications, the measurement of diastolic function worsened progressively during metabolic syndrome [13], indicating an adverse cardiac remodeling independent of cardiac necrosis and potentially related to the soluble mediators increased during the disease. In line with these results, different studies have confirmed the development of left ventricular diastolic dysfunction in subjects presenting metabolic syndrome [20, 21, 37, 38].
Interestingly, hypertensive patients with concomitant metabolic syndrome have been shown to present increased left ventricular mass and wall thickness as compared to patients exclusively affected by hypertension [39]. Furthermore, the authors showed that metabolic syndrome might induce an adverse cardiac remodeling via different pathophysiological mechanisms with a multiplicative effect. Metabolic syndrome patients had not only abnormal diastolic left ventricular relaxation, but also increased cardiac hypertrophy [40]. Cardiac hypertrophy predisposes individuals to cardiac arrhythmias, congestive heart failure, and diastolic dysfunction [41]. Consistently, several evidences have revealed a positive association between the metabolic syndrome and the severity of left ventricular hypertrophy [39, 40, 42]. Indeed, metabolic syndrome induces abnormal loading, which may favor the left ventricular hypertrophy. Since pathological cardiac hypertrophy has been associated with sudden cardiac death, heart failure, and stroke [43], it was proposed that this cardiac alteration might further increase cardiovascular risk in metabolic syndrome [44].
Increased myocardial fibrosis and stiffness have been also observed in animal models of obesity and metabolic syndrome [45]. Since collagen and fibrosis determine tissue compliance, cardiac deposition of this protein might promote left ventricular diastolic dysfunction and negatively affect diastolic function [46, 47]. Several other molecular mechanisms (i.e., insulin resistance aggravating asymptomatic myocardial inflammation and association between visceral obesity and myocardial adiposity) have also been suggested to potentially induce such cardiac structural alterations [48, 49].
Most importantly, the renin-angiotensin-aldosterone (RAA) system might also be involved cardiac fibroblast proliferation and collagen synthesis, thereby increasing cardiac fibrosis [50]. The active role for RAA system was confirmed by the finding that pharmacologic inhibition of this pathway ameliorates heart failure [51, 52], also in metabolic syndrome patients [53]. Thus, these studies suggest that RAA system could be a critical player underlying cardiac modifications in hypertensive metabolic syndrome patients.
3. Pathophysiological Mediators of Adverse Cardiac Remodeling in Metabolic Syndrome
Cardiac structural remodeling in metabolic syndrome might be due to different pathological triggers. Hypertrophic growth accompanies heterogeneous metabolic syndrome disorders, including not only diabetes and hypertension, but also coronary heart disease and ischemic cardiac remodeling. Mechanical alterations have been classically described as major causes inducing an adverse cardiac remodelling [54]. However, metabolic syndrome has been associated with left ventricular hypertrophy [40] independently of ischemic cardiac remodeling. Given the upregulation of several hormones and cytokines in metabolic syndrome patients [55], a potential role on cardiac remodeling has been proposed for these molecules [56]. In particular, a hormonal and inflammatory dysregulation might contribute to the development of cardiac hypertrophy and fibrosis [55]. For instance, increased aldosterone plasma levels in patients with metabolic syndrome [57, 58] might be directly associated with the development of left ventricular hypertrophy [59] or cardiac fibrosis [60]. Although clinical studies have reported that aldosterone induces left ventricular hypertrophy [61], the mechanism by which aldosterone promotes cardiac hypertrophy remains unclear. Okoshi and coworkers showed that aldosterone directly induced cardiac hypertrophy and atrial natriuretic peptide (ANP) mRNA expression (a molecular marker of cardiac hypertrophy) in neonatal rat ventricular myocytes [62]. These results were accompanied by enhanced activation of ERK1/2- and JNK-mediated pathways. This critical role of the ERK pathway in the development of cardiac hypertrophy (in response to endothelin-1) was also confirmed by the in vitro abrogation of cardiomyocyte hypertrophy in the presence of the pharmacological inhibitor of ERKs [63, 64].
On the other hand, aldosterone has been shown as a potent inducer of cardiac fibrosis [60, 65, 66]. Therefore, emerging evidence indicates a crucial role for aldosterone in maladaptive cardiac remodeling in metabolic syndrome [67, 68].
Considering the hypothesized inflammatory etiology of the metabolic syndrome [69], it was proposed that elevated circulating levels of cytokines, adipocytokines, and chemokines might actively regulate cardiac remodeling and via the activation of inflammatory signaling pathways [70–72]. Amongst several mediators, tumor necrosis factor (TNF) and interleukin-6 (IL-6) were shown to promote both insulin resistance [73] and cardiac hypertrophy [74], suggesting that inflammation in metabolic syndrome might be a central feature in atherogenesis as well as in cardiac hypertrophy. In particular, TNF expression, which was shown to be increased in response to pressure overload in the adult heart [75], might be one of the most important mediators of cardiac hypertrophy in metabolic syndrome with hypertension [76]. The molecular pathways potentially involved in metabolic syndrome-induced cardiac hypertrophy have been only partially investigated. To summarize, mechanical stress remains the main responsible of adverse cardiac remodelling in metabolic syndrome, such as hypertension. However, considering the potential dysregulation of inflammatory and hormonal systems, the cardiac pathophysiology in metabolic syndrome might be partially influenced also by these soluble molecules. In the following sections, we will review the pathophysiological role of MAPKs in cardiac remodeling in a general context and also particularly in metabolic syndrome.
4. Role of MAPK in Cardiac Remodeling
4.1. Extracellular Signal Regulated Kinases (ERKs)
One of the most studied MAPK pathways is the Ras/Raf/ERKs pathway. Extracellular stimuli such as stress or hormones activate diverse receptors at the cell surface, driving intracellular recruitment and activation of the guanosine triphosphate (GTP) small G-protein (Ras). This, in turn, induces Raf-1 kinase translocation to the plasma membrane and Raf-1-mediated phosphorylation of MEK proteins (MEK1 and 2). Thereafter, ERKs are activated by MEKs and regulate a large number of nuclear and cytosolic proteins [77] that directly modulate numerous intracellular processes.
For instance, pressure overload was shown to influence ERK-mediated intracellular signaling as well as extracellular matrix deposition within the heart [78]. In response to chronic pressure overload, cardiomyocytes start to grow leading to heart enlargement and hypertrophy. Pressure overload induced by transverse aortic constriction (TAC) in rodents was shown to mediate hypertrophic effect through ERK activation [79]. In addition, Esposito and coworkers showed that TAC procedure is associated with the activation of all three major MAPKs (ERK1/2, p38 MAPK, and JNK) in mice [80]. Consistent with the animal studies, clinical researches reported increased cardiac activation of ERK1/2, JNK, and p38 MAPK in failing human hypertrophic hearts [81]. In line with these results, ERK1/2 activation has been shown as a key element directly promoting cardiac hypertrophy [26, 82].
It was proposed that ERK1/2 induces the activation of various transcription factors by modulating their phosphorylation level, hence leading to hypertrophy. Indeed, using a model of phenylephrine-induced cardiac myocyte hypertrophy, Babu and coworkers revealed that ERK1/2 pathway is involved in Elk-1 upregulation in a model of phenylephrine-mediated hypertrophy [83]. Therefore, these studies suggest a central role for ERK in the pathophysiological development of cardiac hypertrophy. The molecular mechanisms downstream of this pathway remains to be clearly defined.
4.2. Janus Kinases (JNKs)
The hypertrophic effects of JNKs are still controversial. Wang and coworkers showed that specific activation of the JNK pathway was associated with hypertrophy in neonatal cardiomyocytes overexpressing MEK7 [84]. In line with these results, transfection of ventricular myocytes with MEK1 (a MAPK-activating JNK) leads to cardiac hypertrophy via JNK activation [85]. By contrast, transgenic mice selectively overexpressing MEK7 in the cardiac tissue were not shown to develop cardiac hypertrophy despite JNK1 and JNK2 upregulation [86]. Although these mice died from congestive heart failure, they had normal cardiomyocytes size and they did not develop ventricular hypertrophy. Importantly, mice overexpressing MEK7 also presented diastolic dysfunction and paradoxically high levels of ANF mRNA, which is considered a marker for cardiac hypertrophy [86]. The reduction in connexin 43 and gap junctions between ventricular cardiomyocytes might explain the absence of hypertrophy in the presence of increased ANF expression [86]. Also in the case of JNK, the exact mechanisms through which it regulates cardiac hypertrophy remains poorly determined.
4.3. p38 MAPK
In addition to JNK and ERK1/2, p38 MAPK was also intensively investigated. There are at least four isoforms of p38 MAPK that have been involved in cardiac remodeling and inflammation [87]. This signaling pathway is predominantly involved in the inflammatory response, and it can be activated by proinflammatory cytokines, chemokines, and hormones [88–90]. MEK 3, 4, and MEK 6 are the upstream kinases that directly activate p38MAPK [82]. Several downstream transcription factors have been identified as potential p38MAPK substrates, including activating transcription factor-1 (ATF-1), ATF-2, Elk-1, serum response factor (SRF), growth arrest, and myocyte enhance factor 2C (MEF 2C) [91–93]. Since a large amount of studies has been performed in vitro in neonatal rat cardiomyocytes, the role of p38 MAPK in cardiac clinical modifications remains to be confirmed. Nevertheless, p38 MAPK has been implicated in the regulation of both cardiac growth and hypertrophy [94]. Indeed, the inhibition of the p38 MAPK pathway via pharmacological inhibitors or adenovirus blunted the hypertrophic effect associated with p38 MAPK activation [94–96]. These hypertrophic effects of p38 MAPK phosphorylation in cardiomyocytes are also supported by another study where specific activation via adenovirus in ventricular muscle cells induced cardiac hypertrophy [84].
By contrast, Choukroun and coworkers have shown that p38 MAPK is not required for agonist-induced hypertrophy in cardiomyocytes [98]. To determine the role of the p38 MAPK pathway in the response to endothelin-1 (ET-1, a hypertrophic agent), neonatal rat cardiomyocytes were concomitantly treated with ET-1 and the selective p38 MAPK inhibitor, SB203580. The results revealed that this inhibitor had no effect on ET-1-induced hypertrophy, hence suggesting that p38 MAPK activation may not be required during cardiac cell hypertrophy [98]. Since these preliminary studies present some limitations (due to the use of the cardiomyocyte model [immature neonatal cells] and the modest specificity of pharmacological inhibitor, that can inhibit also other intracellular pathways), a genetic approach was also performed. Transgenic mice specifically overexpressing MEK 3 and MEK 6 (upstream activators of p38 MAPK) in the heart do not exhibit cardiac hypertrophy, despite the development of ventricular wall thinning and premature death with signs of congestive heart failure [99].
Taken together, these basic research studies suggest that p38 MAPK, ERKs, and JNKs may all be involved in promoting cardiac hypertrophy (Figure 1). In particular, p38 MAPK activation might also contribute to cardiac fibrosis, while ERK and JNK activation might promote cardiomyocyte growth and defects in gap junctions, respectively. The activation of a single MAPK-mediated cascade might not be sufficient to determine a clinically relevant adverse cardiac remodeling.
Figure 1.
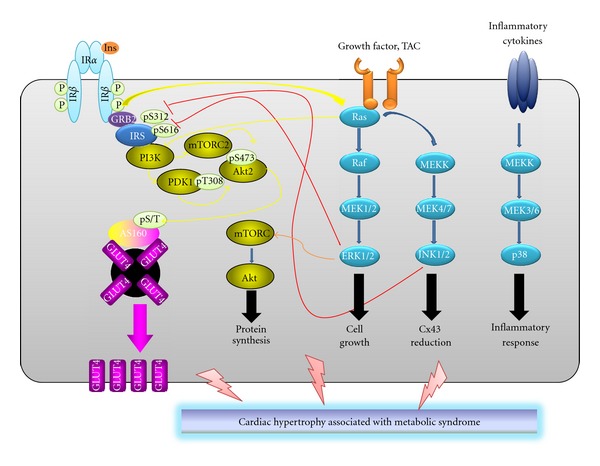
Intracellular pathways mediating hypertrophic cardiac remodelling in metabolic syndrome. Intracellular pathways mediated by phosphatidylinositol 3-kinase (PI3-K) activation play a pivotal role in insulin-mediated glucose transport in cardiomyocytes. Metabolic syndrome is associated with impaired intracellular signaling that could be activated by various stimuli, including inflammatory cytokines. These pathways, mainly dependent on MAPK activation (composed of extracellular signal-regulated kinase [ERK], c-Jun N-terminal kinase [JNK], and p38 MAPK), could be also triggered by chirurgical manipulation (such as transverse aortic constriction [TAC]). MAPK activation might be considered as a critical mechanism aggravating cardiac hypertrophy as well as cardiomyocyte insulin resistance in metabolic syndrome.
5. Potential Role of MAPK in Metabolic Syndrome-Related Cardiac Adverse Remodeling
As suggested above, the alterations in cardiac MAPK activation might be induced in metabolic syndrome by insulin resistance and abnormal inflammation. The association between these molecular dynamics was so relevant that an altered MAPK activation pattern might also be a potential cause of hyperinsulinemia [100]. This pathophysiological role of MAPK in metabolic syndrome-induced heart remodeling was confirmed for JNK activation that was associated with both developments of insulin resistance and cardiac hypertrophy in metabolic syndrome [82, 101]. Assessing the role of ERKs in metabolic syndrome cardiac remodeling is much more complicated since ERK2-knockout mice are not viable [102, 103]. On the other hand, ERK1 knockout mice are viable and fertile. Thus, ERK1-deficient mice were investigated, and they were shown to be protected from diet-induced obesity and insulin resistance [104]. Nonetheless, mice lacking the ERK1 negative regulator p62 presented altered metabolism with increased adipogenesis, reduced energy expenditure, and reduced insulin sensitivity [105]. However, these animals were not investigated on concomitant cardiac remodeling and might be considered a good model to assess metabolic syndrome adverse heart remodeling.
Inflammatory mediators such as TNF are elevated in metabolic syndrome [106]. Condorelli and coworkers showed that Akt and the JNK MAPK mediate TNF-induced hypertrophy in cultured cardiomyocytes [107]. TNF-mediated eccentric cardiac hypertrophy in response to intermittent hypoxia was shown to be mediated by ERK and STAT3 activation in adult rat myocardium [97]. On the other hand, IL-6 levels might also be involved in the determinism of cardiac hypertrophy [22]. Consistent with this hypothesis, Hirota and coworkers demonstrated a concomitant overexpression of both IL-6 and IL-6 receptor in a mouse model of cardiac hypertrophy [108]. Although these observations suggest a potential role for IL-6 in metabolic syndrome, the potential activation of intracellular signaling pathways by this cytokine remains unclear. Cardiotrophin-1 (CT-1), a newly discovered member of the IL-6 family and a key regulator of metabolism were also reported to be upregulated in metabolic syndrome [109, 110]. This cytokine was shown to favor cardiac hypertrophy and fibrosis in mice [23]. Thus, CT-1 might also underlie adverse remodeling in metabolic syndrome and thus represent an attractive pathophysiological target.
Although evidence for the role of p38 MAPK in metabolic syndrome is still lacking (except for some animal studies in diabetic fibrotic cardiomyopathy) [111], enhanced MAPK signaling was shown to increase both insulin sensitivity and promote hypertrophic cardiac remodeling (Table 2). Thus, these proteins might represent a promising target to improve metabolic, inflammatory, and cardiac pathophysiology in metabolic syndrome.
Table 2.
Role of MAPK activation in metabolic syndrome-associated cardiac hypertrophy.
| Author and reference number | Year | Experimental model | MAPK phosphorylation | Cardiac remodeling |
|---|---|---|---|---|
| Okoshi et al. [62] | 2004 | Primary cultures of neonatal rat cardiomyocytes | Increased P-ERK1/2 |
Aldosterone induces hypertrophy through ERK1/2 activation |
|
| ||||
| Yue et al. [63] | 2000 |
Primary cultures of neonatal rat cardiomyocytes | Increased P-ERK1/2 |
ERK1/2 activation mediates endothelin-1- and phenylephrine-induced cardiac hypertrophy |
| Wang and Proud [64] | 2002 | Adult rat ventricular cardiomyocytes | ||
|
| ||||
| Chen et al. [97] | 2007 | Rat myocardium | Increased P-ERK5; P-STAT-3; P-p38 MAPK | Long-term intermittent hypoxia is associated to induced cardiac hypertrophy through the activation of MAPK pathways |
|
| ||||
| Takeishi et al. [79] | 2001 | Guinea pigs | Increased P-ERK1/2, P-p38 MAPK |
Chronic pressure-overload and acute mechanical stretch-induced cardiac hypertrophy is mediated by ERK1/2 and p38 MAPK activation |
|
| ||||
| Esposito et al. [80] | 2010 | Mice left ventricle and white blood cells from mice and hypertensive patients with controlled blood pressure values |
Increased P-ERK1/2, P-p38 MAPK, P-JNK | MAPKs are sensors of pressure overload |
|
| ||||
| Rose et al. [82] | 1998 | Primary cultures of neonatal rat cardiomyocytes | Increased P-JNK and P-p38 MAPK | JNK activation induces hypertrophy, while concomitant activation of p38 MAPK and JNK inhibits hypertrophic response |
|
| ||||
| Wang et al. [84] | 1998 | Primary cultures of neonatal rat cardiomyocytes | Increased P-JNK and P-p38 MAPK | JNK induces hypertrophy, while concomitant activation of p38 MAPK and JNK fails to promote hypertrophic response |
|
| ||||
| Zechner et al. [96] | 1997 | Primary cultures of neonatal rat cardiomyocytes | Increased P-p38 MAPK | Transfection of the cell with constructs activating MAPKs revealed a central role of p38 MAPK activation in cardiac hypertrophy |
|
| ||||
| Choukroun et al. [98] | 1998 | Primary cultures of neonatal rat cardiomyocytes | Increased SAPK and not P-ERK | In contrast to SAPK, ERK activation is not required for hypertrophic response induced by endothelin |
6. Conclusions
In the past decades, relevant progresses have been made in the attempt to define the role of MAPKs in metabolic syndrome and in its clinical manifestations, including heart adverse pathophysiological remodelling. The heart structure has been described to be directly influenced by the mechanical stress, characterizing certain components of the metabolic syndrome (such as hypertension). Importantly, this adverse cardiac remodelling might be partially regulated by elevated hormones and inflammatory cytokines. MAPKs might represent the final pathways commonly activated within cardiomyocytes by both mechanical and soluble determinants. Despite some limitations due to animal and in vitro models, MAPK (mainly JNK and ERK) activation in the peripheral organs (including the heart) in metabolic syndrome was shown to induce insulin resistance and to increase inflammation. On the other hand, although the effect of p38 MAPK and JNK phosphorylation remains controversial, mounting evidence indicates ERK1/2 as responsible for promoting cardiomyocyte growth. We believe that MAPKs might be considered as potential therapeutic targets for drugs aimed to improve metabolic and cardiac dysfunctions in metabolic syndrome. Indeed several inhibitors of MAPK signaling proteins are currently available. Some of them are already being tested in clinical trials for oncological disorders and may theoretically find application also for the treatment or prevention of heart dysfunction in metabolic syndrome. Alternatively, strategies targeting downstream effectors/transcription factors in these cascades could also be a viable therapeutic option.
Acknowledgments
This paper was funded by EU FP7, Grant no. 201668, AtheroRemo to F. Mach. This work was also supported by the Swiss National Science Foundation Grants to F. Mach (no. 310030-118245) and F. Montecucco (no. 32003B-134963/1). This work was supported by a grant from Novartis Foundation to F. Montecucco.
References
- 1.Isomaa B, Almgren P, Tuomi T, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24(4):683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 2.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. The Lancet. 2005;365(9468):1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 3.Armitage JA, Khan IY, Taylor PD, Nathanielsz PW, Poston L. Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? Journal of Physiology. 2004;561(2):355–377. doi: 10.1113/jphysiol.2004.072009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montecucco F, Mach F. Update on therapeutic strategies to increase adiponectin function and secretion in metabolic syndrome. Diabetes, Obesity and Metabolism. 2009;11(5):445–454. doi: 10.1111/j.1463-1326.2008.00986.x. [DOI] [PubMed] [Google Scholar]
- 5.von Bibra H, St John Sutton M. Impact of diabetes on postinfarction heart failure and left ventricular remodeling. Current Heart Failure Reports. 2011;8:242–251. doi: 10.1007/s11897-011-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roden M. Mechanisms of Disease: hepatic steatosis in type 2 diabetes—pathogenesis and clinical relevance. Nature Clinical Practice Endocrinology and Metabolism. 2006;2(6):335–348. doi: 10.1038/ncpendmet0190. [DOI] [PubMed] [Google Scholar]
- 7.Lopaschuk GD. Metabolic abnormalities in the diabetic heart. Heart Failure Reviews. 2002;7(2):149–159. doi: 10.1023/a:1015328625394. [DOI] [PubMed] [Google Scholar]
- 8.Szczepaniak LS, Dobbins RL, Metzger GJ, et al. Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magnetic Resonance in Medicine. 2003;49(3):417–423. doi: 10.1002/mrm.10372. [DOI] [PubMed] [Google Scholar]
- 9.Buckberg G, Hoffman JIE, Mahajan A, Saleh S, Coghlan C. Cardiac mechanics revisited: the relationship of cardiac architecture to ventricular function. Circulation. 2008;118(24):2571–2587. doi: 10.1161/CIRCULATIONAHA.107.754424. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Dong C. Regulatory mechanisms of mitogen-activated kinase signaling. Cellular and Molecular Life Sciences. 2007;64(21):2771–2789. doi: 10.1007/s00018-007-7012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Research. 2002;12(1):9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 12.Hefti MA, Harder BA, Eppenberger HM, Schaub MC. Signaling pathways in cardiac myocyte hypertrophy. Journal of Molecular and Cellular Cardiology. 1997;29(11):2873–2892. doi: 10.1006/jmcc.1997.0523. [DOI] [PubMed] [Google Scholar]
- 13.Peterson LR, Waggoner AD, Schechtman KB, et al. Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. Journal of the American College of Cardiology. 2004;43(8):1399–1404. doi: 10.1016/j.jacc.2003.10.062. [DOI] [PubMed] [Google Scholar]
- 14.Iacobellis G, Ribaudo MC, Leto G, et al. Influence of excess fat on cardiac morphology and function: study in uncomplicated obesity. Obesity Research. 2002;10(8):767–773. doi: 10.1038/oby.2002.104. [DOI] [PubMed] [Google Scholar]
- 15.Wong CY, O’Moore-Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation. 2004;110(19):3081–3087. doi: 10.1161/01.CIR.0000147184.13872.0F. [DOI] [PubMed] [Google Scholar]
- 16.Avelar E, Cloward TV, Walker JM, et al. Left ventricular hypertrophy in severe obesity: interactions among blood pressure, nocturnal hypoxemia, and body mass. Hypertension. 2007;49(1):34–39. doi: 10.1161/01.HYP.0000251711.92482.14. [DOI] [PubMed] [Google Scholar]
- 17.Kortelainen ML. Myocardial infarction and coronary pathology in severely obese people examined at autopsy. International Journal of Obesity. 2002;26(1):73–79. doi: 10.1038/sj.ijo.0801852. [DOI] [PubMed] [Google Scholar]
- 18.Cummings PM, Le BH, Lopes MBS. Postmortem findings in morbidly obese individuals dying after gastric bypass procedures. Human Pathology. 2007;38(4):593–597. doi: 10.1016/j.humpath.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 19.de Simone G, Palmieri V, Bella JN, et al. Association of left ventricular hypertrophy with metabolic risk factors: the HyperGEN study. Journal of Hypertension. 2002;20(2):323–331. doi: 10.1097/00004872-200202000-00024. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes LDL, Brown AL, Mathews SJ, et al. Metabolic syndrome is associated with abnormal left ventricular diastolic function independent of left ventricular mass. European Heart Journal. 2007;28(5):553–559. doi: 10.1093/eurheartj/ehl526. [DOI] [PubMed] [Google Scholar]
- 21.Azevedo A, Bettencourt P, Almeida PB, et al. Increasing number of components of the metabolic syndrome and cardiac structural and functional abnormalities—cross-sectional study of the general population. BMC Cardiovascular Disorders. 2007;7, article 17 doi: 10.1186/1471-2261-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56(2):225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Andres N, Rousseau A, Akhtar R, Calvier L, Inigo C, Labat C, et al. Cardiotrophin 1 is involved in cardiac, vascular, and renal fibrosis and dysfunction. Hypertension. 2012;60:563–573. doi: 10.1161/HYPERTENSIONAHA.112.194407. [DOI] [PubMed] [Google Scholar]
- 24.Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109(3):433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 25.Hannan RD, Jenkins A, Jenkins AK, Brandenburger Y. Cardiac hypertrophy: a matter of translation. Clinical and Experimental Pharmacology and Physiology. 2003;30(8):517–527. doi: 10.1046/j.1440-1681.2003.03873.x. [DOI] [PubMed] [Google Scholar]
- 26.Peterson KL. Pressure overload hypertrophy and congestive heart failure: where is the “Achilles’ Heel”? Journal of the American College of Cardiology. 2002;39(4):672–675. doi: 10.1016/s0735-1097(01)01790-9. [DOI] [PubMed] [Google Scholar]
- 27.Heckbert SR, Post W, Pearson GDN, et al. Traditional Cardiovascular Risk Factors in Relation to Left Ventricular Mass, Volume, and Systolic Function by Cardiac Magnetic Resonance Imaging. The Multiethnic Study of Atherosclerosis. Journal of the American College of Cardiology. 2006;48(11):2285–2292. doi: 10.1016/j.jacc.2006.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Di Mario U, Leonetti F. Adapted changes in left ventricular structure and function in severe uncomplicated obesity. Obesity Research. 2004;12(10):1616–1621. doi: 10.1038/oby.2004.201. [DOI] [PubMed] [Google Scholar]
- 29.Tandri H, Castillo E, Ferrari VA, et al. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia. Sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. Journal of the American College of Cardiology. 2006;48(11):2277–2284. doi: 10.1016/j.jacc.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 30.Tansey DK, Aly Z, Sheppard MN. Fat in the right ventricle of the normal heart. Histopathology. 2005;46(1):98–104. doi: 10.1111/j.1365-2559.2005.02054.x. [DOI] [PubMed] [Google Scholar]
- 31.Kim E, Choe YH, Han BK, et al. Right ventricular fat infiltration in asymptomatic subjects: observations from ECG-gated 16-slice multidetector CT. Journal of Computer Assisted Tomography. 2007;31(1):22–28. doi: 10.1097/01.rct.0000236416.05267.6c. [DOI] [PubMed] [Google Scholar]
- 32.Mazumder PK, O’Neill BT, Roberts MW, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53(9):2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 33.Buchanan J, Mazumder PK, Hu P, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146(12):5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 34.Landau D, Chayat C, Zucker N, et al. Early blood pressure-independent cardiac changes in diabetic rats. Journal of Endocrinology. 2008;197(1):75–83. doi: 10.1677/JOE-08-0040. [DOI] [PubMed] [Google Scholar]
- 35.Bollano E, Omerovic E, Svensson H, Waagstein F, Fu M. Cardiac remodeling rather than disturbed myocardial energy metabolism is associated with cardiac dysfunction in diabetic rats. International Journal of Cardiology. 2007;114(2):195–201. doi: 10.1016/j.ijcard.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 36.Assmann G. Dyslipidaemia and global cardiovascular risk: clinical issues. European Heart Journal, Supplement. 2006;8:F40–F46. [Google Scholar]
- 37.Barylski M, Ciecwierz J, Banach M, Irzmanski R, Pawlicki L, Kowalski J. Left ventricular diastolic dysfunction in subjects with metabolic syndrome. Atherosclerosis Supplements. 2008;9, article 232 [Google Scholar]
- 38.Penjaskovic D, Sakac D, Dejanovic J, Zec R, Zec Petkovic N, Stojsic Milosavljevic A. Left ventricular diastolic dysfunction in patients with metabolic syndrome. Medicinski Pregled. 2012;65:18–22. [PubMed] [Google Scholar]
- 39.Mulè G, Nardi E, Cottone S, et al. Influence of metabolic syndrome on hypertension-related target organ damage. Journal of Internal Medicine. 2005;257(6):503–513. doi: 10.1111/j.1365-2796.2005.01493.x. [DOI] [PubMed] [Google Scholar]
- 40.Chinali M, Devereux RB, Howard BV, et al. Comparison of cardiac structure and function in American Indians with and without the metabolic syndrome (the Strong Heart Study) American Journal of Cardiology. 2004;93(1):40–44. doi: 10.1016/j.amjcard.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Okin PM, Devereux RB, Jern S, et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. Journal of the American Medical Association. 2004;292(19):2343–2349. doi: 10.1001/jama.292.19.2343. [DOI] [PubMed] [Google Scholar]
- 42.Schillaci G, Pirro M, Vaudo G, et al. Metabolic syndrome is associated with aortic stiffness in untreated essential hypertension. Hypertension. 2005;45(6):1078–1082. doi: 10.1161/01.HYP.0000165313.84007.7d. [DOI] [PubMed] [Google Scholar]
- 43.Krauser DG, Devereux RB. Ventricular hypertrophy and hypertension: prognostic elements and implications for management. Herz Kardiovaskuläre Erkrankungen. 2006;31(4):305–316. doi: 10.1007/s00059-006-2819-5. [DOI] [PubMed] [Google Scholar]
- 44.de Simone G, Devereux RB, Chinali M, et al. Metabolic syndrome and left ventricular hypertrophy in the prediction of cardiovascular events: the Strong Heart Study. Nutrition, Metabolism and Cardiovascular Diseases. 2009;19(7):p. 520. doi: 10.1016/j.numecd.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li ZL, Woollard JR, Ebrahimi B, Crane JA, Jordan KL, Lerman A, et al. Transition from obesity to metabolic syndrome is associated with altered myocardial autophagy and apoptosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32:1132–1141. doi: 10.1161/ATVBAHA.111.244061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiological Reviews. 1999;79(1):215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 47.Zhu XY, Daghini E, Rodriguez-Porcel M, et al. Redox-sensitive myocardial remodeling and dysfunction in swine diet-induced experimental hypercholesterolemia. Atherosclerosis. 2007;193(1):62–69. doi: 10.1016/j.atherosclerosis.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 48.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiological Reviews. 2008;88(2):389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meigs JB, Rutter MK, Sullivan LM, Fox CS, D’Agostino RB, Wilson PWF. Impact of insulin resistance on risk of type 2 diabetes and cardiovascular disease in people with metabolic syndrome. Diabetes Care. 2007;30(5):1219–1225. doi: 10.2337/dc06-2484. [DOI] [PubMed] [Google Scholar]
- 50.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium: fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83(6):1849–1865. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 51.Sato A, Saruta T. Aldosterone escape during angiostensin-converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. Journal of International Medical Research. 2001;29(1):13–21. doi: 10.1177/147323000102900103. [DOI] [PubMed] [Google Scholar]
- 52.Struthers AD. The clinical implications of aldosterone escape in congestive heart failure. European Journal of Heart Failure. 2004;6(5):539–545. doi: 10.1016/j.ejheart.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 53.Kosmala W, Przewlocka-Kosmala M, Szczepanik-Osadnik H, Mysiak A, O'Moore-Sullivan T, Marwick TH. A randomized study of the beneficial effects of aldosterone antagonism on LV function, structure, and fibrosis markers in metabolic syndrome. Cardiovascular Imaging. 2011;4:1239–1249. doi: 10.1016/j.jcmg.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. The Lancet. 2006;367(9507):356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 55.Gallagher EJ, Leroith D, Karnieli E. Insulin resistance in obesity as the underlying cause for the metabolic syndrome. Mount Sinai Journal of Medicine. 2010;77(5):511–523. doi: 10.1002/msj.20212. [DOI] [PubMed] [Google Scholar]
- 56.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metabolism. 2011;14(1):9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bochud M, Nussberger J, Bovet P, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48(2):239–245. doi: 10.1161/01.HYP.0000231338.41548.fc. [DOI] [PubMed] [Google Scholar]
- 58.Calhoun DA, Sharma K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiology Clinics. 2010;28(3):517–527. doi: 10.1016/j.ccl.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Gharbawy AH, Nadig VS, Morley Kotchen J, et al. Arterial pressure, left ventricular mass, and aldosterone in essential hypertension. Hypertension. 2001;37(3):845–850. doi: 10.1161/01.hyp.37.3.845. [DOI] [PubMed] [Google Scholar]
- 60.Brilla CG, Matsubara LS, Weber KT. Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary and secondary hyperaldosteronism. Journal of Molecular and Cellular Cardiology. 1993;25(5):563–575. doi: 10.1006/jmcc.1993.1066. [DOI] [PubMed] [Google Scholar]
- 61.Matsumura K, Fujii K, Oniki H, Oka M, Iida M. Role of aldosterone in left ventricular hypertrophy in hypertension. American Journal of Hypertension. 2006;19(1):13–18. doi: 10.1016/j.amjhyper.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 62.Okoshi MP, Yan X, Okoshi K, et al. Aldosterone directly stimulates cardiac myocyte hypertrophy. Journal of Cardiac Failure. 2004;10(6):511–518. doi: 10.1016/j.cardfail.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Yue TL, Gu JL, Wang C, et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. Journal of Biological Chemistry. 2000;275(48):37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Proud CG. Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circulation Research. 2002;91(9):821–829. doi: 10.1161/01.res.0000041029.97988.e9. [DOI] [PubMed] [Google Scholar]
- 65.Robert V, Van Thiem N, Cheav SL, Mouas C, Swynghedauw B, Delcayre C. Increased cardiac types I and III collagen mRNAs in aldosterone-salt hypertension. Hypertension. 1994;24(1):30–36. doi: 10.1161/01.hyp.24.1.30. [DOI] [PubMed] [Google Scholar]
- 66.Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. Journal of Clinical Investigation. 1994;93(6):2578–2583. doi: 10.1172/JCI117269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sowers JR. Metabolic risk factors and renal disease. Kidney International. 2007;71(8):719–720. doi: 10.1038/sj.ki.5002006. [DOI] [PubMed] [Google Scholar]
- 68.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Annals of Internal Medicine. 2009;150(11):776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emanuela F, Grazia M, Marco de R, Maria Paola L, Giorgio F, Marco B. Inflammation as a link between obesity and metabolic syndrome. Journal of Nutrition and Metabolism. 2012;2012 doi: 10.1155/2012/476380.476380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(12):7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 73.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sack MN, Smith RM, Opie LH. Tumor necrosis factor in myocardial hypertrophy and ischaemia—an anti-apoptotic perspective. Cardiovascular Research. 2000;45(3):688–695. doi: 10.1016/s0008-6363(99)00228-x. [DOI] [PubMed] [Google Scholar]
- 75.Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL. Hemodynamic regulation of tumor necrosis factor-α gene and protein expression in adult feline myocardium. Circulation Research. 1997;81(2):187–195. doi: 10.1161/01.res.81.2.187. [DOI] [PubMed] [Google Scholar]
- 76.Nakamura K, Fushimi K, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-α and angiotensin II. Circulation. 1998;98(8):794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 77.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiological Reviews. 1999;79(1):143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 78.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature Reviews Molecular Cell Biology. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 79.Takeishi Y, Huang Q, Abe JI, et al. Src and multiple MAP kinase activation in cardiac hypertrophy and congestive heart failure under chronic pressure-overload: comparison with acute mechanical stretch. Journal of Molecular and Cellular Cardiology. 2001;33(9):1637–1648. doi: 10.1006/jmcc.2001.1427. [DOI] [PubMed] [Google Scholar]
- 80.Esposito G, Perrino C, Schiattarella GG, et al. Induction of mitogen-activated protein kinases is proportional to the amount of pressure overload. Hypertension. 2010;55(1):137–143. doi: 10.1161/HYPERTENSIONAHA.109.135467. [DOI] [PubMed] [Google Scholar]
- 81.Haq S, Choukroun G, Lim H, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103(5):670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 82.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiological Reviews. 2010;90(4):1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Babu GJ, Lalli JM, Sussman MA, Sadoshima JI, Periasamy M. Phosphorylation of Elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. Journal of Molecular and Cellular Cardiology. 2000;32(8):1447–1457. doi: 10.1006/jmcc.2000.1185. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Su B, Sah VP, Brown JH, Han J, Chien KR. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. Journal of Biological Chemistry. 1998;273(20):5423–5426. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 85.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, et al. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart: p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circulation Research. 1996;79(2):162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 86.Petrich BG, Eloff BC, Lerner DL, et al. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. Journal of Biological Chemistry. 2004;279(15):15330–15338. doi: 10.1074/jbc.M314142200. [DOI] [PubMed] [Google Scholar]
- 87.Frantz S, Behr T, Hu K, et al. Role of p38 mitogen-activated protein kinase in cardiac remodelling. British Journal of Pharmacology. 2007;150(2):130–135. doi: 10.1038/sj.bjp.0706963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ono K, Han J. The p38 signal transduction pathway Activation and function. Cellular Signalling. 2000;12(1):1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 89.Waehre A, Halvorsen B, Yndestad A, et al. Lack of chemokine signaling through CXCR5 causes increased mortality, ventricular dilatation and deranged matrix during cardiac pressure overload. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0018668.e18668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lacerda-Miranda G, Soares VM, Vieira AKG, Lessa JG, Rodrigues-Cunha ACS, Cortez E, et al. Ghrelin signaling in heart remodeling of adult obese mice. Peptides. 2012;35:65–73. doi: 10.1016/j.peptides.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 91.Besnard A, Galan-Rodriguez B, Vanhoutte P, Caboche J. Elk-1 a transcription factor with multiple facets in the brain. Frontiers in Neuroscience. 2011;5, article 35 doi: 10.3389/fnins.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Darragh J, Soloaga A, Beardmore VA, et al. MSKs are required for the transcription of the nuclear orphan receptors Nur77, Nurr1 and Nor1 downstream of MAPK signalling. Biochemical Journal. 2005;390(3):749–759. doi: 10.1042/BJ20050196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han J, Molkentin JD. Regulation of MEF2 by p38 MAPK and its implication in cardiomyocyte biology. Trends in Cardiovascular Medicine. 2000;10(1):19–22. doi: 10.1016/s1050-1738(00)00039-6. [DOI] [PubMed] [Google Scholar]
- 94.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. Journal of Molecular and Cellular Cardiology. 2003;35(12):1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 95.Nemoto S, Sheng Z, Lin A. Opposing effects of Jun kinase and p38 mitogen-activated protein kinases on cardiomyocyte hypertrophy. Molecular and Cellular Biology. 1998;18(6):3518–3526. doi: 10.1128/mcb.18.6.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zechner D, Thuerauf DJ, Hanford DS, McDonough PM, Glembotski CC. A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. Journal of Cell Biology. 1997;139(1):115–127. doi: 10.1083/jcb.139.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen LM, Kuo WW, Yang JJ, et al. Eccentric cardiac hypertrophy was induced by long-term intermittent hypoxia in rats. Experimental Physiology. 2007;92(2):409–416. doi: 10.1113/expphysiol.2006.036590. [DOI] [PubMed] [Google Scholar]
- 98.Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. Journal of Clinical Investigation. 1998;102(7):1311–1320. doi: 10.1172/JCI3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao P, Georgakopoulos D, Kovacs A, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(21):12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Montecucco F, Steffens S, Mach F. Insulin resistance: a proinflammatory state mediated by lipid-induced signaling dysfunction and involved in atherosclerotic plaque instability. Mediators of Inflammation. 2008;2008 doi: 10.1155/2008/767623.767623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116(12):1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Satoh Y, Endo S, Ikeda T, et al. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. Journal of Neuroscience. 2007;27(40):10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bélanger LF, Roy S, Tremblay M, et al. MEK2 is dispensable for mouse growth and development. Molecular and Cellular Biology. 2003;23(14):4778–4787. doi: 10.1128/MCB.23.14.4778-4787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bost F, Aouadi M, Caron L, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54(2):402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 105.Lee SJ, Pfluger PT, Kim JY, et al. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Reports. 2010;11(3):226–232. doi: 10.1038/embor.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monteiro R, Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators of Inflammation. 2010;2010 doi: 10.1155/2010/289645.289645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Condorelli G, Morisco C, Latronico MVG, et al. TNF-α signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-α receptor. FASEB Journal. 2002;16(13):1732–1737. doi: 10.1096/fj.02-0419com. [DOI] [PubMed] [Google Scholar]
- 108.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(11):4862–4866. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moreno-Aliaga MJ, Perez-Echarri N, Marcos-Gomez B, Larequi E, Gil-Bea FJ, Viollet B, et al. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metabolism. 2011;14:242–253. doi: 10.1016/j.cmet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 110.Natal C, Fortuño MA, Restituto P, et al. Cardiotrophin-1 is expressed in adipose tissue and upregulated in the metabolic syndrome. American Journal of Physiology. 2008;294(1):E52–E60. doi: 10.1152/ajpendo.00506.2007. [DOI] [PubMed] [Google Scholar]
- 111.Thandavarayan RA, Watanabe K, Ma M, et al. Dominant-negative p38α mitogen-activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. American Journal of Physiology. 2009;297(3):H911–H919. doi: 10.1152/ajpheart.00124.2009. [DOI] [PubMed] [Google Scholar]