

Abstract
The influenza A virus M2 proton channel (A/M2) is the target of the antiviral drugs amantadine and rimantadine, whose use has been discontinued due to widespread drug resistance. Among the handful of drug-resistant mutants, S31N is found in more than 95% of the currently circulating viruses and shows greatly decreased inhibition by amantadine. The discovery of inhibitors of S31N has been hampered by the limited size, polarity, and dynamic nature of its amantadine-binding site. Nevertheless, we have discovered small-molecule drugs that inhibit S31N with potencies greater than amantadine’s potency against WT M2. Drug binding locks the protein into a well-defined conformation, and the NMR structure of the complex shows the drug bound in the homotetrameric channel, threaded between the side chains of Asn31. Unrestrained molecular dynamics simulations predicted the same binding site. This S31N inhibitor, like other potent M2 inhibitors, contains a charged ammonium group. The ammonium binds as a hydrate to one of three sites aligned along the central cavity that appear to be hotspots for inhibition. These sites might stabilize hydronium-like species formed as protons diffuse through the outer channel to the proton-shuttling residue His37 near the cytoplasmic end of the channel.
Keywords: M2-S31N mutant structure, membrane protein structure, M2-S31N inhibitor
The influenza A virus M2 proton channel (A/M2) is the target of the antiviral drugs amantadine and rimantadine (1–3), which bind directly to the pore of the channel (2–4). Although amantadine has been widely used for several decades, drug resistance has curtailed the use of this family of drugs. Many amantadine-resistant influenza viruses can be selected in cell culture (5, 6). A subset of these mutations is found in infected patients undergoing treatment with amantadine (7), and reverse-engineered viruses harboring various pore-lining mutations are competent to replicate in the mouse (8). However, many of these mutations give rise to somewhat attenuated viruses that are less transmissible than WT virus, and they tend to revert in the absence of drug pressure (6, 9). Indeed, large-scale sequencing of transmissible viruses isolated as early as 1918 showed that mutations to pore-lining residues are allowed only within the first turn of the transmembrane (TM) helix at positions 26, 27, and 31 (10). S31N has long been the predominant amantadine-resistant mutation in M2 (11–14). It predominated in 98–100% of the transmissible amantadine-resistant H1N1, H5N1, and H3N2 strains isolated from humans, birds, and swine in the past decade. V27A and L26F are less frequent mutations (10, 11, 15). Extensive studies of point mutations to the pore-lining residues of M2 have been conducted to understand the paucity of natural variants (16, 17). Numerous mutants in the N-terminal aqueous pore retained the ability to conduct protons selectively over other ions, although the magnitude and pH dependence of their conduction varied. However, only a few mutations at the most distal sites, V27A, S31N, and L26F, had properties very similar to WT M2. These same mutants comprise more than 99.9% of reported resistance in transmissible viruses. Thus, the stringency of the sequence conservation in M2 reflects tight functional restraints of the pore-lining residues, where a single mutation to a monomer causes four changes within a very constricted area of the tetrameric pore.
These studies underscore the importance of discovering inhibitors of S31N. However, numerous medicinal chemical efforts directed toward discovery of small-molecule inhibitors of resistant M2 mutants (18–21) have shown little progress until recently (4, 22, 23). For example, we designed a spiroadamantane inhibitor of V27A and L26F mutants (23), but this compound did not inhibit S31N.
Notwithstanding this progress, the inhibition of the remaining predominant S31N mutant has remained a major challenge. The cavity available for binding drugs in WT M2 is relatively small, and this mutation increases both the bulk and polarity of the channel-lining residues, reducing the space available for interaction with drugs. Moreover, this mutation increases the dynamic nature of the protein, which precluded the determination of a single, high-resolution solution NMR structure for S31N (24). Thus, until now, the best known compound targeting the A/M2-S31N mutant is (3,7-dimethyl-1-bisnoradamantyl) amine (25), with a potency similar to amantadine (IC50 = 252 μM and 200 μM, respectively). Recently, we discovered a series of inhibitors of both WT and S31N M2. Here, we begin with one of these early inhibitors and improve the affinity of the compound for S31N in several rounds of design, synthesis, pharmacological testing, and molecular dynamics (MD) simulations. The resulting compounds have sufficiently high affinity for the TM domain of S31N to lock it into a well-defined conformational state, enabling structure determination by solution NMR. The drug contains an ammonium group that appears to mimic a solvated hydronium ion, and it receives special stabilization in the channel by interacting with the carbonyl groups in the backbone atoms of Val27 and the side chain of Asn31. By comparing the position of the ammonium group with that found in earlier drug complexes, it is possible to define a likely trajectory and series of stabilizing interactions that help guide protons to the His37 residues involved in shuttling protons through the M2 channel.
Results and Discussion
Discovery of Potent Inhibitors of S31N.
Amantadine-like compounds functionalized with aryl groups retain potency in a viral replication assay (26), but we have found them to be inactive against the S31N mutant. Nevertheless, we have discovered that conjugation of a -CH2-heteroaryl (Table 1) group to the amine of amantadine leads to significant inhibition of S31N. For example, the 5-methyl–substituted isoxazole (M2WJ369) or 1,2,4-oxadiazole (M2WJ405) causes ∼50% inhibition of proton flux (Table 1) against S31N at 100 μM in a two-electrode voltage clamp (TEVC) assay (22). We therefore synthesized isoxazole or 1,2,4-oxadiazole derivatives bearing a 1-(1-adamantylamino)-methylene group in either the 3- or 5-position of the ring (Fig. S1). Their potencies were tested on A/M2 channels heterologously expressed in Xenopus laevis oocytes (Table 1 and Table S1). The activities were expressed as the percentage of current reduction after 2 min of incubation with 100 μM compounds at pH 5.5. Compounds that displayed more than 80% inhibition at 100 μM were also assayed at 30 μM. Because M2 blockers show slow on/off rates for binding, the IC50 values in TEVC assays (Fig. S2) systematically underestimate their binding constants or EC50 in plaque assays (22, 23).
Table 1.
Structure-activity relationship of isoxazole-, 1,2,4-oxadiazole–, and isoxazoline-containing compounds
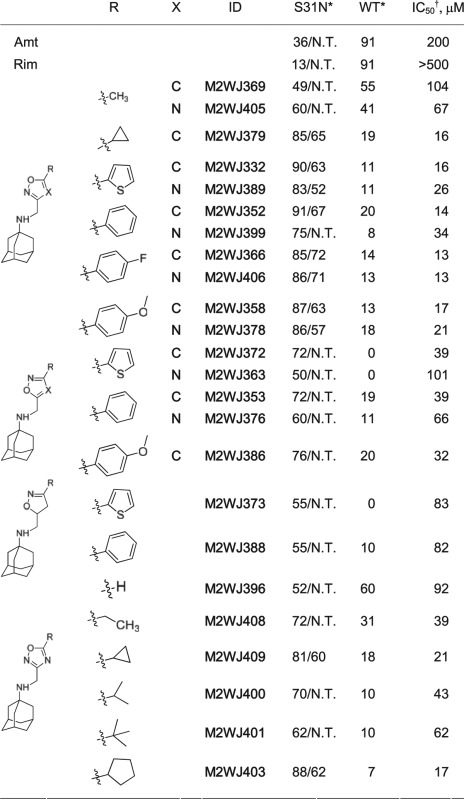 |
Amt, amantadine; N.T., not tested; R, substitutions; Rim, rimantadine; X, N or C.
*Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. All compounds were initially tested at 100 μM. Compounds that showed greater than 80% inhibition at 100 μM were further tested at 30 μM. The data are presented as % inhibition at 100 μM/% inhibition at 30 μM.
†S31N IC50s were calculated based on the equation derived from nonlinear regression curve fitting of a set of compounds with experimentally measured IC50s and % inhibition at 100 μM (more details are provided in SI Materials and Methods).
A phenyl or 2-thienyl enhanced the activity when it was placed in the variable position of the isoxazole and 1,2,4-oxadiazole. The 1-(1-adamantylamino)-methylene group was best placed at the 3-position of the heterocycle. In 4-substituted phenyl series, the activities were sensitive to the p-substituent; fluoride and methoxyl were tolerated, but trifluoromethyl, chloride, and bromide were not (Table 1 and Table S1). Isoxazoles tended to be more active than the corresponding oxadiazoles, and the nonaromatic isoxazoline was much less active. A number of the compounds have IC50 values against S31N on a par with or better than that seen for the inhibition of WT by amantadine or rimantadine.
Plaque Reduction Assay.
Compounds with varying potency in the electrophysiological assay were evaluated in a plaque reduction assay using influenza A virus A/WSN/33, which contains the S31N mutation (Fig. 1). Compounds were assayed at 50 μM and 10 μM with amantadine as a control. At a concentration of 50 μM, amantadine reduced both the number and the size of the plaques (Fig. S3), but almost no inhibition was observed at 10 μM (Fig. 1A). By contrast, compounds that inhibit A/M2-S31N with an IC50 ≤ 16 μM (as derived by electrophysiology) completely inhibited plaque formation at 10 μM. There was a rough correlation between the plaque count and the electrophysiological IC50 (Fig. 1B).
Fig. 1.

Plaque reduction assay. (A) Amantadine (AMT)-resistant influenza virus (A/WSN/33) containing S31N mutation in the M2 TM domain was assayed for plaque formation on Madin-Darby canine kidney epithelial cells (MDCK) in the presence or absence of different drugs at 10 μM. Amantadine was used as a control. (B) Plaque count plot with 10 μM drugs. (C) Plaque reduction assay EC50s for two selected compounds M2WJ332 and M2WJ379; amantadine (Ama) was used as a control.
The electrophysiological assay is conducted under conditions of kinetic rather than thermodynamic control (22, 27), and it appears to “bottom out” at a limiting value of about 10–15 μM drug concentration. Therefore, two compounds, M2WJ332 and M2WJ379, each with an electrophysiological IC50 of 16 μM, were selected for EC50 measurement in the plaque reduction assay (Fig. 1C). The EC50 values for M2WJ332 and M2WJ379 are 153 nM and 1.01 μM vs. S31N, respectively (Fig. 1C), in comparison to 328 nM for amantadine vs. WT M2. These results confirm the expectation that the compounds would be more effective under conditions of longer incubation and demonstrate that M2WJ332 is more potent against S31N than amantadine is against the WT M2. To confirm this conclusion, we measured the time course for recovery of the current after removing drug from the buffer bathing an oocyte expressing M2-S31N. Only 36% of the current was recovered after 20 min at pH 5.5 (Fig. S2C), showing that the drug dissociates with a relatively slow off rate.
Structure M2-S31N (19–49) Complexed to M2WJ332.
The NMR structure of the complex of M2WJ332 with the channel was solved for a peptide spanning from 19 to 49, which includes the TM and portions of the cytoplasmic domain and ectodomain. Two-dimensional 15N heteronuclear single quantum coherence (HSQC) spectra for this peptide showed multiple sets of peaks of weak intensity, indicative of multiple conformations. On addition of excess drug, the total intensity increased and only one set of peaks per subunit was observed (Fig. 2A). Assignments were obtained using doubly and triply labeled samples and standard methods (28, 29) (SI Materials and Methods). The main chain chemical shifts for this complex were very similar to those seen for an earlier complex of a drug bound to the WT M2 protein (30), except near Asn31 (residues 24–36; Fig. S4A), where drugs bind WT. To simplify the spectrum and obtain unambiguous NOE assignments, we also synthesized peptides with 15N-13C–labeled amino acids selectively incorporated at the interhelical interfaces and the drug-binding site (Fig. 2B and Fig. S5). A 2D 13C-edited NOESY experiment identified strong NOEs between the heterocyclical rings and Val27, as well as between the adamantane and Cα of G34 and N31 (Fig. 2B and Fig. S6). The interaction was confirmed with a 15N-edited NOESY at a mixing time of 150 ms recorded for a fully deuterated sample, which reports distances up to 8 Å (31) (Fig. S5C). Representative strips of interhelical NOEs are shown in Fig. S6. The structure was determined with Xplor-NIH (32) using 215 NOE-based distance restraints per monomer (including 28 interhelical and 23 drug–protein NOEs) and 40 torsional angle restraints (Fig. S7C). For comparison, previous work on WT protein used 7 interhelical and 7 drug–protein NOEs in the TM region (33). The S31N structure was well defined from residues 26–44, with a backbone rmsd of 0.47 Å to the mean structure (Fig. 2 C and D and Fig. S7C).
Fig. 2.

NMR studies of M2-S31N (19–49) in the presence of drug. (A) Two-dimensional 15N HSQC (Left) and transverse relaxation optimized spectroscopy (TROSY)-HSQC (Right) spectra acquired for 2 mM (monomer) M2-S31N (19–49) in the absence and presence of M2WJ332 at pH 6.8 at 313 Kelvin in 100 mM DPC and 50 mM Na phosphate in 10% D2O in H2O. (B) Two-dimensional 13C-(1H)-1H NOESY experiment for VANIG sample with a mixing time of 150 ms. (C) Twenty final conformers with the lowest energy representing the NMR solution structure are shown after superposition of the backbone of 26–45. (D). Side chains of the TM are also well defined, and the drug (spheres) locates between V27 and G34.
The structure (Fig. 3A) is closely related to previous structures of the WT M2 channel (Fig. 3B), showing most similarity to the drug-bound channel structure solved by solid-state NMR (SSNMR) (2KQT, backbone rmsd of 0.67 Å for TM residues 25–44) (Fig. S7). At the N-terminal end of the bundle, the helices in the drug-bound S31N are slightly expanded by ∼1.0 Å relative to WT M2 structures, allowing the drug to penetrate between the Val27 residues (Fig. 3B). The thienyl group lies with the Cγ methyls of Val27, snuggly fit in the middle of the ring (Fig. 3A). The adamantyl lies lower in the channel, in a pocket lined by the Cα of Gly34 and the Cβ atoms of Asn31 and Ala30. The ammonium group lies near the center of the channel, just below the carbonyl oxygen of Val27. We found that the conformational form studied is maximal at pH 6.8 and becomes less populated at higher and lower pH as assessed from 15N HSQC spectra (Fig. S4C). Interestingly, the backbone 13C chemical shifts for the form studied here show a good correlation with those of only one of two distinct conformers seen by SSNMR of S31N (18–60) in phospholipid bilayers (R2 = 0.88), indicating that the drug-bound conformation seen here is similar to one of the two conformational forms seen in phospholipid bilayers in the absence of an inhibitor (34). It would be interesting to determine whether the population of the two conformational forms seen by SSNMR is also pH-dependent. The pH dependence seen in our study suggests that compound M2WJ332 binds to a form of M2-S31N in which the His37 residues are partially protonated. This behavior is different from that of WT M2, which gives good spectra at a higher pH when complexed with drugs (35). The difference between the two systems might relate to the fact that the ammonium group of amantadine projects toward His37 in WT but is directed away from His37 in the complex of S31N with M2WJ332. Amantadine and rimantadine are nearly radially symmetrical, allowing these ligands to rotate very rapidly (subnanoseconds) about their axes (2), imparting fourfold symmetry to the time-averaged structure of the complexes previously solved with these drugs. Rotation of M2WJ332 is also sufficiently rapid to average the chemical shifts of N31, indicating that the drug rotates within the binding site on a time scale more rapid than the millisecond regime. However, the intensities of the backbone amide peaks in the 15N HSQC spectrum were significantly weaker for the region where the drug binds (residues 24–33) than for other portions of the channel. Moreover, although the side chain carboxamide amide of Asn31 was resolved in the drug-free spectrum, it disappeared in the complex, suggesting that it might undergo conformational exchange, although further experiments are needed to define the rate.
Fig. 3.

M2-S31N drug inhibition mechanism. (A) Binding site of M2WJ332 in the NMR structure: The side chains V27, A30, N31, and H37 and the molecule are shown as sticks; other side chains are shown as transparent space filling; the backbone of the protein (one monomer not shown for clarity) is shown as opaque ribbons; and residues 19–24 in the NMR ensemble are shown altogether as transparency. (B) Comparison of the N-terminal region, beginning with residue 25, is shown from the present NMR structure (red, 2LY0) with other drug-bound structures. A Cα trace of the main chain is shown, and the side chain of V27 is shown in sticks. Two structures of the WT protein in the absence of drug are shown in green (2LOJ) (41) and blue (3LBW) (36). The structure of the amantadine complex (2KQT) (2) of WT is shown in yellow. The N-terminal regions of the helices shift to accommodate the larger drug bound to the pore in S31N. (C) Drug–protein interactions: One of the Asn side chains forms bidendate interactions with the drug, and the carbonyls from another two Asns form a water-mediated hydrogen bond with the ammonium from the drug. (D–G) Comparison between amine sites from three different M2 inhibitors. The backbone is shown as a cartoon, and the side chains of residues 27, 31, 37, and 41 and the pore-lining carbonyls between 27 and 31 are shown in sticks. (D) Position of the three nitrogen sites within the pore: Light blue spots indicate the MD-calculated density of the nitrogen atom, contoured at 1σ (68%), in the three complexes shown in E–G. (E) Complex between WT-M2TM and amantadine (yellow). (F) Complex between WT-M2TM and spiro-adamantane-amine (23). (G) Complex between S31N-M2TM and M2WJ332.
Structure-Based Drug Design.
The variable substituent in M2WJ332 lies in a pocket that is rimmed by Val27 and open to solvent on one side, explaining the tolerance to substitution in the structure-activity relationship (SAR). It appeared that branched alkyl or cycloalkyl groups might similarly form a hydrophobic interaction with Val27 side chain when placed in the variable position. Indeed, a systematic study of lower alkane substituents showed that the activity was optimal when the substituent was a cyclopropyl or cyclopentyl group as in M2WJ409 and M2WJ403 (Table 1), nicely fitting the dimensions of the cavity.
MD Simulations.
MD simulations of the complexes between M2WJ332 and the TM (25–46) domain of A/M2 (M2TM), provided important insights into the mechanism of binding. Calculations of the complex were initiated before the solution NMR structure was determined; therefore, we used the high-resolution X-ray structure of the apo-form of the channel (3LBW) (36) with amantadine positioned as in 2KQT (2) in a fully hydrated 64-nm2 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer (23, 36, 37). We also conducted a 20-ns all-atom simulation in the same bilayer, starting with the NMR structure once it became available. The two MD ensembles were identical within the thermal fluctuations of the system; thus, we present here the results of the initial unrestrained simulation. An S31N model was generated by simulating the mutated protein for about 100 ns (23). The Asn31 side chains, initially at the interface between helices, all point toward the lumen in simulations with and without drug. For each mutant of M2 (WT and S31N), the drug was tested with the aromatic headgroup either facing toward His37 (amine-down) or away (amine-up).
The molecule reached a more ordered position in the amine-up simulations (matching the NMR structure) than in the amine-down simulations (Fig. S8). In all simulations in which the M2WJ332 molecule was prepared in the amine-up configuration, the molecule shifted toward Asn31. Amine-down simulations showed more variability (SD > 0.5 Å), and the mean position of the amine was shifted by 0.5 Å from the amine-up configuration. The amine-up configuration was unexpected, because amantadine, rimantadine, and other drugs bind WT with their amines down (23, 30). Nevertheless, the trajectory matches the structure independently measured by solution NMR. The adamantane group has a snug fit with the channel lumen, and it rotates about the N-C(1) bond on a nanosecond time scale, as in earlier amantadine simulations. However, unlike amantadine, the rotational motion of the amine is constrained by a stable pattern of water-mediated H-bonds with two Asn31 side chains. Each pattern of H-bonds persisted for tens of nanoseconds; after that, the molecule abruptly rotates by 90° around the axis of the M2 channel, forming an identical H-bond pattern with a new protein monomer. In simulations of S31N-M2TM with and without a bound molecule, the Asn31 side chain pointed inward in most frames, toward the lumen of the channel.
In summary, MD simulations suggested stable hydrogen bonding networks between M2WJ332 and the four Asn31 side chains, three at a time. The drug is clamped into a position by three Asn side chains. Two connect to the ammonium group via high-occupancy water molecules that have highly preferred positions (Fig. 3C). The third Asn31 side chain engages the isoxazole in a bidentate interaction in which the carbonyl receives a hydrogen bond from the ammonium group, whereas the N of the isoxazole receives a hydrogen bond from Asn31 carboxamide (Fig. 3C). The bidentate interaction was found in all members of the ensemble, suggesting that it is relatively strong and specific and that it contributes to the driving force for binding. This interaction explains the lower potencies of the reversed isoxazole (M2WJ372) in which the O and N switch places in the ring, forcing the less basic heteroatom to interact with the amide NH group. Exchange between neighboring pairs of Asn31 is suggested to occur at a time scale faster than microseconds. Most importantly, the position of the amine, interacting with residue 31, was observed without using any experimental restraint, evolving from the amantadine-like position; this fact clearly indicates the presence of favorable interactions between M2WJ332 and the Asn31 side chains.
Conclusions
Here, we describe potent inhibitors of A/M2-S31N. The compounds described in this paper are good starting points for the development of drugs to address the problem of amantadine resistance. As a tool to determine the structure of S31N, we developed compounds specific for this mutant. Given that S31N is by far the most predominant drug-resistant mutant, it might be possible to combine an S31N-specific compound with rimantadine in a manner similar to the combination drugs typically used to address drug resistance in HIV and other infectious diseases.
In establishing an assay for S31N, we found it important to verify that the assay identified true M2 inhibitors, because we found that vesicle assays and viral inhibition assays are complicated by the fact that amantadine-like compounds are proton carriers. Proton carriers, such as chloroquine, are good inhibitors of virus in vitro but lack efficacy in animal and human trials (38). However, by conducting electrophysiological assays in parallel with antiviral assays, it was possible to eliminate many false-positives that acted as proton carriers to delay acidification of the endosome.
S31N binds M2WJ332, with its adamantane ring in approximately the same location as in amantadine/rimantadine, but the ammonium group binds up toward the carbonyl of Val27 and Asn31 rather than down as seen previously (30). Indeed, examination of the location of the ammonium in experimental complexes and MD simulations shows that the ammonium binds to one of three locations (Fig. 3 D–G). In each case, the ammonium locates near the center of the channel and is stabilized by water molecules, which, in turn, hydrogen-bond to carbonyl groups of the channel. Thus, the channel appears to stabilize ammonium hydrates specifically, which we suggest mimic intermediates in the conduction of protons to His37. From a more practical perspective, the presence of multiple sites of interaction for an ammonium group provides a series of starting points for design of new drugs, particularly ones that access the most highly conserved region adjacent to H37 and W41.
The adamantyl group has a snug fit with the walls of the pore, stabilized by van der Waals interactions and the hydrophobic effect. The polarity and composition of the channel walls, particularly in WT M2, are similar to those of an organic solvent, such as ethyl acetate, explaining the favorable partitioning of the adamantane from water to the channel. The structure of the α-helix is particularly favorable for interacting with amantadine-like drugs, because the C = O bonds in solvent-exposed helices are about half-hydrated (39). Thus, they can easily either stabilize an ammonium group when hydrated or dehydrate to stabilize apolar groups, such as adamantane, when dehydrated.
Materials and Methods
TEVC Assay.
The inhibitors were tested via a TEVC assay using X. laevis frog oocytes microinjected with RNA expressing the A/M2 protein as in a previous report (22). The potency of the inhibitors was expressed as the percentage inhibition of A/M2 current observed after 2 min of incubation with 100 μM compounds. The compounds displaying more than 80% inhibition at a concentration of 100 μM were also tested at 30 μM. The IC50 values were calculated from nonlinear regression fitting of percentage inhibition at 100 μM or both 100 μM and 30 μM for these compounds (Fig. S2).
Plaque Reduction Assay.
Selected compounds were tested for inhibition of influenza virus strain wt A/WSN/33 containing the A/M2-S31N ion channel by plaque reduction assays as in a previous report (25).
Protein Expression.
Uniformly 15N-2H–labeled A/M2-S31N (19–49) with the sequence SNDSSDPLVVAANIIGILHLILWILDRLFFK was made by trypsin digestion of the full-length Udorn M2 expressed as in the study by Leiding et al. (40).
Peptide Synthesis.
Selective isotope-labeled A/M2-S31N (19–49) peptides were manually synthesized with Rink Amide Chemmatrix resin (Matrix Innovation, Inc.) using Fmoc chemistry as in the study by Cady et al. (30).
NMR Spectroscopy and Structure Calculations.
Spectra at 313 Kelvin were recorded on Bruker 800- or 900-MHz spectrometers equipped with cryogenic probes. 1H-1H upper distance constraints for structure calculations were extracted from different types of NOESY spectra with the same mixing time of 150 ms [3D 15N-edited, 3D 13C-edited, 3D 13C-edited, 13C-15N–filtered, 2D 15N-(1H)-1H, and 2D 13C-(1H)-1H] (31). Backbone dihedral angle constraints were derived from chemical shifts, and structures were computed using Xplor-NIH (32).
MD Simulations.
The simulations were begun from 3LBW (36), as in the study by Wang et al. (23). The N31 side chains were arranged as in previous long simulation runs of the drug-free channel. The drug molecule was initialized with its secondary amine as close as possible to the position of the primary amine of amantadine in 2KQT (30). Two different orientations were initialized, with the aromatic headgroup pointing to the viral exterior (“up”) and the viral interior (“down”), respectively. Position restraints were initially set on the protein and drug, and they were gradually released during the first 24 ns until the distribution of pore waters reached equilibrium.
Supplementary Material
Acknowledgments
We thank Belgin Canturk and Hyunil Jo for preliminary results. Y.W. thanks Jeff Pelton and Mark Kelly for help with NMR experiments. W.F.D. was supported by NIH Grants GM56423 and AI74571.
Footnotes
Conflict of interest statement: Jizhou Wang is an employee of Influmedix, Inc. W.F.D., R.A.L., M.L.K., and L.H.P. are members of the scientific advisory board of Influmedix, Inc.
This article is a PNAS Direct Submission.
Data deposition: The structure and chemical shift assignments have been deposited in the Protein Data Bank, www.pdb.org (accession no. 2LY0) and BioMagResBank, www.bmrb.wisc.edu (accession no. 18706), respectively.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216526110/-/DCSupplemental.
References
- 1.De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006;5(12):1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cady SD, et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463(7281):689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451(7178):596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, et al. Exploring organosilane amines as potent inhibitors and structural probes of influenza a virus M2 proton channel. J Am Chem Soc. 2011;133(35):13844–13847. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4(11):3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grambas S, Hay AJ. Maturation of influenza A virus hemagglutinin—Estimates of the pH encountered during transport and its regulation by the M2 protein. Virology. 1992;190(1):11–18. doi: 10.1016/0042-6822(92)91187-y. [DOI] [PubMed] [Google Scholar]
- 7.Shiraishi K, et al. High frequency of resistant viruses harboring different mutations in amantadine-treated children with influenza. J Infect Dis. 2003;188(1):57–61. doi: 10.1086/375799. [DOI] [PubMed] [Google Scholar]
- 8.Abed Y, Goyette N, Boivin G. Generation and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutations. Antimicrob Agents Chemother. 2005;49(2):556–559. doi: 10.1128/AAC.49.2.556-559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki H, et al. Emergence of amantadine-resistant influenza A viruses: Epidemiological study. J Infect Chemother. 2003;9(3):195–200. doi: 10.1007/s10156-003-0262-6. [DOI] [PubMed] [Google Scholar]
- 10.Furuse Y, Suzuki A, Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob Agents Chemother. 2009;53(10):4457–4463. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krumbholz A, et al. High prevalence of amantadine resistance among circulating European porcine influenza A viruses. J Gen Virol. 2009;90(Pt 4):900–908. doi: 10.1099/vir.2008.007260-0. [DOI] [PubMed] [Google Scholar]
- 12.Deyde V, et al. Genomic events underlying the changes in adamantane resistance among influenza A(H3N2) viruses during 2006-2008. Influenza Other Respir Viruses. 2009;3(6):297–314. doi: 10.1111/j.1750-2659.2009.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deyde VM, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196(2):249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 14.Simonsen L, et al. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol Biol Evol. 2007;24(8):1811–1820. doi: 10.1093/molbev/msm103. [DOI] [PubMed] [Google Scholar]
- 15.Schmidtke M, et al. Amantadine resistance among porcine H1N1, H1N2, and H3N2 influenza A viruses isolated in Germany between 1981 and 2001. Intervirology. 2006;49(5):286–293. doi: 10.1159/000094244. [DOI] [PubMed] [Google Scholar]
- 16.Stouffer AL, et al. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16(7):1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balannik V, et al. Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel. Biochemistry. 2010;49(4):696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lagoja IM, De Clercq E. Anti-influenza virus agents: Synthesis and mode of action. Med Res Rev. 2008;28(1):1–38. doi: 10.1002/med.20096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duque MD, et al. Inhibitors of the M2 channel of influenza A virus. Recent Advances in Pharmaceutical Sciences. 2011:35–64. [Google Scholar]
- 20.Severson WE, et al. High-throughput screening of a 100,000-compound library for inhibitors of influenza A virus (H3N2) J Biomol Screen. 2008;13(9):879–887. doi: 10.1177/1087057108323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, et al. Exploring the Requirements for the Hydrophobic Scaffold and Polar Amine in inhibitors of M2 from Influenza A Virus. ACS Med Chem Lett. 2011;2(4):307–312. doi: 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balannik V, et al. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48(50):11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, et al. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J Am Chem Soc. 2011;133(32):12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA. 2009;106(18):7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duque MD, et al. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J Med Chem. 2011;54(8):2646–2657. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao X, Li C, Zeng S, Hu W. Discovery of highly potent agents against influenza A virus. Eur J Med Chem. 2011;46(1):52–57. doi: 10.1016/j.ejmech.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J Virol. 1993;67(9):5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sattler M, Schleucher J, Griesinger C, Smith ME, Eck ERHv. Heteronuclear Multidimensional NMR Experiments for the Structure Determination of Proteins in Solution Employing Pulsed Field Gradients. Amsterdam: Elsevier; 1999. [Google Scholar]
- 29.Venters RA, Farmer BT, 2nd, Fierke CA, Spicer LD. Characterizing the use of perdeuteration in NMR studies of large proteins: 13C, 15N and 1H assignments of human carbonic anhydrase II. J Mol Biol. 1996;264(5):1101–1116. doi: 10.1006/jmbi.1996.0699. [DOI] [PubMed] [Google Scholar]
- 30.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J Am Chem Soc. 2011;133(12):4274–4284. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Amsterdam, Boston: Academic; 2007. [Google Scholar]
- 32.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 33.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451(7178):591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andreas LB, Eddy MT, Chou JJ, Griffin RG. Magic-angle-spinning NMR of the drug resistant S31N M2 proton transporter from influenza A. J Am Chem Soc. 2012;134(17):7215–7218. doi: 10.1021/ja3003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93(1):276–283. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acharya R, et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci USA. 2010;107(34):15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carnevale V, Fiorin G, Levine BG, Degrado WF, Klein ML. Multiple Proton Confinement in the M2 Channel from the Influenza A Virus. J Phys Chem C Nanomater Interfaces. 2010;114(48):20856–20863. doi: 10.1021/jp107431g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paton NI, et al. Chloroquine for influenza prevention: A randomised, double-blind, placebo controlled trial. Lancet Infect Dis. 2011;11(9):677–683. doi: 10.1016/S1473-3099(11)70065-2. [DOI] [PubMed] [Google Scholar]
- 39.Walsh ST, et al. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 2003;12(3):520–531. doi: 10.1110/ps.0223003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leiding T, Wang J, Martinsson J, DeGrado WF, Arsköld SP. Proton and cation transport activity of the M2 proton channel from influenza A virus. Proc Natl Acad Sci USA. 2010;107(35):15409–15414. doi: 10.1073/pnas.1009997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma M, et al. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330(6003):509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.