

Abstract
We investigate ps-ns dynamics of the Rho-GTPase Binding Domain (RBD) of plexin-B1, which plays a key role in plexin-mediated cell signaling. Backbone 15N relaxation data of the dimeric RBD are analyzed with the model-free (MF) method, and with the slowly relaxing local structure/molecular dynamics (SRLS-MD) approach. Independent analysis of the MD trajectories, based on the MF paradigm, is also carried out. MF is a widely popular and simple method, SRLS is a general approach, and SRLS-MD is an integrated approach we developed recently. Corresponding parameters from the RBD dimer, a previously studied RBD monomer mutant, and the previously studied complex of the latter with the GTPase Rac1, are compared. The L2, L3 and L4 loops of the plexin-B1 RBD are involved in interactions with other plexin domains, GTPase binding, and RBD dimerization, respectively. Peptide groups in the loops of both the monomeric and dimeric RBD are found to experience weak and moderately asymmetric local ordering centered approximately at the axes, and ns backbone motion. Peptide groups in the α-helices and the β-strands of the dimer (the β-strands of the monomer) experience strong and highly asymmetric local ordering centered approximately at the axes (N–H bonds). N–H fluctuations occur on the ps time-scale. An allosteric pathway for GTPase binding, providing new insights into plexin function, is delineated.
Keywords: protein dynamics, NMR relaxation in proteins, molecular dynamics simulations, model free, slowly relaxing local structure
1. Introduction
Over the last two decades, NMR spin relaxation has effectively revealed the dynamic nature of proteins.1–7 However, the analysis of the experimental spin-relaxation data per se offers in many cases only partial insight into the details of protein dynamics, as physically realistic dynamic models often contain parameters that cannot be determined from the typically limited experimental data sets. This was realized early on, and a Model-Free (MF) method8,9 was developed. MF features a small number of generalized parameters that broadly relate to the amplitude (S2) and timescale (τe) of ps-ns internal motions, whilst the global motion of the protein is treated separately. The MF approach has become more complex,10 but the issue of mode separability (as opposed to mode-coupling), as well as the asymmetric nature of the local motions which are not accounted for in MF, stimulated a number of researchers to look for alternative methods.11–15
The attention of several groups has turned to computer simulations, which can reproduce MF-type generalized order parameters, and even experimental data.16–19 Combining predictive calculations, such as molecular dynamics (MD) simulations, with experimental relaxation parameters has also become a strategy for interpreting the experimental data (e.g. see refs 20 and 21). However, so far these approaches have not analyzed the relaxation data from a tensorial perspective (required to account for features of asymmetry), and have not accounted for dynamical mode-coupling (the separability concept in ref 20 is based on geometric considerations).
The two-body coupled-rotator approach called slowly relaxation local structure (SRLS) has been developed for comprehensive analysis of molecular motion.22–24 SRLS can be regarded as the generalization of MF, yielding the latter in simple limits. As applied to NMR relaxation in proteins,25–28 it allows for general asymmetric properties of the global diffusion of the protein, the local diffusion of the probe, and the local ordering at the site of the motion of the probe. Thus, the restricted local motion is characterized by a local ordering tensor, S, and a local diffusion tensor, D2. In addition, SRLS accounts rigorously for mode-coupling.
In the last decade, the SRLS approach has been applied extensively to 15N relaxation data from proteins.25–28 It emerged as a method that describes protein motions at a physically realistic level, often not achievable with simpler methods such as MF. However, the SRLS calculations are often tedious, the number of parameters to be determined is in some cases large, and the uncertainty in the best-fit parameters determined with data-fitting might be considerable.
To address properly these important matters we developed recently a combined computational-experimental approach that integrates SRLS and MD; we called this method SRLS-MD.29 Similar to SRLS, SRLS-MD describes the restricted local motion in terms of a local ordering tensor, S, and a local diffusion tensor, D2. The principal values ( and ) of S are determined with MD simulations29 extending over tens of ns. The coefficients of the coupling potential derived from these parameters, and known geometric features, are then fixed. This leaves the local diffusion rates, D2,⊥ and D2,||, and the orientation of the main ordering axis in the dipolar frame as the only variables in the data-fitting process. Varying only three parameters reduces significantly parameter uncertainty. As shown below, the physically well-defined (fixed and varied) parameters distinguish among loops, α-helices and β-strands. Importantly, they reveal features associated with biological function.
In this report, the briefly delineated SRLS-MD methodology (see ref 29 for details) is applied to 15N relaxation data from the plexin-B1 Rho-GTPase binding domain (RBD) dimer,30,31 a protein that exhibits complex dynamics.32,33 In parallel, these data are also subjected to MF analysis. Finally, MD trajectories of the dimer structure are analyzed in terms of the MF paradigm.
The RBD, which is part of the intracellular region of the plexin-B1 trans-membrane receptor,34 has a fold that is similar to ubiquitin.35 Like ubiquitin (Figure 1a), the RBD comprises a long helix that lies across a five-stranded β-sheet, and a shorter helix located close to the C-terminus (Figure 1b). Unlike ubiquitin, which contains short loops and tight turns that connect secondary structural elements, the RBD contains several long loops, L1–L4.30,35 The L2 loop (residues K50 – T62) is thought to be important for interactions with neighboring domains.36 The L3 loop (residues S69 – G73) is involved in Rho-GTPase binding. The L4 loop (residues L77 – L89) is associated with dimer formation (Figure 1c).
Figure 1.

Ribbon diagram of a) human ubiquitin (PDB ID UBQ), b) the Rho-GTPase binding domain of human plexin-B1 (RBD) (PDB ID 2JPH) with principal loops, L1–L4, labeled, and c) the RBD dimer (modeled using the NMR structure of the monomer and X-ray crystallography structure of the dimerization interface, accession code 2R2O).
The plexin-B1 RBD has been associated with the inhibition of receptor function by stabilizing an inactive dimeric state.31,36 The loop regions, the Rho GTPase binding interface and the dimerization region partly overlap in the dimer structure. It has been shown that GTPase binding and RBD dimerization oppose one another.31,37 This suggested a model for receptor activation that involves destabilization of the dimeric RBD form by GTPase binding, in synergy with extracellular ligand binding.31,34,37 Thus, the Rac1-bound form of the monomer may be considered a mimic of the activated receptor. The dynamic properties of the various RBD forms, including the monomer, the dimer and GTPase-bound form are thus of great interest, in particular given the suggestion that an allosteric network is associated with the signal transduction mechanism.32,38,39
A monomeric form of the RBD, stabilized by the W90F mutation in the L4 loop,40 was studied previously with MF, MD and SRLS-MD.29,32,33 Several RBD loops of this construct have been shown to be outstandingly mobile. The complex of the monomer mutant with the Rac1 GTPase was also studied previously with MF and MD.32,33 On the other hand, the wild type dimeric RBD has not been investigated so far. Here, we carried out MD simulations of this RBD form, and analyzed the MD trajectories according to the MF paradigm. Furthermore, we acquired 15N T1, T2 and 15N–{1H} NOE at magnetic fields of 14.1 and 18.8 T, and 298 K, and analyzed these data with MF and SRLS-MD.
Our long-term objective is to establish the relation between RBD structural dynamics and plexin function. To this end, we compare herein the MF, MD and SRLS-MD results obtained for the various RBD forms, with particular emphasis on loop dynamics. As shown below, the emerging picture constitutes a significant advancement toward achieving the goal set.
2. Materials and Methods
Sample preparation, NMR experiments and relaxation data analysis
The Rho-GTPase Binding Domain (RBD) of human plexin-B1 comprises residues 1742–1862, and two N-terminal lysine residues (here numbered as residues 1 – 122). This protein was expressed from a pet11a plasmid, and purified as described previously.30 The NMR sample contained 750 μM protein, 4 mM DTT, 4 mM MgCL2 and 10% (v/v) D2O in a pH 6.8 buffer that mimics physiological conditions.32 15N longitudinal relaxation rates R1 = 1/T1, 15N transverse relaxation rates R2 = 1/T2, and steady-state heteronuclear 15N–{1H} NOEs were acquired at 298K on Bruker DRX instruments equipped with cryoprobes, operating at magnetic fields of 14.1 and 18.8 T. Previously developed pulse sequences41,42 were used.
Eleven R1 experiments were carried out with delays ranging from 2 to 2000 ms. Eight R2 experiments were carried out with delays ranging from 12.8 to 153.6 ms. The heteronuclear 15N–{1H} NOE was acquired in an interleaved manner, with a 4.5 s long proton saturation period. Recycle delays of 5 s were used in all of the experiments. The spectra were processed using nmrPipe.43 Peak intensities were determined using Sparky.44 The resonance assignments obtained previously for the RBD monomer35 were used to assign the RBD dimer. Except for several exchange-broadened resonances in the dimerization interface, the chemical shifts of the monomer are virtually preserved. 15N R1 and R2, and the respective uncertainties based on triplicate experiments for three of the timepoints, were determined with the computer program Curvefit.45 Uncertainties in the experimental 15N–{1H} NOEs were determined from the baseline noise of the respective NMR spectra.
3. Theoretical Background
3.1. Model-Free
Based on the assumption that the global motion of the protein and the local motion of the probe are independent (i.e., decoupled), the total time correlation function is expressed as:8
| (1) |
where Ct(t) is the total time correlation function, Cg(t) = exp(−t/τm) is the time correlation function for global motion, and is the time correlation function for local/internal motion. τm is the correlation time for (isotropic) rotational reorientation of the protein. S2 is the squared generalized order parameter defined as the value of Ci(t) at long times. where τe is the effective correlation time for local motion, defined as the area below Ci(t) (required to be exact) divided by (1 − S2). Fourier transformation of eq 1 yields the measurable MF spectral density:8
| (2) |
If τe is much smaller than τm, one has .
The extended MF spectral density (EMF) is given by:10
| (3) |
The parameter is the (effective) correlation time for fast local motion. The parameter is the (effective) correlation time for slow local motion. and are squared generalized order parameters associated with these motions. The third term of eq 3 is often omitted under the assumption that is very small, obtaining thereby the reduced EMF formula.
3.2. Molecular Dynamics
Similar to our study of the RBD monomer,29 we carried out molecular dynamics simulations with the NAMD molecular dynamics package using the all-atom empirical potential energy function CHARMM22 with the CMAP correction.46 A 12 Å cut-off for non-bonded forces was used. Long-range electrostatic forces were accounted for with the standard particle-mesh Ewald method. This set-up, depicted in Table S1 of the Supporting Information, is similar to that used in ref 29, with the following new features. First, the RBD dimer was assembled by fusing the NMR structure of the W90F RBD monomer mutant (PDB accession code 2JPH) to the dimerization mini-β-sheet of the dimer X-ray crystallographic structure (PDB accession code 2R2O). The NMR structure was chosen for consistency with previous work,29 because several regions (including the long L1 loop) are not seen in the electron density map of the dimer crystal structure, and because the extensive loop regions might be affected by crystal contacts. Second, a larger simulation box, with explicit water treated with the TIP3P model, was used. Simulations of 55 ns in length were carried out in quadruplicate, using different seeds to increase sampling efficiency.
To eliminate the global tumbling from the analysis, the snapshots were superimposed on a reference frame, taken as the starting structure. Because the relative orientation of the monomers in the dimer changes slightly in the course of the simulations, the snapshots of each monomer were superimposed on their respective starting structure. S2 and τe were derived from 5 – 55 ns long segments of the MD trajectories. S2 was set equal to the value of Ci(t) for t = 5 ns, as most Ci(t) functions reached a plateau value at that time.
This assessment is based on the following considerations. For residues located in secondary-structure elements, virtually the same results were obtained in the time-regime extending from 5 to 10 ns. For residues located in loops and end-chain segments, convergence at later times is absorbed in most cases by larger errors in S2 (see below). The errors in S2 were estimated in terms of the differences among MD trajectories differing in their starting values (e.g., see Figure S2 of the Supporting Information). Agreement of the MD-based S2 profile with the dimer structure (see below) supports this scheme.
3.3. SRLS-MD
SRLS treats two rotators, protein and probe (given in this case by the N–H bond in the context of the peptide-bond plane), associated with the diffusion tensors D1 and D2, respectively.22–24 The rotators are coupled by a local potential of mean torque, U, which represents the spatial restrictions exerted by the immediate protein surroundings at the site of the motion of the NMR probe.25–28 The local order parameters (i.e., the principal values of the local ordering tensor, S) are defined in terms of the potential U (see eqs 4 and 5 below). In the limit where the rotators are time scale separated (decoupled), the tensors D1 and D2 are isotropic, and the spatial restrictions are either very strong or very weak, axially symmetric, and centered at the Z-axis of all the magnetic tensors taken axial and collinear, one has D2 = 1/(6τe) and (where is the axial order parameter). Thus, as already mentioned, in certain limiting cases the SRLS parameters yield the MF parameters.
In the general case, D1, D2 and S are rank 2 tensors allowed to have an arbitrary orientation and rhombic symmetry. Dynamical coupling between the rotators is accounted for rigorously.22 Figure 2 illustrates the main features of the SRLS frame scheme (the complete frame scheme appears in Figure 2 of ref 29). ZD1 is the principal axis of the global diffusion tensor (Figure 2a). The Principal Axes System (PAS) of the local ordering tensor, S, called OF, is distributed with respect to the local director, given in this case by the equilibrium orientation of the N–H bond, NHeq. The Euler angles ΩNHeq–OF are the coordinates associated with this distribution; the angle βNHeq–OF is shown in Figure 2b. The orientation of the OF frame with respect to the dipolar frame, DF, which points along the N-H bond, is given by the Euler angles (αD, βD, 0), where γD is set equal to 0 because the DF frame is axially symmetric. The orientation of the local diffusion frame with respect to the OF frame is given by the Euler angles (αO, βO, 0), where γO is set equal to 0 because the D2 tensor has been taken axially symmetric. Typical values of the angles βD and βO are illustrated in Figure 2c. SRLS allows for mutually tilted magnetic 15N–1H dipolar and (axial) 15N chemical shift anisotropy (CSA) tensors.
Figure 2.
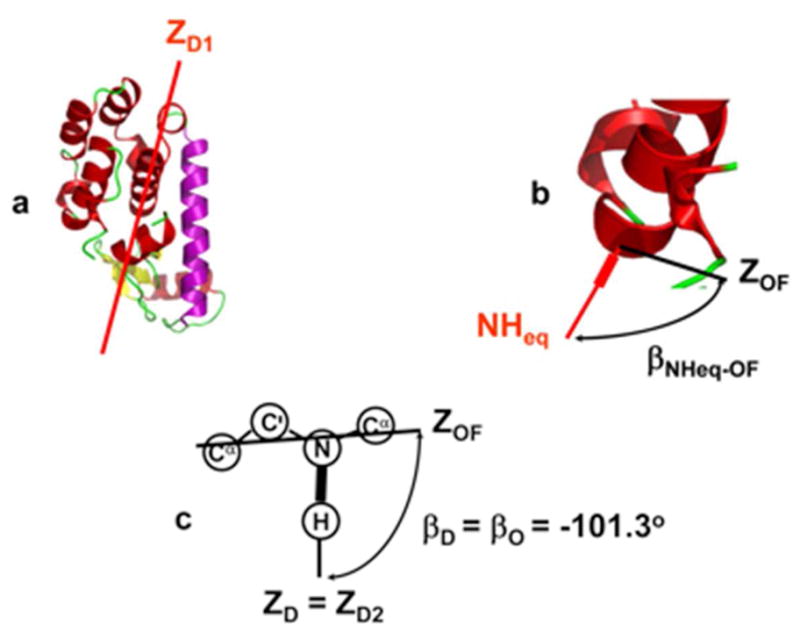
The main features of the SRLS frame scheme depicted on a typical protein structure. Given that SRLS (and its MF limit) relate to the motion of two bodies – the global motion of the protein and the restricted motion of the spin-bearing moiety (the probe) – the coordinate frames of the various tensors are fixed either in the protein, or in the probe. (a) Principal axis of the global diffusion tensor, ZD1, fixed in the protein. (b) Local director, NHeq, fixed in the protein. Main local ordering axis, ZOF, fixed in the probe. The Euler angles Ω NHeq–OF describe the distribution of the OF frame with respect NHeq (the angle β NHeq–OF is shown). (c) Main local ordering axis, ZOF, principal axis of the local diffusion tensor, ZD2, and principal axis of the dipolar tensor, ZD (which points along the N–H bond). All three axes are fixed in the probe. The angles βD and βO are shown, with their typical value of −101.3°.
The potential, U(ΩNHeq–OF), is expanded in the complete basis set of the Wigner rotation matrix elements. When only the lowest (L = 2) terms are preserved, one has:23
| (4) |
where evaluates the strength of the local potential, and its non-axiality.
The order parameters are defined as:23
| (5) |
For a local director with at least three-fold symmetry and a local ordering frame with at least two-fold symmetry, only the order parameters and survive.
In this study, the principal values of the local ordering tensor, and , are derived from MD trajectories. The physical parameters and can be related to the MF parameter S2 (see Supporting Information). The coefficients of the potential, and , are calculated from and using eq 5, as outlined in ref 29 for the RBD monomer mutant. It was shown that the local potential in terms of which and are defined constitutes a good approximation to the local potential calculated from the MD trajectory.29 The estimated error in and is approximately 7%. “Data-fitting” consists of minimizing the sum of the squared differences between corresponding experimental and calculated relaxation parameters divided by the respective experimental error.25 To calculate the magnetic tensor components, the values of rNH = 1.02 Å and 15N CSA σCSA = −170 ppm, have been used.32 The calculated relaxation parameters are obtained by solving the Smoluchowski equation for the appropriate set of parameters. A typical parameter set includes , αD, αO and βO fixed, and D2,⊥, D2,|| and βD allowed to vary.
Note that over-fitting is avoided by typically starting the analysis with parameter values corresponding to the MF limit, and adding variables only when the results are not tenable physically. In addition, we ascertain that the variables used are statistically independent.
4. Results and discussion
4.1. Experimental NMR relaxation parameters
15N R1, R2 and {1H}–15N NOE data are shown in Figure S1 and are listed in Table S2. Only a single set of resonances is seen in the HSQC NMR spectrum. The fact that the chemical environments in the RBD units are identical indicates that on average the dimer structure is symmetrical. A large part of the dimerization loop, L4, and part of the α2 helix are not detected in the HSQC spectrum, most likely because the respective NMR resonances are exchange-broadened beyond detection.
Excluding the terminal chain segments, where 15N R1 decreases monotonically, the average 15N R1 values are 1.55 and 1.3 s−1 at field strengths of 14.1 and 18.8 T, respectively. The average 15N R2 values for the secondary structure elements are 18.0 and 22.5 s−1 at 14.1 and 18.8 T, respectively. The loops, in particular L1 and L3, and the terminal chain segments, have significantly smaller 15N R2 values. Such clear 15N R2-based discrimination between secondary structure elements and loops is associated with complex internal (most likely loop-related) dynamics. Most globular proteins do not contain long loops. Plexin-B1 RBD is quite unusual in this respect. The average 15N–{1H} NOE values for the secondary structure elements are 0.85 and 0.95 at 14.1 and 18.8 T, respectively. The loops L1 and L3 are associated with substantially smaller NOEs. In the L2 loop, the NOEs are similar to the average NOE of the secondary structure elements.
4.2. MF analysis
Global motion
The computer program RotDif,47 which employs experimental 15N R2/R1 ratios of “rigid” N–H bonds (i.e., N–H bonds that experience negligible local motion) and a 3D structure, was applied. The NMR structure of the RBD monomer was used to assemble the dimer structure (see above). This initial analysis yielded a global correlation time, τm, of 9.8 – 10.8 ns and a ratio A = D1,||/D1,⊥ of 1.14 – 1.21 for the extent of axiality of the global diffusion tensor. These parameters are similar to the corresponding parameters for the monomer, where τm = 8.6 ± 0.4 ns and A =1.26 ± 0.12.32 This result is difficult to rationalize, given the differences in size between the two forms of the RBD. Furthermore, the complete 15N relaxation data set of the dimer form could not be fit with the MF-based computer program “Dynamics”48,49 with these values of τm and A. It could be fit with τm = 16.5 ± 0.83 ns and A ~ 1. This value of τm is consistent with the size of the dimer. A ~ 1 is consistent with relatively fast monomer fluctuations in the dimer, implied by mobility at the monomer-monomer interface. On average, this yields a spherical solution structure that differs from the non-spherical crystal structure. A similar scenario was encountered for the RBD monomer, as discussed in detail in ref 29, and for the proteins E. coli adenylate kinase and the ribonuclease binase.27,28 Since approximately 90% of the protein is in its dimeric state, we ignore effects associated with the monomer-dimer equilibrium (analysis of the data in terms a monomer-dimer equilibrium, as described by Fushman,48 did not yield substantially improved results). Based on the evidence and considerations outlined above, we used isotropic global diffusion with τm = 16.5 ± 0.83 ns in all of the calculations described below.
Local motion
The squared generalized order parameter, S2, derived by the MF approach, is interpreted as amplitude of local N–H motion with values ranging from 0 (unconstrained motion of the N–H bond) to 1 (N–H bond fixed in the protein).8,9 The program Dynamics48,49 was used to analyze the data shown in Figure S1. This data-fitting scheme features four models based on the MF formula (eq 2), and four models based on the EMF formula (eq 3). Out of 99 N–H bonds accessible with 15N relaxation measurements, 51% were fit with model 1 (only S2 allowed to vary in the data-fitting process), 19% with model 2 (S2 and τe allowed to vary), and 25% with model 5 ( , and τs allowed to vary). The best-fit values obtained are given in Table S3. A conformational exchange contribution (to R2), Rex, of 7.0 s−1 was required for residue T83. For the monomer, N–H bonds located near to, or within, the dimerization L4 loop, required Rex contributions.
SRLS accounts for conformational exchange contributions in the same manner as MF – by adding a Rex term to the expression for 1/T2. When Rex is large, hence the credibility of conformational exchange is high, we exclude the respective N–H bond from the analysis. There is a one such case for the dimer – T38. MF found smaller Rex contributions for several residues of the monomer. SRLS ascribed to these residues (as it did for most residues) anisotropic spatial restrictions (Figure 5) and realistic local geometry (Figure 7), rationalizing thereby the augmented experimental 1/T2 values.
Figure 5.

Best-fit values of the order parameters (a) and (b) obtained for the RBD monomer mutant (black circles) and the RBD dimer (red squares) with MD simulations, as described in the text. The black bars on the right denote the average error of 7% in and .
Figure 7.

Best-fit values of the Euler angle αD (a), βD (b), αO (c) and βO (d) obtained for the RBD monomer mutant (black circles) and the RBD dimer (red squares) with SRLS-based data-fitting, as described in the text. The average error of 10% does not exceed the symbol height.
S2 is shown in Figure 3a for both the monomer and the dimer. For the secondary structure elements, the monomer values exceed the dimer values by 0.05 – 0.08. The largest differences are observed for the β1 and β5 strands, the α2 helix and the tight β2/α1 turn. For the loops, S2 for the dimer is similar to S2 for the monomer. The dimer shows quite a few sporadic spikes, which are not seen in the corresponding MD-derived S2 profiles (Figure 4a). While these features may result from a number of sources, including fixed N–H bond lengths and 15N CSA,50 we ascribe them primarily to the utilization of oversimplified MF spectral densities incommensurate with the dynamic complexity of the plexin-B1 RBD.
Figure 3.
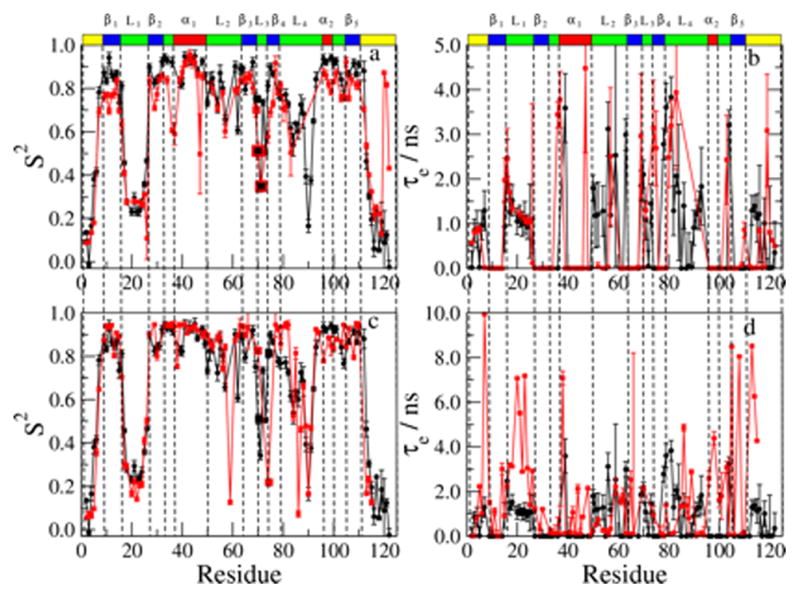
Best-fit values of S2 and τe obtained with MF analysis. (Note that both τe and τs are denoted τe) S2 (a) and τe (b) for the RBD monomer mutant (black circles) and dimer (red squares). S2 (c) and τe (d) for the RBD monomer mutant (black circles) and its complex with the Rac1 GTPase (red squares). The data for the monomer mutant and its complex with the Rac1 GTPase were taken from ref 32.
Figure 4.

Average S2 (a) and τe (b) values obtained with MD simulations for the RBD dimer as outlined in the text (shown as red squares). Average S2 and τe values obtained with MD simulations for the RBD monomer mutant are shown as black circles for comparison. The data of the monomer mutant were taken from ref 32.
The effective correlation time for local motion is shown in Figure 3b for both RBD forms. As mentioned above, the experimental data were fit predominantly with models 1, 2 and 5. Model 1 pre-sets τe = 0, model 2 is expected to yield τe ≪ τm (i.e., ps τe values), and model 5 is expected to yield ns τe values (in this case, “τe” denotes “τs”; the same symbol is used to facilitate comparison with the similarly derived MD local motional correlation times). In practice, except for the terminal chain segments, model 2 yielded (similar to model 5) effective local motional correlation times on the order of several ns. Altogether, 50% of the N–H sites, including virtually all of the secondary structure elements and quite a few N–H bonds located in the L2 and L3 loops and the terminal chain segments, were fit with model 1. The remainder of the polypeptide chain, including L1, parts of L2 and L3, and part of the terminal chain segments, has τe in the 0.5 – 4.5 ns range.
In general, the correlation times for local motion are somewhat larger for the dimer as compared to the monomer. In particular, the N-, and C-terminal chain segments and the L1 and L3 loops have similar, or slightly larger, τe values for the dimer. The L2 loop, comprising residues K50 – T55, wraps around one side of the protein. While the S2 values for the dimer and the monomer are similar in this loop, ns local motions are only experienced by the monomer.
Figure 3c shows the S2 values for the monomer, superimposed on the S2 values for its complex with the Rac1 GTPase (re-plotted from ref 32). In general, the differences between monomer and complex (Figure 3c) are small; they are smaller than the differences between monomer and dimer (Figure 3a). The L3 loop, associated with GTPase binding, is an exception. For example, the S2 values of G70 and A72 are substantially larger for the complex than for the monomer (Figure 3c); the corresponding differences between dimer and monomer are within experimental error. This is consistent with the fact that residues G70 and A72 are not part of the dimerization interface.
The τe values for the monomer and the RBD-Rac1 complex are shown in Figure 3d. Similar to the τe data for the dimer, the τe data for the complex show a large number of spikes. For quite a few residues of the L1 loop, τe for the complex exceeds significantly τe for the monomer. This indicates that the L1 loop is affected considerably by Rac1 binding. By contrast, the τe values for the dimer are virtually the same as the corresponding values for the monomer (Figure 3b).
The N-terminal part of the L2 loop (residues K50 – T55) experiences significantly larger changes in τe upon dimerization than upon Rac1 binding. A similar pattern was observed for changes in S2. This is interesting, since the chain segment K50 – T55, as well as the L1 loop, are at considerable distance from both the dimerization interface and the GTPase binding site.
4.3. MD analysis
The squared generalized order parameter, S2, and the effective correlation time for local motion, τe, were derived from time correlation functions as described in section 2. They are shown superimposed on the corresponding monomer data in Figure 4. In the secondary structure elements, corresponding S2 values are virtually the same. In the L4 loop and the first part of the L2 loop, S2 for the dimer is larger than S2 for the monomer. In much of the L1 loop and the terminal chain segments, S2 for the dimer is smaller than S2 for the monomer. Unlike the MF-based S2 profile, the MD-based S2 profile does not show sporadic spikes along the sequence.
The MD-derived τe values range from 0.2 to 1.8 ns (Figure 4b). Within the first part of the L2 loop, τe for the dimer is on average smaller than τe for the monomer. This is consistent with the changes in S2, indicating reduced (faster smaller-amplitude) fluctuations. In the C-terminal segment of the polypeptide chain, τe for the dimer is larger than τe for the monomer. In the L4 loop, τe for the dimer is substantially smaller than τe for the monomer.
The precision of the MD-derived parameters is low (see Figure S2). However, both S2 and τe are consistent with the secondary structure of the RBD and the location of its loops. Upon dimerization the L4 loop, which forms the interface between the monomer units in the dimer, becomes more rigid (larger S2) and less mobile (smaller τe). On the other hand, in the C-terminal segment of the L1 loop S2 is smaller for the dimer. One might conjecture that dimerization is accompanied by an allosteric mechanism involving loop regions of compensating rigidity and mobility.
The MD analysis features several important benefits versus the MF analysis. Thus, the MD analysis treats all of the N–H bonds in the same manner employing a single correlation function form. In comparison, the MF analysis, applied to the experimental data, uses several different models (predominantly 1, 2 and 5), i.e., employs several different spectral density functions. The MD analysis eliminates the global motion consistently based on a specific procedure. In comparison, the MF analysis models the global motion using filtered experimental data; this procedure is based on empirical filtering criteria. Finally, the MF analysis is prone to force-fitting (good statistics but inaccurate best-fit parameters, which have absorbed unaccounted for factors). As pointed out above, force-fitting accounts at least for some, if not the majority, of the sporadic spikes visible in Figure 3. Nevertheless, MF is useful in comparing (qualitatively) results from different systems (see below).
4.4. Comparison between MF and MD analyses
It is useful to compare corresponding MF (Figure 3) and MD (Figure 4) parameters. Superimposed S2 and τe τe profiles for the monomer and the dimer are shown in Figure S3, after having eliminated sporadic spikes from the MF data. For the monomer, the S2 profiles from MF and MD analyses are generally similar. Relatively small differences are observed in the C-terminal regions of the L1 and L4 loops, and the C-terminal chain segment. They can be ascribed to limited sampling of the MD trajectories (shown in Figure S2) and/or force-fitting in the MF analysis. For the dimer, the S2 profiles from MF and MD are virtually the same, except for small differences in the C-terminal chain segment. The τe profiles from MF (also shown in Figure 3b) differ from their counterparts from MD (also shown in Figure 4b). MD yields τe of 1 – 3 ns for the loops (except for L2) and terminal chain segments, and τe of 0.2 – 0.8 ns for the secondary structure elements. For the latter MF yields mostly τe = 0; these values are clearly artefactual. Quite remarkably, for the L1 loop τe from MF is similar to τe from MD. For the L2 loop MF yields irregular τe patterns for the monomer and τe = 0 for the dimer.
The features delineated above indicate that more elaborate methods of analysis are required to extract fully and properly the information inherent in the experimental data. We show below that significant progress is made along these lines by using SRLS-MD as method of analysis.
SRLS-MD analysis
The global diffusion was taken isotropic with τm = 16.5 ns (see above).
Local ordering
In the first step of the analysis, and are derived from the MD trajectories, as described briefly in section 2, and in detail in ref 29. is a physical measure of local ordering strength and is a physical measure of local ordering rhombicity (asymmetry). To calculate and using eq 5 one has to define the Euler angles ΩNHeq–OF = (αNHeq–OF, βNHeq–OF, 0). NHeq is given by the N–H orientation in the 3D (reference) structure. The orientation of the OF frame with respect to the known dipolar tensor frame, DF, is given by the Euler angles ΩOF–DF = (αD, βD, 0). In previous SRLS studies of 15N spin relaxation in proteins,26–28 we found with data-fitting that βD = −101.3°. This corresponds to the main axis of the local ordering tensor, ZOF, pointing along . Main local ordering axes parallel to are encoded in other methods of 15N relaxation analysis, such as the 3D Gaussian Axial Fluctuations model.14 Crankshaft backbone motions revealed by MD studies51 are consistent with acting as main ordering axis. Based on these considerations, we set βD = −101.3° and αD = −90° (consistent with the stereo-chemistry of the peptide-bond plane) in the calculation of and
In the next step of the analysis, and are calculated as described in ref 29 from and based on eqs 4 and 5. Thus, the MD component of the SRLS-MD analysis provides for every experimentally accessible N–H bond the parameters and , which are used as fixed input to the SRLS-based data-fitting calculation. Additional parameters that are pre-set in this calculation include αO = βO = −90° (αO and βO define the orientation of the D2 tensor with respect to the S tensor). In the SRLS frame scheme, this corresponds to the principal axis of D2 pointing along the N–H bond, which is a physically tenable geometric scenario.
Thus, only the local diffusion rates, D2,⊥ and D2,||, and the tilt angle, βD, between the principal axes of the local ordering and dipolar tensors, are left as variables in the data-fitting calculation. With βD = −101.3° as starting value, this scheme yielded good statistics for approximately 75% of the data. For the remaining 25% of the data, to obtain good statistics we also allowed αD, αO and βO to vary from starting values of −90°. Two cycles of optimization, allowing the set D2,⊥, D2,|| and βD and the set αD, αO and βO to vary sequentially, were carried out. The best-fit values obtained are given in Tables S4 and S5.
The order parameters and are shown in Figure 5. The axial order parameter, (Figure 5a), is negative. values approaching −0.5 correspond to strong perpendicular ordering. This agrees with the angle βD being in most cases close to −101.3° (see Table S5), representing a main ordering axis approximately perpendicular to the local director (NHeq). Except for the C-terminal chain segment, where the monomer exhibits stronger ordering that the dimer, the two RBD forms exhibit similar ordering strength. In general, the local ordering is strong in the secondary structure elements and substantially weaker in the loops and the terminal chain segments. In particular, very strong and uniform local ordering prevails in the α1 and α2 helices, and the β1, β3 and β5 strands.
Weaker and quite diversified local ordering prevails in the β2 and β4 strands, which are adjacent to the C-termini of the L1 and L3 loops, respectively. Thus, the ordering properties of the L1 and L3 loops extend to their C-terminal secondary structure neighbors. This finding agrees with the fact that the β2 and β4 strands are located at the exterior of the protein, with every other N–H group facing the solvent. This spatial disposition is likely to impart to the β2 and β4 strands ordering properties akin to flexible loops. The L1 loop, and for the monomer the L4 loop, exhibit similar patterns, with the weakest ordering occurring in their middle. A different pattern is observed for the L2 and L3 loops, where the weakest ordering occurs near the N-, and C-termini, respectively. The local ordering at the N–H sites of the L2 loop and the C-terminal chain segment is weaker in the dimer as compared to the monomer. Thus, insightful information on the strength of the local ordering is provided by the SRLS-MD analysis.
The rhombic order parameter, , is shown in Figure 5b. The largest theoretical value of is . Negative means (in the SRLS frame scheme) that ordering perpendicular to the peptide-bond plane is preferential to ordering along the N–H bond. is large and quite uniform in the α-helices and most β-strands, and relatively small in the L1 and L3 loops, and for the monomer also in the L4 loop. For both RBD forms, the central part of the L2 loop exhibits relatively large rhombicity, comparable to that of the secondary structure elements.
The following comment is in order. The MF-versus-MD comparison based on the squared generalized order parameter is illustrated in Figure S3. An analogous SRLS-versus-MD comparison is not feasible because SRLS provides the axial, , and rhombic, , components of the local ordering tensor, S, whereas MD provides the squared generalized order parameter, S2. However, it is appropriate to compare distribution functions, Peq. We carried out a detailed comparison in this spirit for the monomer mutant.29 Figure 8 of that article shows that the potential of eq 4, which describes the spatial restrictions at the site of the motion of the N–H bond, is a good approximation to the corresponding part of the CHARMM potential energy function used in the MD simulations. This result also applies to the plexin-B1 dimer, as it relates to the local spatial restrictions at the N–H sites of the plexin-B1 RBD.
Figure 8.
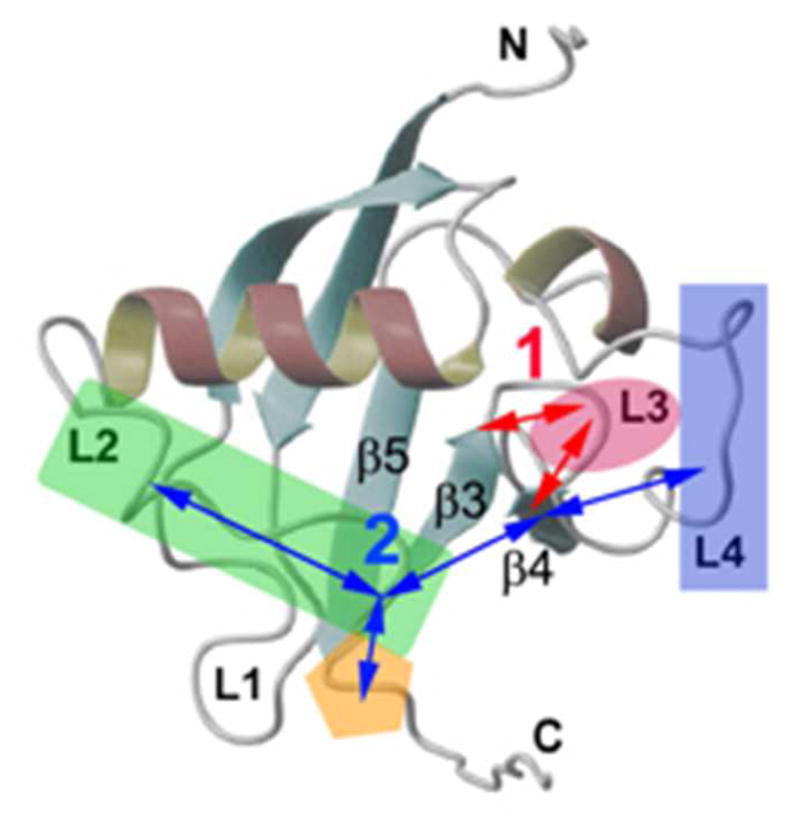
Illustration of allosteric pathways that are suggested by this study of RBD mainchain dynamics. The RBD is shown as ribbon diagram, as in Figure 1. Possible pathways 1 and 2 are shown. Dimerization engages pathway 1 (L4 to β4, L2 and C-terminus), whereas GTPase binding (associated with the L3 loop) engages both pathway 1 and 2 (entropy compensation occurs between L3 and surrounding regions, e.g., the termini of β3 and β4).
The axial, , and rhombic, , coefficients of the SRLS potential of eq 4 are derived from and (eq 10 of ref 29). For strong local ordering the components of the local ordering tensor can be determined with higher accuracy than the coefficients of the local potential.26,27 The errors in and were estimated at 7% (see above, and caption of Figure 5). Based on previous work,26,27 we estimate the errors in and at 10%. Further discussion of error estimation in SRLS-based data-fitting, in general, appears in refs 26 and 27.
Local diffusion
Figure 6a shows the logarithm of the best-fit values of D2,⊥. This represents slow backbone motion, comparable to the global tumbling. On the right we show corresponding values of τ2,⊥ = 1/(6 D2,⊥)*. Several log(D2,⊥) values within the β-strands are smaller than log(D1); further studies are required to clarify this matter. Within the L1, L2 and L3 loops and the C-terminal chain segment, log(D2,⊥) is approximately 7.5. This corresponds to a correlation time, τ2,⊥, of 5 ns, which is approximately 3 times faster than the correlation time for global tumbling of 16.5 ns. log(D2,⊥) is increased relative to D1 in the L2 loop and for several N–H bonds located near the C-terminus of the polypeptide chain. This indicates that relatively fast ns backbone motions occur at these sites.
Figure 6.
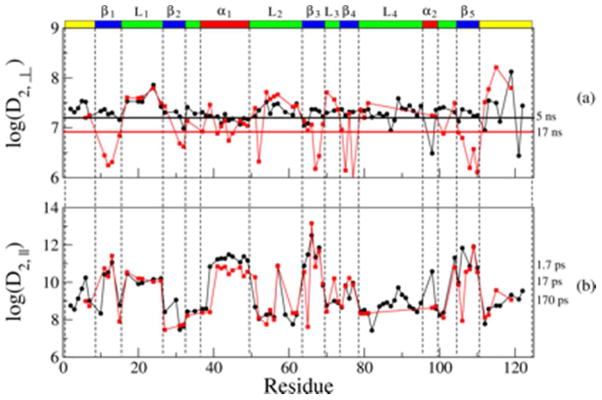
Best-fit values of the local motional rates, D2,⊥ (a) and D2,|| (b), obtained for the RBD monomer mutant (black circles) and the RBD dimer (red squares) with SRLS-based data-fitting, as described in the text. On the right we show the correlation times, τ2,⊥ = 1/(6 D2,⊥) and τ2,|| = 1/(D2,||), that correspond to typical diffusion rate values, D2,⊥ and D2,||. The error in the motional rates D1, D2,⊥ and D2,|| is estimated at 10%.
The logarithm of D2,||, which represents N–H fluctuations, is shown in Figure 6b. The dynamic range of this parameter is large. The N–H fluctuations are particularly fast in the α1 helix and the β1, β3 and β5 strands, where <τ2,||> = <1/(6 D2,||)> ~ 10 ps. The strands β1, β3 and β5 represent the core of the protein; fast N–H fluctuations are consistent with a relatively rigid core. The correlation time τ2,|| is on average 50 ps for the L1 loop and particularly large for the β2 strand and most of the L2 loop, where <τ2,||> = 1 ns.
Clearly, SRLS provides insightful information on local motional modes in the plexin-B1 RBD.
Local geometry
The angles αD and βD (S tensor orientation) and αO and βO (D2 tensor orientation) constitute what we call “the local geometry”. These parameters are shown in Table S5, and are depicted pictorially in Figure 7. For the dimer, αD is close to −90° (Figure 7a) and βD is close to −101.3° (Figure 7b), indicating that is, within a good approximation, the main local ordering axis. For the monomer, βD is zero for many β-strands, indicating that the main local ordering axis points along the N–H bond. For both the monomer and the dimer, αO = βO = −90° for the α-helices and the loops, and αO = βO = 0 for the β-strands (Table S5 and Figures 7c,d). That SRLS-MD reveals differences in the local geometry between monomer and dimer is of particular interest. The diversity of tensor orientations in the β-strands is more extensive for the monomer than for the dimer, in particular near their N- and/or C-termini. This suggests the establishment of geometric regularity by long-range allosteric effects associated with dimerization. Note that in MF and MF-based MD analyses, αD = βD = αO = βO = 0 is implicit. Hence, these methods cannot provide information on features of local geometry.
4.6. Allosteric pathways for GTPase binding and dimerization revealed by N–H bond dynamics
A pictorial representation appears in Figure 8. As pointed out above, the RBD dimer is associated with the stabilization of inactive receptor states31,34 whereas the Rac1-bound form of the monomer may be considered a mimic of the activated receptor. Two regions of the protein structure are of particular interest in this context. They include the GTPase binding region (the L3 loop, colored magenta in Figure 8) and the dimerization region (mainly the L4 loop, colored blue in Figure 8). Although the C-terminal part of L4 is not detected with 15N relaxation experiments, both X-ray crystallography and MD describe it as relatively rigid. By contrast, the L3 loop, associated with GTPase binding, is substantially more flexible in the dimer (and monomer) as compared to the RBD-GTPase complex. This is evident by examining the S2 profiles obtained with MF and MD (Figures 3c and 4a). The regions flanking the L3 loop are more rigid in the dimer. Proceeding toward the N-, and C-termini of the polypeptide chain, one encounters chain segments that are more flexible in the dimer (e.g., the β1 and β5 strands and the a2 helix).
In this context, “flexibility” is substantiated as follows by the SRLS-MD analysis. On average, the L3 loop of the dimer exhibits the strongest local ordering (largest ), the smallest potential rhombicity (smallest ) (Table S4), the slowest backbone motion (smallest D2,⊥), and the slowest N–H fluctuations (smallest D2,||) (Figure 6) among all of the RBD loops. Thus, despite the relatively strong and nearly axial local restrictions experienced, the N–H bonds of loop L3 move relatively slowly both at the collective-backbone level, and at the individual-local level. This might be associated with the nature of their surroundings (cf. Figure 8).
The β4 strand, which flanks the L3 loop at its C-terminus, represents the “intersection” between the allosteric pathways 1 and 2 depicted in Figure 8. The local ordering is strong and rhombic in this β-strand. It decays to weak and nearly axial ordering in the C-terminal chain segment. The local ordering is also strong and rhombic in the β3 strand, which flanks L3 at its N-terminus. This β-strand is part of the allosteric pathway 1. The decay to weak and nearly axial ordering in the N-terminal chain segment occurs through oscillations between weak and moderately rhombic ordering in L1, L2 (colored green in Figure 8) and L3, and strong and substantially rhombic ordering in α1, β1 and β2. The N–H fluctuations (D2,||), which are limited in L3, are substantial in β5 (toward the C-terminus) and β3, α1, L1 and β1. Dips in D2,|| can also be seen in L2 and β2. The L3 loop and the β3 strand are associated with the allosteric pathway 1. SRLS-MD provides an insightful (mesoscopic) description of the structural dynamics of the dimeric RBD form.
Based on this analysis, we conjecture that the GTPase binds to the RBD dimer at an entropically favored site – the flexible L3 loop – through a mechanism that involves an allosteric pathway that appears to materialize through variations in rigidity. Thus, the entropy lost at the L3 loop is regained in other regions of the protein, in the manner specified above. This mechanism requires further support. However, other authors have described similar scenarios in the context of protein-protein interactions (cf. refs 32, 52–54).
Let us examine the implications of the MF analysis in the context of allostery. We have shown previously that Rac1 binding affects the dynamic state of the L2 loop, despite this region being a great distance away from the GTPase binding site.32 The L2 loop (Figure 8) is also affected by RBD dimerization, with ns motion experienced by the monomer being quenched by this process. MD simulations reveal substantial changes in S2 and τe upon dimer formation (cf. L4 loop in Figures 4 and 8). Another region that shows (complex) differences between monomer and dimer is the chain segment comprising residues 112 – 116, flanking the β5 strand at its C-terminus (colored orange in Figure 8). Here, the polypeptide chain is fluctuating with increasing speed as it emerges from the tightly packed region of the RBD structure. Local motions on the ns time scale are no longer encountered, as shown by the τe values, and further elaborated by the SRSL local motional component D2,⊥.
The local ordering in the L2 loop and at the C-terminus of the polypeptide chain are relatively weak in both RBD forms (Figures 5 and 8). The tensor orientations are largely similar (Figures 7 and 8), and the motions captured by D2,⊥ are quite a bit faster for the dimer (Figures 6 and 8). These results point to an allosteric change that is largely dynamic in nature, without substantial alteration of the local geometry/structure.55–58 This finding is consistent with the lack of chemical shift perturbations in this region, revealed by comparing NMR spectra of the monomer and the dimer.40
We may speculate about allosteric features/biological function emerging from this study as follows. Changes per se in L3 are implied exclusively by GTPases binding. However, they correlate with long-range dynamic chances in other regions of the protein, extending up to the L2 loop (pathway 2 in Figure 8). In addition, entropy compensation occurs between L3 and the termini of the β3 and β4 strands (pathway 1 in Figure 8). Dimerization has no effect on L3, possibly because this region still has to be available for binding the GTPase. The latter process is likely to be the first step of a mechanism that destabilizes the dimer (indeed, a dimeric GTPase-bound form has been crystallized36). Both dimerization and more so GTPase binding have an allosteric effect on L2, rigidifying this loop (pathway 2 in Figure 8, involving both L4 and L3). As mentioned above, this region of the RBD domain is of particular importance, as it makes contacts with the adjacent catalytic domains of plexin-B1. Further studies, including molecular dynamics simulations of the GTPase-bound state, are required to ascertain the validity of the allosteric mechanism described above.
5. Conclusions
Picosecond-nanosecond structural dynamics of the RBD of plexin-B1 in monomeric, dimeric and GTPase-bound forms has been investigated with 15N relaxation analyzed with the MF and SRLS-MD methods. Traditional MD simulations have also been carried out. The MF and MD analyses provide a semi-quantitative picture singling out the loops as being more flexible than the secondary structure elements. The MD analysis shows that upon dimerization a monomer-monomer interface with increased rigidity and reduced ns mobility is generated. SRLS-MD describes structural dynamics at every N–H site in terms of a rhombic local ordering tensor, S, and an axial local diffusion tensor, D2. The former connotes “structure” and the latter connotes “motion”.
The local ordering is described by the physical order parameters, and , in the context of realistic geometry of main ordering axis pointing along . represents ordering strength and represents ordering rhombicity. The local diffusion is described by the components D2,⊥ and D2,|| of the local diffusion tensor, D2, the principal axis of which is in most cases parallel to the N–H bond. D2,|| represents fast, but not necessarily very fast, N–H fluctuations. D2,⊥ represents ns backbone motion coupled automatically to the global tumbling. The , D2,⊥ and D2,|| profiles are highly discriminating. An allosteric pathway that communicates GTPase binding and dimerization to other regions of the protein is revealed by these physical descriptors.
Supplementary Material
Acknowledgments
This work of M.B. is supported by the NIH grant 1R01GM092851. Some of the dynamics calculations and analyses were carried out at the Case Western Reserve High Performance Cluster and at Lonestar (Austin, Texas) via a TeraGrid award to M.B. A.P. acknowledges support provided by the Ministero dell’Istruzione, Università e Ricerca (MIUR), grant 2008J9RNB3 PRIN 2008 TIME and by the University of Padova, grant STPD08RCX5 “Progetto Strategico” HELIOS. M.Z. gratefully acknowledges the hospitality of the Department of Physiology and Biophysics, Case Western Reserve University School of Medicine, Cleveland OH, were most of this work was carried out. This work was also supported by the Israel Science Foundation (grant No. 437/11 to E. M.), the Binational Science Foundation (grant No. 2010185 to E.M. and Jack H. Freed), and the Damadian Center for Magnetic Resonance at Bar-Ilan University, Israel. E.M. gratefully acknowledges the hospitality of the Department of Computational and Systems Biology, University of Pittsburgh School of Medicine, where part of this work was carried out.
Footnotes
τ2,⊥ represents a “correlation time” in the sense of the inverse of the K = 0 eigenvalue of the quantum mechanical symmetric top only in absence of an orienting potential. In the presence of a potential it represents an effective correlation time associated with D2,⊥.
Supporting Information Available. A section showing the algebraic relation between S2, on the one hand, and and , on the other hand. A paragraph delineating the MF models featured by the computer program Dynamics.48 A figure showing the experimental 15N T1, T2 and 15N–{1H} NOE values for the dimer acquired at 14.1 and 18.8 T. A figure showing 4 MD trajectories for the plexin dimer. A figure showing S2 and τe from MF and MD. A table summarizing the parameters used in the MD simulations. A table showing the experimental 15N T1, T2 and 15N–{1H} NOE values for the dimer acquired at 14.1 and 18.8 T. A table showing the best-fit vlues of S2 and τe obtained with MF analysis. A table showing the best-fit values of , and obtained with SRLS analysis. A table showing the best-fit values of D2,||, D2,⊥, αO, βO, αD and βD obtained with SRLS analysis. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Palmer AG. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 2.Mittermaier A, Kay E. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 3.Igumenova TI, Frederick KK, Wand AJ. Chem Rev. 2006;106:1672–1699. doi: 10.1021/cr040422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarymowycz VA, Stone MJ. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 5.Hall JB, Fushman D. J Am Chem Soc. 2006;128:7855–7870. doi: 10.1021/ja060406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loth K, Pelupessy P, Bodenhausen G. J Am Chem Soc. 2005;127:6062–6068. doi: 10.1021/ja042863o. [DOI] [PubMed] [Google Scholar]
- 7.Wang T, Weaver DS, Cai S, Zuiderweg ERP. J Biomol NMR. 2006;36:79–102. doi: 10.1007/s10858-006-9047-4. [DOI] [PubMed] [Google Scholar]
- 8.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 9.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 10.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 11.LeMaster DM. J Am Chem Soc. 1999;121:1726–1742. [Google Scholar]
- 12.Idiyatullin D, Daragan VA, Mayo KH. J Phys Chem B. 2003;107:2602–2609. [Google Scholar]
- 13.Brüschweiler R, Wright PE. J Am Chem Soc. 1994;116:8426–8427. [Google Scholar]
- 14.Bremi T, Brüschweiler R. J Am Chem Soc. 1997;119:6672–6673. [Google Scholar]
- 15.Prompers JJ, Brüschweiler R. J Am Chem Soc. 2001;123:7305–7313. doi: 10.1021/ja0107226. [DOI] [PubMed] [Google Scholar]
- 16.Buck M, Karplus M. J Am Chem Soc. 1999;121:9645–9658. [Google Scholar]
- 17.Trbovic N, Kim B, Friesner RA, Palmer AG., III Proteins. 2008;71:684–694. doi: 10.1002/prot.21750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maragakis P, Lindorf-Larssen K, Eastwood MP, Dror RO, Klepeis JL, Arkin IT, Jensen MO, Xu H, Trbovic N, Friesner RA, et al. J Phys Chem B. 2008;112:6155–6158. doi: 10.1021/jp077018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, et al. Proc Natl Acad Sci USA. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 20.Prompers JJ, Brüschweiler R. J Am Chem Soc. 2002;124:4522–4534. doi: 10.1021/ja012750u. [DOI] [PubMed] [Google Scholar]
- 21.Dhulesia A, Abergel D, Bodenhausen G. J Am Chem Soc. 2007;129:4998–5006. doi: 10.1021/ja067429w. [DOI] [PubMed] [Google Scholar]
- 22.Polimeno A, Freed JH. Adv Chem Phys. 1993;83:89–206. [Google Scholar]
- 23.Polimeno A, Freed JH. J Phys Chem. 1995;99:10995–11006. [Google Scholar]
- 24.Liang Z, Freed JH. J Phys Chem B. 1999;103:6384–6396. [Google Scholar]
- 25.Tugarinov V, Liang Z, Shapiro YE, Freed JH, Meirovitch E. J Am Chem Soc. 2001;123:3055–3063. doi: 10.1021/ja003803v. [DOI] [PubMed] [Google Scholar]
- 26.Meirovitch E, Shapiro YE, Polimeno A, Freed JH. J Phys Chem A. 2006;110:8366–8396. doi: 10.1021/jp056975t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meirovitch E, Shapiro YE, Polimeno A, Freed JH. Prog NMR Spectrosc. 2010;56:360–405. doi: 10.1016/j.pnmrs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meirovitch E, Polimeno A, Freed JH. Encyclopedia of Magnetic Resonance. John Wiley; Chichester, U.K: 2011. Protein dynamics by NMR spin relaxation: the slowly relaxing local structure perspective. [Google Scholar]
- 29.Zerbetto M, Buck M, Meirovitch E, Polimeno A. J Phys Chem B. 2011;115:376–388. doi: 10.1021/jp108633v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong Y, Hota PK, Hamaneh MB, Buck M. Structure. 2008;16:246–258. doi: 10.1016/j.str.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong Y, Chugha P, Hota PK, Alviani RS, Li M, Tempel W, Shen L, Park HW, Buck M. J Biol Chem. 2007;282:37215–37224. doi: 10.1074/jbc.M703800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouguet-Bonnet S, Buck M. J Mol Biol. 2008;377:1474–1487. doi: 10.1016/j.jmb.2008.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamaneh MB, Zhang L, Buck M. Biophys J. 2011;101:196–204. doi: 10.1016/j.bpj.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hota PK, Buck M. Cell Mol Life Sciences. 2012 doi: 10.1007/s00018-012-1019-0. published on-line. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong Y, Buck M. J Biomol NMR. 2005;31:369–370. doi: 10.1007/s10858-005-0943-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong Y, Hota PK, Penachioni JY, Hamaneh MB, Kim S, Alviani RS, Shen L, He H, Tempel W, Tamagnone L, et al. J Biol Chem. 2009;284:35962–35972. doi: 10.1074/jbc.M109.056275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hota PK, Buck M. Protein Sci. 2009;18:1060–1071. doi: 10.1002/pro.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Bouguet-Bonnet S, Buck M. Methods in Molecular Biology: Allostery: Methods & Protocols. Vol. 796. Humana Press; New York, USA: 2011. Combining NMR and molecular dynamics studies for insights into the allostery of small GTPase-protein interactions; pp. 235–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Hota PK, Tong Y, Li B, Shen L, Nedyalkova L, Borthakur S, Kim S, Tempel W, Buck M, et al. J Biol Chem. 2011;286:26093–26106. doi: 10.1074/jbc.M110.197053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong Y, Hughes D, Placanica L, Buck M. Structure. 2005;13:5–17. doi: 10.1016/j.str.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grzesiek S, Bax A. J Am Chem Soc. 1993;115:12593–12594. [Google Scholar]
- 42.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 43.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 44.Goddard TD, Kneller DG. SPARKY. Vol. 3. University of California; San Francisco: http://production.rbvi.ucsf.edu/home/sparky/ [Google Scholar]
- 45.Viles JH, Duggan BM, Zaborowski E, Schwarzinger S, Huntley JJ, Kroon GJ, Dyson HJ, Wright PE. J Biomol NMR. 2011;21:1–9. doi: 10.1023/a:1011966718826. [DOI] [PubMed] [Google Scholar]
- 46.Buck M, Bouguet-Bonnet S, Pastor RW, MacKerell AD. Biophys J. 2006;90:L36–L39. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker O, Varadan R, Fushman D. J Magn Reson. 2004;168:336–345. doi: 10.1016/j.jmr.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 48.Fushman D, Cahill S, Cowburn D. J Mol Biol. 1997;266:173–194. doi: 10.1006/jmbi.1996.0771. [DOI] [PubMed] [Google Scholar]
- 49.Fushman D. Methods Mol Biol. 2012;831:485–511. doi: 10.1007/978-1-61779-480-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Case DA. Acc Chem Res. 2002;35:325–331. doi: 10.1021/ar010020l. [DOI] [PubMed] [Google Scholar]
- 51.Fadel AR, Jin DQ, Montelione GT, Levy RM. J Biomol NMR. 1995;6:221–226. doi: 10.1007/BF00211787. [DOI] [PubMed] [Google Scholar]
- 52.Wand AJ. Nat Struct Biol. 2001;8:926–931. doi: 10.1038/nsb1101-926. [DOI] [PubMed] [Google Scholar]
- 53.Finerty PJ, Mittermaier AK, Muhandiram R, Kay LE, Forman-Kay JD. Biochemistry. 2005;44:694–703. doi: 10.1021/bi048641k. [DOI] [PubMed] [Google Scholar]
- 54.Bracken C, Carr PA, Cavanagh J. J Mol Biol. 1999;285:2133–2146. doi: 10.1006/jmbi.1998.2429. [DOI] [PubMed] [Google Scholar]
- 55.Changeux JP, Edelstein SJ. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 56.Popovych N, Sun S, Ebright RH, Kalodimos CG. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcos E, Crehuet R, Bahar I. PLoS Comput Biol. 2011;7:e1002201. doi: 10.1371/journal.pcbi.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Del Sol A, Tsai CJ, Ma B, Nussinov R. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.