

Abstract
Neuroendocrine prostate cancer (NEPC), also referred to as anaplastic prostate cancer, is a lethal tumor that most commonly arises in late stages of prostate adenocarcinoma (PCA) with predilection to metastasize to visceral organs. In the current study, we explore for evidence that Aurora kinase A (AURKA) and N-myc (MYCN) gene abnormalities are harbingers of treatment-related NEPC (t-NEPC). We studied primary prostate tissue from 15 hormone naïve PCAs, 51 castration-resistant prostate cancers, and 15 metastatic tumors from 72 patients at different stages of disease progression to t-NEPC, some with multiple specimens. Histologic evaluation, immunohistochemistry, and fluorescence in situ hybridization were performed and correlated with clinical variables. AURKA amplification was identified in overall 65% of PCAs (hormone naïve and treated) from patients that developed t-NEPC and in 86% of metastases. Concurrent amplification of MYCN was present in 70% of primary PCAs, 69% of treated PCAs, and 83% of metastases. In contrast, in an unselected PCA cohort, AURKA and MYCN amplifications were identified in only 5% of 169 cases. When metastatic t-NEPC was compared to primary PCA from the same patients, there was 100% concordance of ERG rearrangement, 100% concordance of AURKA amplification, and 60% concordance of MYCN amplification. In tumors with mixed features, there was also 100% concordance of ERG rearrangement and 94% concordance of AURKA and MYCN co-amplification between areas of NEPC and adenocarcinoma. AURKA and MYCN amplifications may be prognostic and predictive biomarkers, as they are harbingers of tumors at risk of progressing to t-NEPC after hormonal therapy.
Introduction
The development of neuroendocrine prostate cancer (NEPC, also referred to as anaplastic prostate cancer) is thought to drive approximately 25% of the nearly 34,000 cases per year of lethal prostate cancer in the United States [1]. However, data from autopsy studies suggest that the incidence of NEPC may be significantly underestimated [2]. It is known that the amount of neuroendocrine differentiation increases with disease progression and correlates with patient exposure to long-term androgen deprivation therapy. Preclinical studies also support transformation of prostate adenocarcinoma (PCA) cells into neuroendocrine cells when depleted of androgen in vitro and in xenograft models [3–7]. Therefore, with the introduction of new highly potent androgen receptor (AR)-targeted therapies into the clinic, the incidence of treatment-related NEPC (t-NEPC) might escalate. Patients who develop t-NEPC have an aggressive clinical course and often develop visceral metastases, and most survive less than 1 year [8]. Because neuroendocrine cells do not express AR or secrete prostate-specific antigen (PSA), the PSA level tends to be low or does not elevate in proportion to clinical progression. Elevated serum markers of neuroendocrine differentiation, such as chromogranin A and neuron-specific enolase (NSE), may support the diagnosis.
The prostate canser-specific ERG gene rearrangement occurs in approximately 50% of t-NEPC [9–11] and there is concordance of ERG status and other molecular abnormalities between PCA and t-NEPC foci of mixed tumors [11]; these data strongly suggest that there is a similar cell of origin of PCA and t-NEPC. This also distinguishes t-NEPC from neuroendocrine tumors arising from other anatomic sites and suggests that molecular studies of PCA may provide insight into events that occur early before the development of t-NEPC.
We recently demonstrated that the cell cycle kinase, Aurora kinase A, and the transcription factor, N-myc, cooperate to induce neuroendocrine differentiation in prostate cancer [12]. Furthermore, treatment of NEPC models with an Aurora kinase inhibitor resulted in significant tumor shrinkage and reversal of the neuroendocrine phenotype, thereby providing rationale for clinical evaluation of an Aurora kinase inhibitor for patients with t-NEPC. In that study, we also showed that concurrent overexpression and amplification of the Aurora kinase A gene (AURKA) and N-myc gene (MYCN) in metastatic t-NEPC was significantly higher (40%) when compared to an unselected cohort of localized PCA (5%). Interestingly, one of the patients who progressed from PCA to t-NEPC had amplification of AURKA and MYCN in his primary hormone naïve PCA, suggesting that these molecular events occur early in the disease [12].
In the current study, we examined the histologic spectrum of t-NEPC and evaluated AURKA and MYCN amplifications in primary prostate tumors and metastases from 72 patients who developed lethal t-NEPC.
Materials and Methods
Case Selection
Pathology material from 72 patients who clinically developed NEPC was evaluated. Clinical parameters for the diagnosis of NEPC included rapid progression of the disease with visceral metastases in the setting of low (≤10 ng/ml) or modestly rising PSA and/or elevated neuroendocrine serum markers (chromogranin A > 5x upper limit of normal, NSE > 2x upper limit of normal). All patients received androgen deprivation therapy before disease progression toward NEPC. Cases were identified at different collaborating institutions under Institutional Review Board (IRB)-approved protocols for the purpose of this study. The clinical information collected for each patient included the age at diagnosis of PCA, clinical stage, type of primary and systemic therapy, interval of time between initial diagnosis and castration-resistant state, interval of time between castration-resistant prostate cancer (CRPC) and metastatic disease including sites of metastases, and death.
For comparison purposes, an unselected cohort of 169 patients with localized PCA who underwent radical prostatectomy (RP) at Weill Cornell Medical Center was used. In addition, two pathology specimens from patients with primary (de novo) mixed small cell carcinoma with prostatic adenocarcinoma were assessed, and six prostatectomy cases of hormone naïve, localized PCA with Paneth cell-like neuroendocrine change were included in the study as separate controls of low-grade neuroendocrine differentiation [13].
A summary of clinical characteristics of patients included in the study is presented in Table 1.
Table 1.
Clinicopathologic Characteristics of Patients Who Developed t-NEPC and Controls.
| Archival material studied from patients who developed t-NEPC (n = 72), 100% |
| Primary hormone naïve PCA only (n = 11), 15.3% |
| Primary hormone naïve PCA, treated PCA, and subsequent metastases (n = 1), 1.4% |
| Primary hormone naïve PCA and subsequent metastases (n = 3), 4.2% |
| Gleason score of localized PCA: 3+3=6 to 5+5=10 |
| Pathologic stage of localized PCA: pT2c N0 to pT3a N1 |
| Treated PCA only (n = 49), 68.0% |
| Treated PCA and subsequent metastases (n = 1), 1.4% |
| Metastases only* (n = 7), 9.7% |
| Age at diagnosis of PCA: 42 to 84 years (median = 65 years) |
| Time interval to progression to CRPC: 2 to 10 years (median = 4 years) |
| Overall survival after clinical diagnosis of NEPC: 8 to 14 months (median = 12 months) |
| Treatments received as monotherapy or in combination: RP, radiation therapy, androgen deprivation therapy including MDV3100 or abiraterone, chemotherapy (carboplatin + taxol, carboplatin + etoposide, docetaxel, irinotecan, docetaxel + radium-223 chloride) (Table W1) |
| Unselected cohort of patients with localized PCA who underwent RP (n = 169), 100% |
| Age at diagnosis of PCA: 42 to 75 years (median = 62 years) |
| Gleason score of localized PCA: 3+3=6 to 4+5=9 |
| Pathologic stage of localized PCA: pT2a N0 to pT3b N0 |
| Primary (de novo) NEPC (mixed small cell carcinoma and PCA) (n = 2), 100% |
| Age at diagnosis of de novo NEPC: 65 and 67 years |
| Pathologic stage (RP performed): pT3a N0 both cases |
| Localized PCA with Paneth cell-like neuroendocrine differentiation (n = 6), 100% |
| Age at diagnosis of PCA: 54 to 74 years (median = 65 years) |
| Gleason score of localized PCA: 3+3=6 to 4+5=9 |
| Pathologic stage of localized PCA: pT2a N0 to pT3b N1 |
Fifteen metastatic sites included retroperitoneum (one), colon (one), bladder (three), brain (two), pleura (one), pelvic soft tissue (two), liver (one), and bone (four).
Pathologic Evaluation
Formalin-fixed paraffin-embedded tissue of the aforementioned cases was available. Regarding archival material from the 72 patients who clinically progressed to t-NEPC, different specimens were available corresponding to different stages of disease (see below). Hematoxylin and eosin (H&E)-stained slides from surgical resections and biopsies were reviewed by study pathologists (J.M.M., K.P., B.D.R., and M.A.R.). Pathologic evaluation included Gleason score of untreated tumors (prostate biopsy, transurethral resection, and/or prostatectomy specimens), histologic examination of metastases and treated prostate tumors, and pathologic tumor stage. Classification of the spectrum of neuroendocrine tumors, both primary and metastatic PCAs, was applied using the definitions used in lung classification [14,15]. Briefly, tumors with neuroendocrine morphology were small cell carcinoma (pure or combined) and large cell neuroendocrine carcinoma, and non-small cell carcinomas were poorly differentiated adenocarcinomas of the prostate with or without neuroendocrine differentiation. Poorly differentiated adenocarcinomas were considered to have neuroendocrine differentiation when more than 30% of tumor cells were positive for synaptophysin or chromogranin A.
Archival tissue from the 72 patients who developed t-NEPC included 11 primary hormone naïve PCA cases only (matched treated PCA or metastasis unavailable), 1 case of hormone naïve PCA with available tissue of treated PCA and metastases, 3 cases of hormone naïve PCA and subsequent metastases after treatment (matched treated PCA unavailable), 49 treated PCA only (matched hormone naïve PCA or metastases unavailable), 1 treated PCA and subsequent metastases (matched hormone naïve PCA unavailable), and 7 cases of metastases only (matched hormone naïve or treated PCA unavailable) (Table 1 and Figure W1A). A total of 15 metastatic tumors from 12 patients were interrogated. Sites of metastases included retroperitoneum (n = 1), colon (n = 1), bladder (n = 3), brain (n = 2), pleura (n = 1), pelvic soft tissue (n = 2), liver (n = 1), and bone (n = 4). Some patients presented with synchronous metastases at other anatomic locations including peritoneum, lungs, and stomach.
Overall, the assessed pathology material of these patients, some with multiple specimens from different stages in progression toward t-NEPC, included 15 hormone naïve clinically localized PCAs and 66 treated tumors: 51 treated PCAs and 15 metastases, the latter being from 12 patients (see Figure W1A).
A subset of 19 neuroendocrine tumors from non-prostate origin was also interrogated for AURKA and MYCN amplifications and included primary small cell carcinoma of lung (n = 12) and bladder (n = 2), metastatic small cell carcinoma of lung to cerebellum (n = 1), well-differentiated neuroendocrine tumor (“typical carcinoid”) of bowel (n = 1), metastatic well-differentiated neuroendocrine tumor (“typical carcinoid”) of bowel to liver (n = 1) and lung (n = 1), and mammary ductal carcinoma in situ with neuroendocrine differentiation (n = 1).
Fluorescence In Situ Hybridization
To assess AURKA and MYCN amplifications and PTEN status, we used a locus-specific probe plus reference probe fluorescence in situ hybridization (FISH) assays as previously described [12,16]. The reference probe was located at 10q25 (BAC RP11431P18), spanning a stable region of the chromosome. Amplification was defined as the presence of four or more copies on average for gene-specific (AURKA or MYCN) signals per nucleus compared to two reference signals. ERG rearrangement was assessed using dual-color break-apart interphase FISH assay as described previously [17,18]. At least 50 nuclei were evaluated per tissue section using a fluorescence microscope (Olympus BX51; Olympus Optical, Tokyo, Japan).
Immunohistochemistry
Immunohistochemistry (IHC) stain was performed in a subset of 44 cases using antibodies for synaptophysin (Clone SP11 from Lab Vision/Thermo Fisher Scientific, Kalamazoo, MI) and chromogranin A (Clone LK2H10 from Biogenex, Fremont, CA), following vendors' specified optimal dilutions for IHC. In a subset of 15 cases (primary PCA of patients who developed t-NEPC), IHC for Aurora kinase A was performed (ab13824 from Abcam Inc, Cambridge, MA; 1:800 dilution).
Results
Clinical Characteristics of Patients Who Progressed to t-NEPC
Full clinical information was available in 43 of 72 patients and partial clinical information in the remaining 29 patients. The age at diagnosis of PCA ranged from 42 to 84 years (median = 65 years). Time interval to progression to CRPC ranged from 2 to 10 years (median = 4 years), time on androgen deprivation therapy ranged from 1 to 11 years (median = 4 years), and overall survival after clinical diagnosis of NEPC ranged from 8 to 14 months (median = 12 months).
Treatment modalities received since diagnosis of PCA encompassed one or more of the following: RP, radiation therapy (external beam radiation and brachytherapy), androgen deprivation therapy such as luteinizing hormone-releasing hormone analogs, luteinizing hormone-releasing hormone antagonists, and anti-androgens including MDV3100 and abiraterone, and chemotherapy protocols with carboplatin + paclitaxel, carboplatin + etoposide, docetaxel, irinotecan, docetaxel + radium-223 chloride.
Detailed treatment received including hormonal and chemotherapy in a subset of patients along with survival, pathology, and FISH data is presented in Table W1.
The age at diagnosis of PCA in the unselected cohort of 169 patients used for comparison (RP only) ranged from 42 to 75 years (median = 62 years).
Histopathology
Microscopic evaluation of 51 treated PCA cases and 15 metastases demonstrated three major histologic groups: 1) pure neuroendocrine prostate carcinoma, which included small cell carcinoma (n = 18) and large cell neuroendocrine carcinoma (n = 1); 2) poorly differentiated adenocarcinoma with (n = 21) or without (n = 8) neuroendocrine differentiation; 3) mixed neuroendocrine carcinoma and adenocarcinoma (n = 18). Among the latter group of 18 cases with mixed morphology, the neuroendocrine carcinoma component included areas of small cell carcinoma (n = 15) and large cell neuroendocrine carcinoma (n = 3) (Table 2). The spectrum of NEPC, primary and metastatic, is illustrated in Figure 1.
Table 2.
ThreeHistologicGroups of Treated PCA and Metastases from Patients Who Developed t-NEPC.
Treated PCA cases (n = 51) and metastatic sites (n = 15) examined (n = 66), 100%
|
Figure 1.

Morphologic spectrum of t-NEPC. (A) Small cell carcinoma of the prostate. The tumor is composed of sheets of uniform cells with scant cytoplasm, hyperchromatic nuclei, coarse chromatin, and unapparent nucleoli. (B) Large cell neuroendocrine carcinoma of the prostate. Tumor is composed of sheets and ribbons of cells with abundant cytoplasm, large nuclei with coarse chromatin, brisk mitotic activity, and foci of necrosis; pseudorosettes are also apparent. (C) Metastatic poorly differentiated adenocarcinoma of the prostate without neuroendocrine differentiation, treated (metastatic CRPC). Sheets of tumor cells with pale eosinophilic cytoplasm and abundant mitotic figures are seen within fibroadipose tissue. (D) Poorly differentiated adenocarcinoma of the prostate with neuroendocrine differentiation, treated (CRPC). Note the vaguely organoid pattern of tumor cells, which have amphophilic cytoplasm and prominent nucleoli. (E) Poorly differentiated adenocarcinoma of the prostate with focal areas of neuroendocrine differentiation, treated (CRPC). Areas of tumor cells with neuroendocrine differentiation are interspersed and demonstrate basophilic appearance. (F) Mixed t-NEPC and adenocarcinoma of prostate, treated (CRPC). Areas of small cell carcinoma and poorly differentiated adenocarcinoma are seen (H&E stain, original magnification, x200).
The prostate specimens from two patients who presented with primary (de novo) NEPC corresponded to one case of mixed small cell carcinoma with areas of PCA Gleason score 5 + 4 = 9 (prostate needle biopsies) and one case of mixed large cell neuroendocrine carcinoma with areas of PCA with ductal features (transurethral resection of prostate).
The Gleason scores of 15 hormone naïve PCAs (i.e., initial diagnosis of patients who later developed t-NEPC) ranged from 3 + 3 = 6 to 5 + 5 = 10, and their pathologic tumor stage ranged from pT2c N0 to pT3a N1. Areas of benign prostate tissue were also identified in these 15 specimens.
The Gleason scores of the unselected cohort of 169 cases of localized PCA ranged from 3 + 3 = 6 to 4 + 5 = 9, and their pathologic tumor stage ranged from pT2a N0 to pT3b N0. In addition, archival material from this control cohort also included 50 benign prostate tissue samples.
Gleason scores of the six PCA cases with Paneth cell-like neuroendocrine differentiation ranged from 3 + 3 = 6 to 4 + 5 = 9 (Table 1).
FISH and IHC Results
In the group of primary hormone naïve PCA cases from patients who clinically progressed to t-NEPC, AURKA amplification was identified in 10 of 15 (67%) cases, seven of which (70%) also had concurrent MYCN amplification. Protein overexpression of Aurora kinase A was confirmed by IHC in five of such seven cases. Aurora kinase A overexpression was seen as multifocal and scattered positive nuclei, as illustrated in Figure W2. The presence of AURKA or MYCN amplification was not associated with neuroendocrine marker (synaptophysin and chromogranin A) expression in these primary PCA cases.
Among t-NEPC cases, AURKA amplification was identified in 29 of 46 (63%) treated PCAs and in 12 of 14 (86%) metastases that were assessable. Concurrent MYCN amplification was present in 20 of 29 treated tumors (69%) and in 10 of 12 metastases (83%) that were evaluable (see Figure W1B).
In only two of all 75 specimens assessable by FISH did MYCN gain occurred without concurrent AURKA amplification; both cases previously treated with hormonal therapy, one was obtained from prostate and the other from a bladder mass.
In contrast, AURKA amplifications were identified only in 5% (8 of 169 cases) of the unselected PCA cohort, with concurrent MYCN amplification identified in 7 of 169 of cases (not seen in absence of AURKA amplification). Particularly noteworthy is the fact that AURKA amplification was detected in all six cases of PCA with Paneth cell-like neuroendocrine differentiation, one of them with concurrent MYCN amplification (Figure 2).
Figure 2.

Prostate cancer with Paneth cell-like neuroendocrine differentiation harbors AURKA amplification. (A–F) Six cases of localized prostate cancer with Paneth cell-like change were identified and used as separate controls of low-grade neuroendocrine differentiation. On H&E stain, tumor cells with Paneth cell-like neuroendocrine differentiation are easily identified and contain distinct large eosinophilic granules in the cytoplasm. One case (A) demonstrated AURKA and MYCN amplifications. The other five cases (B–F) harbored AURKA amplification only (insets). ERG rearrangement, one through insertion (D) and one through deletion (A), is identified in two of these cases (insets). Clusters of tumor cells with Paneth cell-like neuroendocrine differentiation are located around asterisks, and more focal areas are marked with arrowheads (H&E stain, original magnification, x400; FISH images, original magnification, x600).
Of the two de novo NEPC cases, one showed AURKA amplification with concurrent MYCN polysomy, and the other one was negative for either amplification. The results of AURKA and MYCN amplifications by FISH are summarized in Table 3.
Table 3.
Results of AURKA and MYCN Amplifications by FISH.
| Group | Assessable Cases/Total Cases | AURKA Amplification | Concurrent MYCN Amplification |
|---|---|---|---|
| Hormone naïve PCA of patients who developed t-NEPC | 15/15 | 67% (10/15) | 70% (7/10) |
| Treated PCA (CRPC and t-NEPC) | 46/51 | 63% (29/46) | 69% (20/29) |
| Metastatic t-NEPC | 14/15 | 86% (12/14) | 83% (10/12) |
| Control cohort of patients with localized PCA | 169/169 | 5% | 100% (8/8) |
| Primary (de novo) NEPC | 2/2 | 50% (1/2) | 100% (1/1) |
| PCA with Paneth cell-like neuroendocrine differentiation | 6/6 | 100% (6/6) | 17% (1/6) |
AURKA and MYCN amplifications were detected in more than 95% of nuclei evaluated on each positive case. No AURKA or MYCN amplification was detected in benign prostate tissue (n = 50). In all cases of primary PCA, the presence of AURKA and MYCN amplifications was independent of other clinical prognostic features (Gleason grade, serum PSA, and stage) including neuroendocrine marker expression (chromogranin A and synaptophysin) by IHC.
In the five cases where metastatic t-NEPC was compared to primary PCA from the same patient, either hormone naïve or treated PCA, there was 100% concordance (five of five matching cases) of AURKA amplification. MYCN amplification was present in three of five cases (60% concordance), with metastatic t-NEPC demonstrating MYCN amplification at all times. Histologic and molecular findings of three of these cases are illustrated in Figure 3. In prostate tumors with mixed features, there was 94% concordance in AURKA/MYCN amplification between areas of neuroendocrine carcinoma and adenocarcinoma. An example of such combined histomorphology is highlighted in Figure 4, with both areas showing AURKA and MYCN amplifications.
Figure 3.

AURKA and MYCN amplifications in primary prostatic adenocarcinoma predict the development of t-NEPC. (A–D) Top panel illustrates several specimens from a patient at different stages of disease progression to t-NEPC. (A and B) Images of hormone naïve prostate cancer with areas of Gleason score 3 + 3 = 6 (A) and 4 + 5 = 9 (B) at initial diagnosis. Concurrent AURKA (upper inset) and MYCN (middle inset) amplifications are present in both areas. (C) Subsequent metastasis/local recurrence in the bladder demonstrates poorly differentiated adenocarcinoma without neuroendocrine differentiation, exhibiting both AURKA and MYCN amplifications (upper and middle insets, respectively). (D) Five years after treatment, the patient presents with metastatic large cell neuroendocrine carcinoma in pelvic soft tissue. The tumor has organoid appearance focally forming pseudorosettes, and cells have abundant cytoplasm and prominent nucleoli. The tumor has both AURKA and MYCN amplifications (upper and middle insets, respectively). Clonal origin is confirmed by ERG rearrangement through translocation in all tumors (lower inset). (E and F) Center panel illustrates prostatectomy specimen from a patient with initial diagnosis of PCA Gleason score 4 + 5 = 9 (E), which has concurrent AURKA and MYCN amplifications (upper and middle insets, respectively). A liver biopsy 7 years after (F) shows metastatic small cell carcinoma, which harbors AURKA and MYCN coamplification as well. Clonal origin is confirmed by ERG rearrangement through deletion in both tumors (lower inset). (G and H) Lower panel illustrates needle biopsies from a patient with initial diagnosis of (G) PCA Gleason score 3 + 4 = 7 with intraductal spread (IDC-P) with amplification of AURKA (upper inset) but not MYCN (middle inset). Eight years after initial diagnosis and intermittent treatment, the patient developed pancytopenia and bone lytic lesions, which biopsy demonstrates (H) metastatic small cell carcinoma (frozen tissue artifact present), consistent with spread from known prostatic primary. In addition to AURKA amplification (upper inset), clonal origin is confirmed by ERG rearrangement through translocation in both tumors (lower inset). The metastatic tumor demonstrates MYCN amplification (middle inset) (H&E stain, original magnification, x200; FISH images, original magnification, x600).
Figure 4.

Concordance of AURKA and MYCN amplifications in tumors with mixed areas of neuroendocrine carcinoma and poorly differentiated adenocarcinoma. Representative image of local recurrence of castration-resistant prostatic carcinoma with areas of mixed small cell carcinoma (right) and adenocarcinoma (left). Both areas demonstrate concordance of AURKA and MYCN amplifications (upper and middle insets, respectively). Clonal origin is supported by ERG rearrangement through translocation in both areas (lower inset) (H&E stain, original magnification, x200; FISH images, original magnification, x600).
Overall, ERG rearrangement was observed in 29 of 69 (42%) assessable tumors (PCA and metastases) in the t-NEPC cohort, 10 through insertion and 19 through deletion. PTEN deletion was observed in 14 of 49 (29%) assessable cases, eight of which were also ERG rearranged. There was no association between AURKA/MYCN amplification and ERG rearrangement or PTEN deletion status. Among the five cases of metastatic t-NEPC with matching PCA (hormone naïve or treated), four were positive for ERG rearrangement in the PCA and corresponding metastases, and one was negative for such gene rearrangement in both sites. This 100% concordance of ERG rearrangement is compatible with a clonal origin of t-NEPC, identical to those findings observed in tumors with mixed features (Figure 4).
In neuroendocrine tumors from non-prostate origin, AURKA amplification was detected in 10 of 11 (91%) assessable primary small cell carcinomas of lung including the metastasis to cerebellum and in one assessable primary small cell carcinoma of the bladder. MYCN amplification was detected in seven of these cases (64%), always in the presence of AURKA amplification. In contrast, AURKA/MYCN amplifications were not seen in well-differentiated neuroendocrine tumor (“typical carcinoid”) of bowel and metastases or in ductal carcinoma in situ with neuroendocrine differentiation.
Discussion
The findings of the current study suggest that a broader definition of NEPC is desirable to capture the wide spectrum of this disease. Although the occurrence of de novo NEPC is rare, the incidence of NEPC that secondarily arises with disease progression and after therapy appears common and is associated with poor clinical outcome [19–25]. Given emerging preclinical and clinical evidence supporting promotion of neuroendocrine transformation by androgen deprivation therapies [3–7,26,27], we propose that these secondary NEPC tumors should be termed t-NEPC. Widely under-recognized, especially as patients with advanced stage disease are rarely biopsied to make the diagnosis, progression toward an AR-negative neuroendocrine phenotype is one proposed mechanism by which tumors acquire resistance to hormonal therapies. Thus, the landscape of advanced prostate cancer is evolving as novel potent AR-targeted therapies enter widespread clinical use (e.g., abiraterone acetate, MDV3100, TAK700) and patients develop resistance, and the incidence of t-NEPC will presumably escalate. Alternative mechanisms of resistance also include up-regulation or continued activation of the AR [28]. Recognition of resistance associated with the development of an AR-negative t-NEPC versus an AR-activated CRPC is essential, as this affects how patients may respond to subsequent therapies; for instance, patients with t-NEPC would be less likely to respond to hormonal agents and may better respond to chemotherapy or enrollment in a clinical trial.
Although there are some limitations regarding clinical data presented in the current study, this retrospective cohort represents the largest tissue collection of t-NEPC reported to date. The main purpose of this study was to generate hypotheses regarding novel biomarkers and insight into the pathogenesis of t-NEPC. This work will be the basis for future clinical studies, including the prospective evaluation of AURKA and MYCN in primary tumors and metastases from patients with t-NEPC. If validated in prospective trials, these biomarkers would be useful for clinical decision making. For this purpose, defining the histomorphology and molecular characterization of t-NEPC become critical steps toward understanding the spectrum of disease, especially as efforts are made to incorporate the latest discoveries into the clinic, either as potential targets or in the form of significant biomarkers, both for diagnostic and prognostic uses. Planned clinical trials incorporating metastatic tumor biopsies for patients with CRPC including those patients with NEPC treated with an Aurora kinase inhibitor will be valuable for prospective correlation of pathologic findings and genomic sequencing results with clinical features and will help further define t-NEPC.
Former studies have described the phenotypic features of metastatic hormone-refractory PCA to bone [29] and visceral sites [30,31], and of pure small cell carcinoma of prostate, which occurrence is rare (less than 1% of PCA cases) [32]. Here, we highlight that most treated prostate tumors and metastases from patients who clinically developed NEPC, including de novo NEPC cases, are part of a morphologic spectrum that encompasses pure neuroendocrine histology and mixed tumors with areas of poorly differentiated adenocarcinoma. Although the term anaplastic small cell carcinoma has also been used clinically to describe tumor progression in patients with CRPC [33,34], the histologic features of treated prostate tumors from patients who progress to NEPC, both primary and metastatic, are heterogeneous. The term t-NEPC may be a more useful descriptor for the clinical scenario of patients with CRPC with rapid disease progression (visceral and/or lytic bone metastases) and low serum PSA, especially in the setting of potent androgen deprivation therapy.
Some molecular characteristics of CRPC (e.g., ERG, PTEN status) have been described earlier [30,35–38]. In our series, ERG rearrangement was observed in 42% of cases, which is at similar frequency as reported in other cohorts. However, ERG rearrangement occurred more often through deletion (as opposed to translocation), a finding that has been previously associated with an aggressive behavior [37,39]. These findings also support clonal origin of t-NEPC and acinar prostate cancer, previously demonstrated by concordance of ERG rearrangement in tumors with mixed features [9–11] and in primary PCA and metastatic NEPC [8] (Figure 4). This has important clinical implications, as it suggests that adenocarcinomas may harbor molecular lesions before the development of t-NEPC, which is relevant toward the development of novel biomarkers and identifying patients at high risk for progression.
We previously identified higher expression and amplification of AURKA and MYCN in NEPC in contrast to hormone naïve PCA [12]. In the current study, AURKA amplification was also detected in overall 65%primary PCAfrompatients that later developed t-NEPC, with concurrent MYCN amplification in a substantial proportion of cases. This is highly significant, especially when compared to 5% frequency of AURKA and MYCN amplifications observed in an unselected primary PCA population. Furthermore, these alterations were independent of Gleason score, PSA level, or pathologic tumor stage at initial diagnosis, suggesting that they add prognostic value. In the current study, we also did not observe any correlation with Gleason score. Importantly, AURKA and MYCN amplifications were also seen in low-grade Gleason 3 + 3 = 6 tumors, the type of tumors that might be considered for active surveillance. Because AURKA and MYCN alterations arise early, other genetic changes are clearly also important for disease progression. There was also high concordance of AURKA and MYCN amplifications (100%and 60%, respectively) when ensuing t-NEPC or later metastases were also interrogated (Figure 3); this suggests that AURKA and MYCN alterations are acquired early and persist during disease progression. The importance is two-fold: AURKA is targetable (i.e., Aurora kinase A inhibitors), and AURKA and MYCN amplifications may represent new prognostic and predictive biomarkers because, as demonstrated in our current study, their presence identifies patients with PCA who are at risk of progressing to t-NEPC after androgen deprivation therapy. These patients may therefore benefit from early intervention with an Aurora kinase A inhibitor.
Paneth cell-like change in PCA has been suggested to represent a low-grade neuroendocrine differentiation with generally favorable prognosis [13]. Noteworthy, all six cases of PCA with these features harbored AURKA amplification. This particular histomorphology of PCA may be enriched for AURKA amplification, with resultant significant clinical implication (Figure 2). This might therefore suggest that these tumors may not be low grade. In the prior study by Tamas and Epstein [13], patients with PCA with Paneth cell-like neuroendocrine differentiation including tumors with Gleason pattern 5 had no evidence of progression with mean and median follow-ups ranging from 42.5 to 60.5 months. These criteria were used to assign PCA with Paneth cell-like neuroendocrine differentiation to a category of favorable prognosis. Given the high association with AURKA amplification and the development of t-NEPC illustrated in our current study, future studies with longer follow-up and detailed hormonal therapy are needed. PCA with Paneth cell-like neuroendocrine differentiation should best be viewed for now as uncertain clinical behavior.
AURKA and MYCN amplifications were also identified in small cell carcinomas of non-prostate origin but not in well-differentiated neuroendocrine tumors. This warrants further exploration for the role of AURKA and MYCN in small cell carcinomas of other primary sites. In addition, it demonstrates that both AURKA and MYCN amplifications may be good markers of neuroendocrine differentiation, but unlike the recurrent ETS rearrangements or SPOP mutations [40] they are not PCA specific.
Finally, this study also highlights the divide between the clinical presentation of NEPC and the pathologist's view of NEPC. Surgical pathologists only rarely encounter NEPC in their routine clinical practice due in part to the rare nature of de novo small cell prostate cancer, selection bias toward cases that are amenable to surgery and radiation, and the near universal absence of systematic biopsies for men with CRPC. More recently, clinical protocols are incorporating biopsies during the treatment course of CRPC. As exposed in this study and other recent studies that have evaluated metastatic samples, there is a wider range of morphology than normally encountered in hormone naïve prostate cancer. As a result, there is a significant gap in knowledge as to how treatment alters the course of many solid tumors but particularly prostate cancer, when hormonal or taxane-based therapy can be administered on the basis of clinical symptoms of bone pain, radiology images, or elevated PSA results without first obtaining a tissue diagnosis. We anticipate that given the more aggressive approach to obtaining biopsies on clinical trials for CRPC, we will need to more formally address the pathologic and molecular changes that occur as a result of targeted therapies. Therefore, we would advocate keeping a more encompassing broad view for the definition of NEPC until we better understand the biology and response to treatment through emerging clinical trials with mandatory tissue biopsies.
In summary, t-NEPC is a clinical entity that has an array of histologic features including pure neuroendocrine morphology (small cell carcinoma in most cases) and mixed tumors with poorly differentiated adenocarcinoma component. AURKA and MYCN amplifications occur early and are present in hormone naïve tumors from patients who ultimately progress to t-NEPC after androgen deprivation therapy (Figure 5). Therefore, AURKA and MYCN amplifications may be prognostic and predictive biomarkers, as they are harbingers of tumors at risk of progressing to t-NEPC after hormonal therapy and may identify patients that could potentially benefit from Aurora kinase inhibitor therapy.
Figure 5.
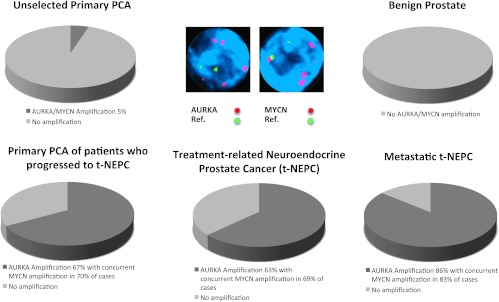
Concurrent AURKA and MYCN gene amplifications are harbingers of lethal t-NEPC. AURKA and MYCN gene amplifications evaluated by FISH are not present in benign prostate tissue and identified only in 5% of unselected primary prostate cancers. In contrast, 67% of primary tumors from patients who clinically develop t-NEPC harbor AURKA amplification, 70% of which also demonstrate concurrent MYCN amplification. Similar frequency of AURKA/MYCN amplification is present in t-NEPC. Metastatic t-NEPC harbors AURKA amplification in 86% of cases, with 83% MYCN co-amplification.
Supplementary Material
Footnotes
This work was supported by NCI R01 CA152057 (M.A.R.), Early Detection Research Network NCI U01 CA111275 (J.M.M. and M.A.R.), and the Prostate Cancer Foundation (H.B. and M.A.R.). M.A.R. is a co-inventor of the patent on the detection of gene fusions in prostate cancer, filed by the University of Michigan and the Brigham and Women's Hospital. The diagnostic field of use for ETS gene fusions has been licensed to Hologic Gen-Probe. The authors declare that there are no relationships that could be construed as resulting in an actual, potential, or perceived conflict of interest with regard to the current manuscript submitted for review.
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Brawn PN, Speights VO. The dedifferentiation of metastatic prostate carcinoma. Br J Cancer. 1989;59(1):85–88. doi: 10.1038/bjc.1989.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ismail AH, Landry F, Aprikian AG, Chevalier S. Androgen ablation promotes neuroendocrine cell differentiation in dog and human prostate. Prostate. 2002;51(2):117–125. doi: 10.1002/pros.10066. [DOI] [PubMed] [Google Scholar]
- 4.Ito T, Yamamoto S, Ohno Y, Namiki K, Aizawa T, Akiyama A, Tachibana M. Up-regulation of neuroendocrine differentiation in prostate cancer after androgen deprivation therapy, degree and androgen independence. Oncol Rep. 2001;8(6):1221–1224. doi: 10.3892/or.8.6.1221. [DOI] [PubMed] [Google Scholar]
- 5.Shen R, Dorai T, Szaboles M, Katz AE, Olsson CA, Buttyan R. Transdifferentiation of cultured human prostate cancer cells to a neuroendocrine cell phenotype in a hormone-depleted medium. Urol Oncol. 1997;3(2):67–75. doi: 10.1016/s1078-1439(97)00039-2. [DOI] [PubMed] [Google Scholar]
- 6.Wright ME, Tsai MJ, Aebersold R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Mol Endocrinol. 2003;17(9):1726–1737. doi: 10.1210/me.2003-0031. [DOI] [PubMed] [Google Scholar]
- 7.Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, Karan D, Batra SK, Lin MF. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13(1):151–167. doi: 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 8.Beltran H, Tagawa ST, Park K, MacDonald TY, Milowsky MI, Mosquera JM, Rubin MA, Nanus DM. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30(36):e386–e389. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 9.Guo CC, Dancer JY, Wang Y, Aparicio A, Navone NM, Troncoso P, Czerniak BA. TMPRSS2-ERG gene fusion in small cell carcinoma of the prostate. Hum Pathol. 2011;42(1):11–17. doi: 10.1016/j.humpath.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, Hicks JL, Meeker AK, Bieberich CJ, De Marzo AM, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24(6):820–828. doi: 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson SR, Zhang S, Yao JL, Huang J, Lopez-Beltran A, Shen S, Osunkoya AO, MacLennan GT, Montironi R, Cheng L. ERG-TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol. 2011;24(8):1120–1127. doi: 10.1038/modpathol.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang YW, Sheikh KL, Terry S, Tagawa ST, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487–495. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamas EF, Epstein JI. Prognostic significance of Paneth cell-like neuroendocrine differentiation in adenocarcinoma of the prostate. Am J Surg Pathol. 2006;30(8):980–985. doi: 10.1097/00000478-200608000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol. 2010).;21(suppl 7):vii65–vii71. doi: 10.1093/annonc/mdq380. [DOI] [PubMed] [Google Scholar]
- 15.Travis WD, Linnoila RI, Tsokos MG, Hitchcock CL, Cutler GB, Jr, Nieman L, Chrousos G, Pass H, Doppman J. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol. 1991;15(6):529–553. doi: 10.1097/00000478-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, Tchinda J, Tomlins SA, Hofer MD, Pienta KG, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66(17):8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 18.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 19.Aprikian AG, Cordon-Cardo C, Fair WR, Zhang ZF, Bazinet M, Hamdy SM, Reuter VE. Neuroendocrine differentiation in metastatic prostatic adenocarcinoma. J Urol. 1994;151(4):914–919. doi: 10.1016/s0022-5347(17)35121-2. [DOI] [PubMed] [Google Scholar]
- 20.Aprikian AG, Cordon-Cardo C, Fair WR, Reuter VE. Characterization of neuroendocrine differentiation in human benign prostate and prostatic adenocarcinoma. Cancer. 1993;71(12):3952–3965. doi: 10.1002/1097-0142(19930615)71:12<3952::aid-cncr2820711226>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 21.Abrahamsson PA. Neuroendocrine cells in tumour growth of the prostate. Endocr Relat Cancer. 1999;6(4):503–519. doi: 10.1677/erc.0.0060503. [DOI] [PubMed] [Google Scholar]
- 22.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999;39(2):135–148. doi: 10.1002/(sici)1097-0045(19990501)39:2<135::aid-pros9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 23.Abrahamsson PA, Falkmer S, Falt K, Grimelius L. The course of neuroendocrine differentiation in prostatic carcinomas. An immunohistochemical study testing chromogranin A as an “endocrine marker”. Pathol Res Pract. 1989;185(3):373–380. doi: 10.1016/S0344-0338(89)80016-0. [DOI] [PubMed] [Google Scholar]
- 24.di Sant'Agnese PA. Neuroendocrine differentiation in prostatic carcinoma: an update. Prostate Suppl. 1998;8:74–79. [PubMed] [Google Scholar]
- 25.di Sant'Agnese PA, Cockett AT. Neuroendocrine differentiation in prostatic malignancy. Cancer. 1996;78(2):357–361. doi: 10.1002/(SICI)1097-0142(19960715)78:2<357::AID-CNCR27>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 26.Berruti A, Mosca A, Porpiglia F, Bollito E, Tucci M, Vana F, Cracco C, Torta M, Russo L, Cappia S, et al. Chromogranin A expression in patients with hormone naïve prostate cancer predicts the development of hormone refractory disease. J Urol. 2007;178(3 pt 1):838–843. doi: 10.1016/j.juro.2007.05.018. quiz 1129. [DOI] [PubMed] [Google Scholar]
- 27.Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45(5):586–592. doi: 10.1016/j.eururo.2003.11.032. discussion 592. [DOI] [PubMed] [Google Scholar]
- 28.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 29.Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003;34(7):646–653. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- 30.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 31.Rubin MA, Putzi M, Mucci N, Smith DC, Wojno K, Korenchuk S, Pienta KJ. Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res. 2000;6(3):1038–1045. [PubMed] [Google Scholar]
- 32.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32(1):65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz LH, LaTrenta LR, Bonaccio E, Kelly WK, Scher HI, Panicek DM. Small cell and anaplastic prostate cancer: correlation between CT findings and prostate-specific antigen level. Radiology. 1998;208(3):735–738. doi: 10.1148/radiology.208.3.9722854. [DOI] [PubMed] [Google Scholar]
- 34.Rubenstein JH, Katin MJ, Mangano MM, Dauphin J, Salenius SA, Dosoretz DE, Blitzer PH. Small cell anaplastic carcinoma of the prostate: seven new cases, review of the literature, and discussion of a therapeutic strategy. Am J Clin Oncol. 1997;20(4):376–380. doi: 10.1097/00000421-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 35.Bismar TA, Yoshimoto M, Duan Q, Liu S, Sircar K, Squire JA. Interactions and relationships of PTEN, ERG, SPINK1 and AR in castration-resistant prostate cancer. Histopathology. 2012;60(4):645–652. doi: 10.1111/j.1365-2559.2011.04116.x. [DOI] [PubMed] [Google Scholar]
- 36.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han B, Mehra R, Suleman K, Tomlins SA, Wang L, Singhal N, Linetzky KA, Palanisamy N, Zhou M, Chinnaiyan AM, et al. Characterization of ETS gene aberrations in select histologic variants of prostate carcinoma. Mod Pathol. 2009;22(9):1176–1185. doi: 10.1038/modpathol.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, Rubin MA, Pienta KJ, Shah RB, Chinnaiyan AM. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68(10):3584–3590. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, Berney D, Foster CS, Fletcher A, Gerald WL, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27(3):253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.