

Abstract
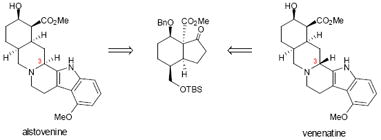
The yohimbinoid alkaloids have received considerable attention from the synthetic community due to their interesting chemical structures and varied biological activity. Although there have been several elegant syntheses of certain members of this group of alkaloids, a truly unified approach has yet to be developed. In short, general approaches to this compound class have been hampered by a lack of complete control in setting the C(3) stereocenter at a late stage. Herein, we report that a functionalized hydrindanone enables a divergent strategy that builds on precedent from Stork, which addresses this long standing challenge. Utilizing an aminonitrile intermediate, the stereochemistry at C(3) of the yohimbinoid skeleton can be effectively controlled in a Pictet-Spengler reaction. This approach has been applied to the first total syntheses of the C(3) epimeric natural products venenatine and alstovenine.
The yohimbine indole alkaloids are among the most studied natural products, primarily owing to their potent and multifarious bioactivity.1,2,3,4,5,6,7,8 While there have been many elegant syntheses to arrive at specific members of this class,9,10,11,12,13,14,15,16,17,18,19,20 especially the archetypal member, reserpine (2, Figure 1), a unified approach to these molecules remains an unsolved problem.21 In the realm of complex molecule assembly, the total synthesis of 2 by R. B. Woodward and co-workers is well recognized as a milestone event.9,10,11 In many ways, reserpine presented a perfect forum to illustrate the power of stereoelectronic considerations in the preparation of the E-ring of this natural product and ultimately arrive at cyclization precursor 1. Unfortunately, in the Woodward synthesis of reserpine, cyclization of amide 1 to forge the C ring of 2 led to the undesired C(3) epimer. As a result, a lactonization and acid-catalyzed epimerization sequence was required to invert the stereochemistry at C(3) to match the natural product (i.e., 2).
Figure 1.
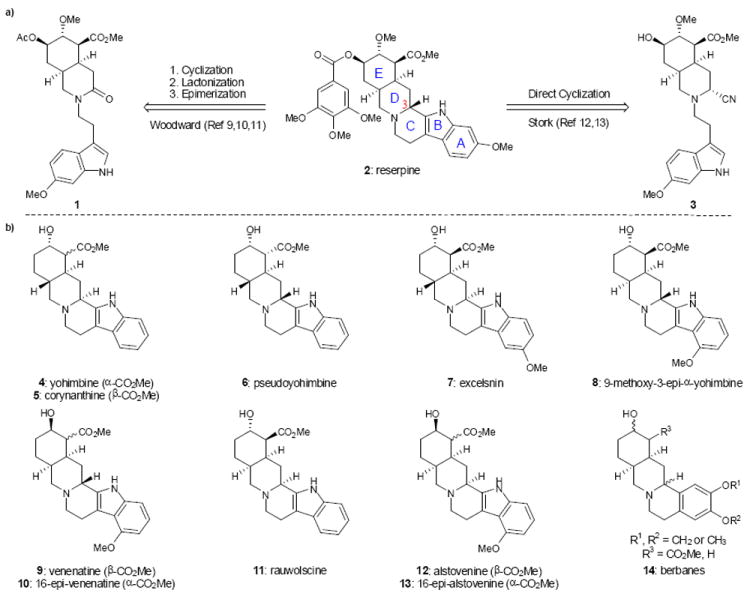
Classic approaches to reserpine and selected yohimbine and berbane alkaloids. a) Classic approaches to closure of the C-ring and setting of the C-3 stereocenter of reserpine. b) Selected yohimbinoid natural products that vary in the substitution pattern of the indole ring. The ability to access a variety of yohimbine natural products from a common precursor would allow for a unified approach to this group of alkaloids. Me, methyl; Ac, acetyl
Following Woodward’s pioneering synthesis of reserpine (2), subsequent syntheses of 2 and related yohimbinoid natural products have appeared in the literature. However, because many of these syntheses adopted a strategy that is similar to the Woodward approach, they were also plagued with a stereoselectivity problem (or in some cases, a regioselectivity issue)1 in installing the C(3) center, which had to be corrected to obtain the desired diastereomer. In 1989, Stork reported an inventive solution to the reserpine C(3) problem, that took advantage of the ionization of an intermediate aminonitrile (i.e., 3, Figure 1).12,13 The subtleties in stereocontrol in the Pictet-Spengler reaction afforded by aminonitriles allowed Stork to selectively access the undesired as well as the desired C(3) epimer necessary for the total synthesis of reserpine. While the Stork approach sets the foundation to address the C(3) problem, it remained unclear whether this tactic could be extended to the synthesis of other yohimbinoid natural products given the significant influence of the indole nucleophile on the diastereoselectivity of the Pictet-Spengler ring closure from an aminonitrile as discovered in our own work (vide infra). A general solution to this problem would yield a strategy for the preparation of the yohimbinoid alkaloids, given that structures in this class vary in the substitution about the indole moiety (see 4-13, Figure 1), and include several members that are epimeric at C(3).22,23,24,25,26,27 Herein, we disclose our efforts toward a general synthesis of the D/E cis-fused members of the yohimbinoid alkaloid family (i.e., 8-13, Figure 1). This approach is showcased in the first total syntheses of the C(3) epimeric natural products venenatine (9) and alstovenine (12) in racemic form.28,29,30,31,32,33
Interestingly, the seemingly subtle difference in the stereochemistry of 9 and 12 (epimers at C(3)) results in a complete reversal of their effects on the central nervous system (CNS). Alstovenine (12) behaves as a CNS stimulant in mice (1 mg/kg) and significantly enhances the analgesic effects of morphine, whereas venenatine (9) displays activity analogous to reserpine (50 mg/kg in mice) and inhibits the analgesic effects of morphine.34 Because the yohimbinoid “family at large” displays such interesting and contrasting activity, a concise and high-yielding synthesis of yohimbine congeners (which include the berbanes, see 14, Figure 1)35,36,37 would facilitate more detailed studies regarding the mode of action of these molecules.
Results and discussion
Our synthetic strategy (Figure 2), inspired by Stork’s elegant synthesis of reserpine, targets a variety of yohimbinoid alkaloid natural products from the elaboration of pentacycles related to 15, which vary only in the indole moiety. Pentacycle 15 could in turn arise from aminonitrile 16, where careful tuning of the reaction conditions would enable cyclization to either series of C(3) epimeric natural products. The aminonitriles related to 16 would arise from a condensation/Strecker sequence with a common intermediate aldehyde (17) and the requisite amine derivative. Aldehyde 17 was expected to arise from oxidative cleavage of hydrindanone 18,38,39,40 which could be constructed from a Diels-Alder reaction between diene 19 and enone ester 20. This cycloaddition would set four contiguous stereocenters, leaving only the C(3) stereocenter to be set at a later stage.
Figure 2.
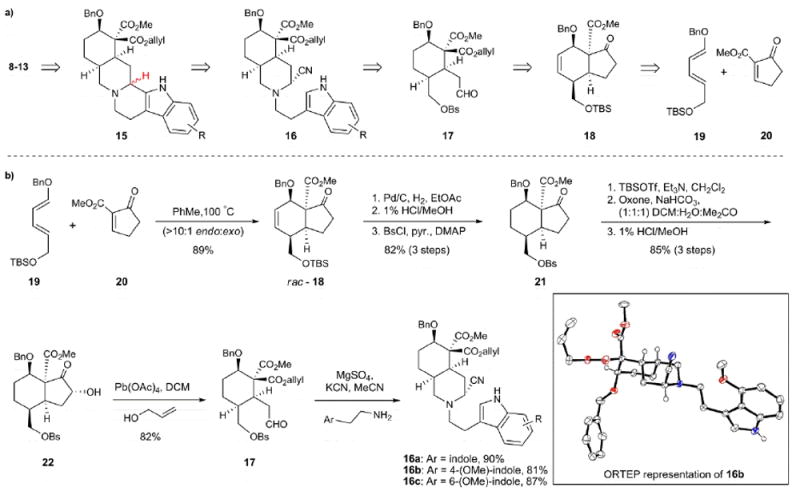
Retrosynthetic plan and synthesis of the aminonitrile Pictet-Spengler substrates. a) A variety of yohimbinoid natural products can be accessed from common hydrindanone precursor 18. b) The Diels-Alder cyclization between diene 19 and dienophile 20 provides hydrindanone 18 setting four contiguous stereocenters. Conversion of hydrindanone 18 into aldehyde 17 sets the stage for rapid formation of aminonitriles (16a-c) with varying substitution on the indole moiety. Bs, benzenesulfonyl; TBS, tert-butyldimethylsilyl; Bn, benzyl; PhMe, toluene; pyr, pyridine; DMAP, 4-dimethylaminopyridine; TBSOTf, tert-butyldimethylsilyl triflate; Oxone, potassium monopersulfate; DCM, dichloromethane.
Our synthetic studies commenced with readily available diene 1940 (Figure 2) and enone ester 20.41 Heating the mixture of these two compounds in toluene at 100 °C afforded hydrindanone 18 in 89% yield as a >10:1 ratio of the endo:exo diastereomers (endo assignment is in reference to the more electron withdrawing ketone moiety), which were readily separated by column chromatography. Of note, the enantiomers of the endo adducts can be readily separated on a preparatory scale using supercritical fluid chromatography and a chiral ODH column (for further details, see the Supporting Information). While access to 18 sets the stage for preparing the natural products in enantioenriched form, the syntheses reported herein were carried out in racemic form. Hydrogenation of 18, silyl ether cleavage and sulfonation of the resulting primary alcohol group yielded benzenesulfonate 21 in 82% yield over three steps. At this juncture, α-hydroxylation followed by oxidative cleavage with lead tetraacetate unveiled aldehyde 17 in 70% yield. Treatment of aldehyde 17 with potassium cyanide in the presence of various tryptamine derivatives then furnished the desired aminonitriles (16a-c) as single diastereomers and in excellent overall yields. The relative stereochemistry of the 4-OMe tryptamine aminonitrile was unambiguously established by X-ray crystallographic analysis (see ORTEP 16b, Figure 2; some hydrogen atoms removed for clarity).42
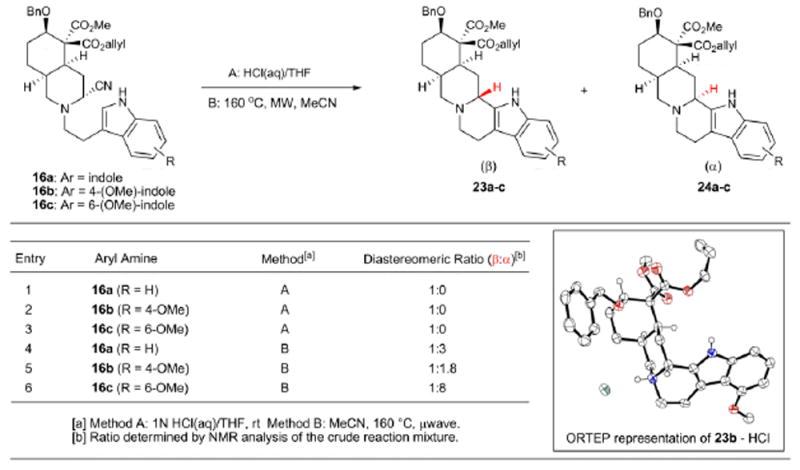
Substrates 16a-c were subjected to Pictet-Spengler cyclization conditions (Table 1), with special attention paid to the role arene nucleophilicity43,44 plays in both the rate and stereoselectivity of the pentacycle formation.45,46,47 Consistent with the elegant studies of the Stork group, treatment of aminonitriles 16a-c with hydrochloric acid (0.1 M in THF) at 23 °C resulted in exclusive formation of the β-diastereomer (i.e., 23a-c; see entries 1-3) for all substrates. The relative stereochemistry was unambiguously assigned for substrate 23b by X-ray crystallographic analysis (see ORTEP, Table 1; some hydrogen atoms removed for clarity).
Table 1.
Role of the indole nucleophile in the stereoselectivity of the Pictet-Spengler cyclization.
|
More surprising, however, was that the inherent diastereoselectivity of the thermal Pictet-Spengler cyclization (heating to 160 °C in acetonitrile; Method B in Table 1) of the substrates was greatly influenced by the nature of the indole fragment (see entries 4-6). Tryptamine-derived aminonitrile 16a cyclized upon heating in acetonitrile to afford a 1:3 (β:α) mixture of diastereomers, whereas 4-methoxytryptamine substrate 16b and 6-methoxytryptamine substrate 16c cyclized to give a 1:1.8 (β:α) and 1:8 (β:α) mixture of diastereomers, respectively. Of note, the latter observation is in line with the observed diastereoselectivity for the thermal cyclization of aminonitrile 3 in refluxing acetonitrile reported by Stork.12,13
Because several yohimbinoid natural products possess the indole substitution pattern present in 16a and 16b, the stereoselectivities for these cyclizations needed to be improved if a versatile approach to the yohimbinoid natural products was to be realized. As such, we embarked upon a campaign to optimize the diastereoselectivity of the Pictet-Spengler cyclization under thermal conditions (i.e., Method B) and chose to focus on substrate 16b as it represented the most challenging selectivity scenario. Stork has previously proposed the formation of a tight ion pair between the departing cyanide and forming iminium ion under thermal Pictet-Spengler conditions for aminonitriles, which leads to attack of the arene nucleophile from the β-face (see 26→24b, Figure 3).12,13 On the basis of this analysis, we reasoned that a solvent with a lower static permittivity (dielectric constant)48 would lead to tighter ion pairing and further enhance the diastereoselectivity of the cyclization. In an initial investigation of solvent effects (entries 3-7, Table 2), only acetonitrile and isopropanol, among solvents of similar polarity, provided the desired diastereomer (24b) as the major product, suggesting a more participatory role of the solvent. Several mechanistic possibilities that may exist are shown in Figure 3.
Figure 3.
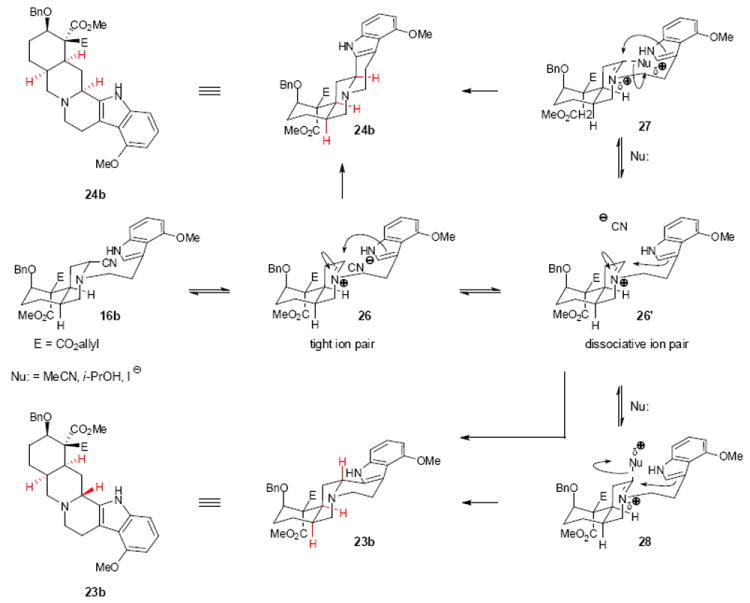
Plausible reaction pathways for the thermal cyclization. Thermal cyclization of aminonitrile 16b leads to a diastereomeric mixture of pentacycles 23b and 24b. The diastereomeric ratio is dependent on the reaction conditions employed with nucleophilic solvents and additives favouring formation of the α-diastereomer 23b. Several mechanistic possibilities are presented. Nu, nucleophile; Bn, benzyl.
Table 2.
Solvent effects in the Pictet-Spengler cyclization.
| ||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (°C)[a] | Additive[b] | Diastereomeric Ratio (β:α)[c] |
| 1 | THF | rt[d] | HCl | 1:0 |
| 2 | MeCN | 82[e] | - | n/r |
| 3 | MeCN | 160 | - | 1:1.8 |
| 4 | acetone | 160 | - | 2.5:1 |
| 5 | i-PrOH | 160 | - | 1:1.3 |
| 6 | i-PrCN | 160 | - | 1.4:1 |
| 7 | PhMe | 160 | - | 1.8:1 |
| 8 | PhMe | 160 | Imidazole | 1:1.6 |
| 9 | PhMe | 160 | DMAP | 1:2.0 |
| 10 | MeCN | 160 | DMAP | 1:4 |
| 11 | MeCN | 160 | NaI | 1:10 |
Reaction carried out in microwave reactor in a sealed vial unless otherwise noted.
2.8 equivalents.
Ratio determined by NMR analysis of the crude reaction mixture.
Reaction carried out in a sealed vial under a N2 atmosphere
Reaction carried out in a sealed vial in an oil bath
In one scenario, an acetonitrile solvent molecule could engage iminium ion 26‘ to form a different electrophilic species (see 27 or 28, Figure 3) that could then be displaced by the indole moiety in a direct intramolecular SN2-like process. As a probe for this mechanistic possibility, nucleophilic additives such as DMAP and imidazole were investigated. Gratifyingly, heating aminonitrile 16b to 160 °C in the presence of DMAP in toluene led to reversal of the stereoselectivity (from a 1.8:1.0 d.r. (β:α) for the thermal cyclization without additives in toluene; entry 7, Table 2) to favor the α diastereomer (1:2 d.r., (β:α); entry 9, Table 2). Furthermore, in acetonitrile, the diastereoselectivity was enhanced (1:4 d.r. (β:α); entry 10, Table 2) relative to cyclization in the absence of additives (1:1.8 d.r. (β:α); entry 3, Table 2). Upon addition of a stronger nucleophile (in the form of NaI; entry 11, Table 2), a greater than 1:10 d.r. (β:α) in favor of the α-diastereomer at C(3) was obtained.
Although the mechanism of the thermal Pictet-Spengler cyclization and the role of the iodide additive is still unclear, it may be that rapid exchange of the cyanide for the iodide occurs to produce a reactive iodo species (analogous to 16b, Figure 3). At this juncture, iminium ion 27 could begin to form with the much larger iodide serving as an intimate counteranion and effectively blocking the α-face and leading, overwhelmingly, to the α-diastereomer (24b) as the major product. Computational studies are currently underway to provide insight on the observed diastereoselectivities of the Pictet-Spengler reaction as well as the role of additives such as NaI.
With optimized cyclization conditions leading to either C(3) epimer in hand, all that remained to complete the synthesis of venenatine (9) and alstovenine (12) was removal of the allyl ester by deallylation/decarboxylation and cleavage of the benzyl ether in 23b and 24b (Figure 4). Toward this end, we have found that treatment of ester 23b with a Pd(II) precatalyst under neutral conditions and elevated temperature facilitates conversion to the desired monoester (29) along with some elimination of the benzyloxy group to give an enoate (30). Hydrogenolysis of the benzyl ether in 29 to give venenatine (9) was fraught with its own challenges. Initial attempts utilizing palladium on carbon and H2 (1 atm) were unfruitful. Neither increasing the H2 pressure (up to 200 psi) nor using acid additives resulted in the desired benzyl ether cleavage. However, treatment of 29 with BBr3 at low temperature selectively cleaved the benzyl ether, leaving both the methyl ester and methyl ether intact, to afford venenatine (9) in 60% yield. The stereochemical assignment of the methyl ester group was determined by NOESY studies (for further details, see Supporting Information). Interestingly, in the case of alstovenine the use of the deallylation/decarboxylation conditions employed for venenatine pentacycle 23b led only to recovered starting material and the product of β-hydroxy elimination. This undesirable outcome could be circumvented through the use of a more homogenous catalyst system (Pd2dba3, pyrrolidine) to afford 31. Lastly, BBr3 promoted cleavage of the benzyl ether yielded alstovenine (12).
Figure 4.
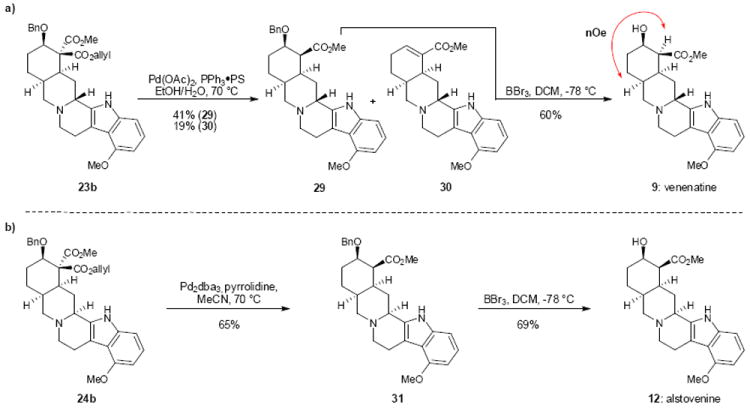
Synthesis of venenatine and alstovenine. Pd catalyzed deallylation/decarboxylation allows for allyl ester removal in 23b and 24b under mild conditions, minimizing β-hydroxy elimination. Treatment of 29 and 31 with BBr3 leads to selective removal of the benzyl protecting group and access to venenatine and alstovenine respectively. PS, polymer supported; nOe, nuclear Overhauser effect; dba, dibenzylideneacetone; Bn, benzyl; DCM, dichloromethane.
While the spectroscopic data for synthetic and natural alstovenine were in reasonable agreement, we were surprised to observe some large inconsistencies (> 4.8 ppm) when the 13C data for synthetic venenatine was compared with that reported for the isolated natural product, especially given the support for our assignment by X-ray and NOESY analysis of key intermediates. In an effort to identify the origin of these discrepancies, computational predictions for the 13C NMR chemical shifts49 of 9 were undertaken since all attempts to obtain an authentic sample for comparison failed. In general, the computed predictions for the 13C NMR chemical shifts (Calculated at the SMD(chloroform)-mPW1PW91/6-311+G(2d,p)//B3LYP/6-31+G(d,p) level with linear scaling; for complete computational details see the Supporting Information) were found to be in better agreement with our synthetic data (mean absolute deviation (MAD) of 1.37 ppm) than with the isolation data (MAD of 1.85 ppm). Furthermore, the computational predictions identified two 13C resonances (at 104.3 ppm and 31.0 ppm) from the isolation data as outliers; both deviating by 5.0 ppm or more from the computational data (A complete line listing comparison can be found in the Supporting Information). Importantly, the outlier at 104.3 ppm reported in the 13C data for isolated venenatine, which was one of the resonances that differed markedly from our synthetic 13C data, also varies significantly from the corresponding 13C resonances reported for closely related congeners including alstovenine, 16-epi-alstovenine, 16-epi-venenatine, and 9-methoxy-3-epi-α-yohimbine, which are all in line with our synthetic data. While no conclusive statement can be made regarding the origin of this discrepancy, it is possible that the reported data for isolated venenatine may have been mistabulated.
In summary, we have identified, an effective, general path to the syntheses of a subset of yohimbinoid alkaloids that are epimeric at C(3). This work, which has resulted in the first total syntheses of the alkaloids venenatine and alstovenine in racemic form, builds on important observations by Stork pertaining to aminonitrile Pictet-Spengler cyclizations. Key to the success of our synthetic studies was the identification of conditions to effectively control the diastereoselectivity of a late-stage Pictet-Spengler cyclization employing a variety of indole nucleophiles. Highlights from this study include: 1) the use of a hydrindanone intermediate 18, which allows rapid, stereoselective synthesis of the yohimbinoid skeleton, 2) the discovery that nucleophilicity of the indole moiety plays a significant role in the inherent diastereoselectivity of the Pictet-Spengler cyclization, and 3) the addition of an exogenous nucleophile (NaI) that leads to the complete reversal of the diastereoselectivity in the installation of the C(3) center.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH (NIGMS RO1 084906), NSF (CHE-0957416 and CHE-030089), and American Cancer Society (RSG-09-017-01 CDD) for support of this work. R.S. is a Camille Dreyfus Teacher-Scholar. T.P.L. thanks the NSERC (Canada) for a postdoctoral fellowship. J.D. thanks the National Science Foundation (NSF) for a predoctoral fellowship and J.L.W. thanks Chevron for a departmental fellowship. C. Kraml and N. Byrne from Lotus Separations are thanked for the separation of the enantiomers of Diels-Alder adduct 18.
Footnotes
Author contributions
T.P.L, J.L.W, and R.S. conceived and designed the synthetic experiments. T.P.L., J.L.W. and J.D. carried out the synthetic work. M.W.L. and D.J.T. carried out the computational work. T.P.L., J.L.W. and R.S. co-wrote the manuscript. All authors discussed the results and commented on the mansuscript.
References
- 1.Chen F-E, Huang J. Reserpine: A challenge for total synthesis of natural products. Chem Rev. 2005;105:4671–4706. doi: 10.1021/cr050521a. [DOI] [PubMed] [Google Scholar]
- 2.Aube J, Ghosh S. In: Advances in heterocyclic natural products. Pearson WH, editor. Vol. 3. JAI Press; Greenwich, CT: 1996. pp. 90–150. [Google Scholar]
- 3.Szantay C, Honty K. In: Monoterpenoid indole alkaloids. Saxton JE, editor. Chapter 4. John Wiley & Sons; Chichester: 1994. pp. 161–216. [Google Scholar]
- 4.Szantay C, Blasko G, Honty K, Dornyei G. In: The alkaoids. Brossi A, editor. Vol. 27. Academic Press; Orlando, FL: 1986. pp. 131–268. [Google Scholar]
- 5.Schlitter E. In: The Alkaloids: chemistry and physiology. Manske RHF, editor. VIII. Academic Press; New York: 1965. pp. 287–334. [Google Scholar]
- 6.Baxter EW, Mariano PS. In: Alkaloids: chemical and biological perspectives. Pelletier SW, editor. Vol. 8. Springer- Verlag; New York: 1992. pp. 197–319. [Google Scholar]
- 7.Goldberg MR, Robertson D. Yohimbine: a pharmacological probe for study of the alpha 2-adrenoreceptor. Pharmacological Reviews. 1983;35:143–180. [PubMed] [Google Scholar]
- 8.Tam SW, Worcel M, Wyllie M. Yohimbine: a clinical review. Pharmacology & Therapeutics. 2001;91:215–243. doi: 10.1016/s0163-7258(01)00156-5. [DOI] [PubMed] [Google Scholar]
- 9.Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. The total synthesis of reserpine. Tetrahedron. 1958;2:1–57. [Google Scholar]
- 10.Woodward RB, Bader FE, Bickel H, Frey AJ, Kierstead RW. The total synthesis of reserpine. J Am Chem Soc. 1956;78:2023–2025. [Google Scholar]
- 11.Woodward RB, Bader FE, Bickel H, Fery AJ, Kierstead RWA. Simplified route to a key intermediate in the total synthesis of reserpine. J Am Chem Soc. 1956;78:2657–2657. [Google Scholar]
- 12.Stork G. The stereospecific synthesis of reserpine. Pure Appl Chem. 1989;61:439–442. [Google Scholar]
- 13.Stork G, Tang PC, Casey M, Goodman B, Toyota M. Regiospecific and stereoselective syntheses of (±)-reserpine and (−)-reserpine. J Am Chem Soc. 2005;127:16255–16262. doi: 10.1021/ja055744x. [DOI] [PubMed] [Google Scholar]
- 14.Wender PA, Schaus JM, White AW. General methodology for cis-hydroisoquinoline synthesis: synthesis of reserpine. J Am Chem Soc. 1980;102:6157–6159. [Google Scholar]
- 15.Wender PA, Schaus JM, White AW. General methodology for cis-hydroisoquinoline synthesis. 3. A sixteen step synthesis of reserpine. Heterocycles. 1987;3:263–270. [Google Scholar]
- 16.Martin SF, Rueger H, Williamson SA, Grzejszczak S. General strategies for the synthesis of indole alkaloids. Total synthesis of (±)-reserpine and (±)-.alpha.-yohimbine. J Am Chem Soc. 1987;109:6124–6134. [Google Scholar]
- 17.Wenkert E, et al. Total synthesis of the yohimbines. J Am Chem Soc. 1979;101:5370–5376. [Google Scholar]
- 18.Sparks SM, Gutierrez AJ, Shea KJ. Preparation of perhydroisoquinolines via the intramolecular Diels Alder reaction of N-3,5-hexadienoyl ethyl acrylimidates: A formal synthesis of (±)-reserpine. J Org Chem. 2003;68:5274–5285. doi: 10.1021/jo0341362. [DOI] [PubMed] [Google Scholar]
- 19.Szantay C, Honty K, Toke L, Szabo L. Über eine einfache synthese der yohimbinalkaloide. Chem Ber. 1976;109:1737–1748. [Google Scholar]
- 20.Toke L, Honty K, Szabo L, Blasko G, Szantay C. Synthesis of yohimbines. I. Total synthesis of alloyohimbine and .alpha.-yohimbine and their epimers. Revised structure of natural alloyohimbine. J Org Chem. 1973;38:2496–2500. [Google Scholar]
- 21.Hudlicky T, Reed JW, editors. The way of synthesis. Wiley-VCH; Weinheim: 2007. pp. 541–571. [Google Scholar]
- 22.Phillips DD, Chadha MS. The alkaloids of Rauwolfia serpentine Benth. J Am Pharm Assoc (Baltim) 1955;44:553–567. doi: 10.1002/jps.3030440912. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee A. J Ind Chem Soc. 1941;18:485. [Google Scholar]
- 24.Karrer P, Salomon H. Uber zwei neue alkaloide aus der yohimberinde. Helv Chim Acta. 1926;9:1059–1062. [Google Scholar]
- 25.Benoin PR, Burnell RH, Medina JD. Alkaloids of Aspidosperma excelsum Benth. Can J Chem. 1967;45:725–730. [Google Scholar]
- 26.Takayama H, Ishikawa H, Kitajima M, Aimi N, Aji BM. A new 9-methoxyyohimbine-type indole alkaloid from Mitragyna africanus. Chem Pharm Bull. 2004;52:359–361. doi: 10.1248/cpb.52.359. [DOI] [PubMed] [Google Scholar]
- 27.Chatterjee A, Majumder PL, Ray AB. Structure of venoxidine, an alkaloid of Alstonia venenata R. Br. Tetrahedron Lett. 1965;42:159–162. doi: 10.1016/s0040-4039(01)99585-1. [DOI] [PubMed] [Google Scholar]
- 28.Govindachari TR, Viswanathan N, Pai BR, Savitri TS. Chemical constituents of Alstonia venenata R. Br. Tetrahedron Lett. 1964;16:901–906. doi: 10.1016/s0040-4020(01)98382-6. [DOI] [PubMed] [Google Scholar]
- 29.Govindachari TR, Viswanathan N, Pai BR, Savitri TS. Chemical constituents of Alstonia venenata R. Br. Tetrahedron. 1965;21:2951–2956. doi: 10.1016/s0040-4020(01)98382-6. [DOI] [PubMed] [Google Scholar]
- 30.Chatterjee A, Roy DJ, Mukhopadhyay S. 16-epivenenatine and 16-epialstovenine, new stereomers from Alstonia venenata. Phytochemistry. 1981;20:1981–1985. [Google Scholar]
- 31.Ray AB, Chatterjee A. Alstovenine, a new indole alkaloid isolated from Alstonia venenatus R. Br. J Indian Chem Soc. 1963;40:1043–1044. [Google Scholar]
- 32.Dutta SC, Ray AB. Indian J Chem. 1975;13:98–100. [Google Scholar]
- 33.Ray AB, Chatterjee A. Further studies on the major alkaloids of the stem-bark of Alstonia venenata R. Br. Structure and stereochemistry of alstovenine and its congeners. J Indian Chem Soc. 1964;41:638–640. [Google Scholar]
- 34.Bhattacharya SK, Ray AB, Dutta SC. Psychopharmacological investigations of the 4-methoxyindole alkaloids of Alstonia venenata. Planta Med. 1975;2:164–170. doi: 10.1055/s-0028-1097779. [DOI] [PubMed] [Google Scholar]
- 35.Vizi ES, et al. Berbanes: a new class of selective .alpha.2-adrenoceptor antagonists. J Med Chem. 1987;30:1355–1359. doi: 10.1021/jm00391a015. [DOI] [PubMed] [Google Scholar]
- 36.Vizi ES, et al. CH-38083, a selective, potent antagonist of alpha-2 adrenoceptors. J Pharmacol Exp Ther. 1986;238:701–706. [PubMed] [Google Scholar]
- 37.Toth I, et al. Investigations on the chemistry of berbanes. 10. Synthesis of raunescinone analogs with hypotensive and antihypertensive activity. J Med Chem. 1984;27:1411–1415. doi: 10.1021/jm00377a006. [DOI] [PubMed] [Google Scholar]
- 38.Jung ME, Light LA. Stereospecific synthesis of substituted cis-hydrindan-5-one and their regiospecific enolization and functionalization: synthetic intermediates for reserpine. J Am Chem Soc. 1984;106:7614–7618. [Google Scholar]
- 39.Ficini J, Guingant A, d’Angelo J. Synthese stereoselective des cis et trans carboxy-4-tetrahydro 3a, 4,5, 6 indanones-1. Tetrahedron Lett. 1983;24:907–910. [Google Scholar]
- 40.Lebold TP, Gallego GM, Marth CJ, Sarpong R. Synthesis of the bridging framework of phragmalin-type limonoids. Org Lett. 2012;14:2110–2113. doi: 10.1021/ol300647k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ley SV, Murray PJ, Palmer BD. Total synthesis of the sesquiterpene (±)-hirsutene using an organoselenium-mediated cyclization reaction. Tetrahedron. 1985;41:4765–4769. [Google Scholar]
- 42.Farrugia LJ. Ortep-3 for Windows - A Version of ORTEP-III with a Graphical User Interface (GUI) J Appl Crystallogr. 1997;30:565. [Google Scholar]
- 43.Otero N, Mandado M, Mosquera RA. Nucleophilicity of indole derivatives: Activating and deactivating effects based on proton affinities and electron density properties. J Phys Chem A. 2007;111:5557–5562. doi: 10.1021/jp0708953. [DOI] [PubMed] [Google Scholar]
- 44.Pratihar S, Roy S. Nucleophilicity and site selectivity of commonly used arenes and heteroarenes. J Org Chem. 2010;75:4957–4963. doi: 10.1021/jo100425a. [DOI] [PubMed] [Google Scholar]
- 45.Han D, et al. Conformational analysis of the cis- and trans-adducts of the Pictet Spengler Reaction. Evidence for the structural basis for the C(1)–N(2) scission process in the cis- to trans-isomerization. J Nat Prod. 2007;70:75–82. doi: 10.1021/np060391g. [DOI] [PubMed] [Google Scholar]
- 46.Deng L, Czerwinski K, Cook JM. Stereospecificity in the Pictet-Spengler reaction kinetic vs thermodynamic control. Tetrahedron Lett. 1991;32:175–178. [Google Scholar]
- 47.Lounasmaa M, Jokela R. Stereoregulation of the C(12b)H-C(2)H relationship in the preparation of 2-substituted 1,2,3,4,6,7,12,12b-octahydro-indolo[2,3-a]quinolizines. Tetrahedron. 1989;45:3975–3992. [Google Scholar]
- 48.Reichardt C. Solvents and solvent effects in organic chemistry. 3. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- 49.Lodewyk MW, Siebert MR, Tantillo DJ. Computational prediction of 1H and 13C chemical Shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.