

ABSTRACT
Objectives: Pulmonary arterial hypertension (PAH) is a rare and severe disease with incompletely under stood pathogenesis. PAH is associated with pulmonary arterial remodeling and inflammation. We evaluated the effects of Sildenafil on the Monocrotaline (MCT) –induced disease in Wistar rats, for potential benefit in the early phases of inflammation and vascular remodeling.
Material and Methods: MCT-injected rats, MCT-injected sildenafil-treated rats (starting day 1 with 2 x 0.2 mg/day; total of 2 mg/kgc/day) and saline-injected control rats were evaluated at day 14 and day 28 following MCT for pulmonary morphological changes – lesions, inflammation (inflammator y index), arterial morphometry (hypertrophy index), immunohistochemistry for smooth muscle cell marker.
Outcomes: The administration of sildenafil following MCT significantly reduced the severity of inflammation in the acute stage of the disease (reduction of the inflammatory index by 6.038% (p <0.05)) and prevented pulmonary arterial remodeling (reduction of the hypertrophy index by 7.306% (p<0.001)). It also improved survival in the early phase with a mortality rate during the first 14 days of 4 in the MCT- exposed rats vs 1 in the MCT-exposed sildenafil-treated rate.
Conclusions: Early administration of sildenafil in the MCT experimental PAH improves inflammation and survival, and prevents pulmonary vascular remodeling. Our study suggests that one of the mechanisms involved, besides vasodilatation and anti-proliferative effect, could be a direct anti-inflammatory effect of sildenafil.
Keywords: pulmonary arterial hypertension, monocrotaline, sildenafil, inflammation, pulmonary arterial remodeling
INTRODUCTION
Pulmonary arterial hypertension (PAH) manifests as a progressive increase of the pulmonary arterial resistance, leading to right heart failure and death. In the absence of treatment, PAH has a very poor prognosis, with a mean survival of 2½ years when NYHA III and of merely 6 months when NYHA IV (1).
PAH is characterized by an increased mean pulmonary artery pressure (mPAP) ≥25 mmHg, with a normal pulmonary artery wedge pressure (2).
The main morphopathological finding in PAH is the remodeling of small pulmonary arteries (muscular arteries and arterioles), with marked hyperplasia and proliferation of the smooth muscle cells, leading to their thickening and hyperconstriction. Intimal hyperplasia also occurs, with marked thickening of the intima, plexiform lesions and vascular fibrinoid necrosis leading to their obstruction. In situ thrombosis is an additonal mechanism, contributing furthermore to the development of PAH (3).
The pathogenesis of PAH remains uncertain (4). It is a multi-factorial disease in which predisposed individuals (genetically - BMPR2 mutation; with PAH associated conditions – sclero- derma, portal hypertension, HIV infection and others), under the influence of environmental factors, develop pulmonary arterial endothelial dysfunction, leading to pulmonary artery remodeling, with increase of pulmonary vascular resistance and PAH.
Elevated pulmonary vascular resistance appears to be the result of an unbalance between vasodilators and vasoconstrictors factors, in addition to a remodeled pulmonary arterial wall. Key vasodilator molecules are the nitric oxide (NO) and prostacyclin. The NO deficit, a potent vasodilator locally produced by the endothelial cell, seems to play an important part in the pathogenesis of PAH (5). Studies have shown that NO is a very active local mediator with numerous roles: - vasodilatation; - anti-thrombotic effect; - mitosis inhibition and anti-proliferative effect over the smooth muscle cells.
Smooth muscle cell NO-relaxation is realized throughout the activation of an intra-cytoplasmic guanil-cyclase which transforms GTP to cyclic GMP (cGMP). High intracellular levels of cGMP are also responsible for the anti-mitogenic effect of NO. The NO acts shortly as it is rapidly inactivated by the binding to the hemoglobin of local erythrocytes. Phosphodiesterase 5 (PDE5) rapidly inactivates the cGMP by changing it into a inactive linear form.
Sildenafil (SIL) is a strong PDE5 inhibitor. PDE5 inhibition leads to a significant increase of intra-cellular cGMP and therefore vasodilatation and probably smooth muscle cells’ mitosis inhibition.
Studies on the monocrotaline (MCT) induced disease in the Wistar rat have shown the beneficial effects of sildenafil regarding the pulmonary arterial remodeling, PAH and right heart failure. The main mechanisms are vasodilatation (6,7) and suppression of vascular remodeling(8). Sildenafil upregulates MKP-1 expression and promotes degradation of phosphorylation of ERK1/2, which suppresses the proliferation of pulmonary artery smooth muscle cells – in a porcine experimental model (9).
Clinical studies in PAH patients have shown that sildenafil improves symptoms, NYHA class, exercise capacity and hemodynamics (10-14).
Inflammation seems to play an important role in PAH pathogenesis (15), both in humans and in the MCT experimental model. Lymphocytes and macrophages infiltrates were found in the plexiform lesions of idiopathic PAH, with increased local expression of chemokines CCL2, CCL5 and CXCL1 (fractalkine) (16-18). PAH is associated with inflammatory states – HIV infection, systemic auto-immune diseases (scleroderma) (15). Pro-inflammatory cytokines levels – including IL1 and IL6, are increased both in idiopathic PAH (19) and MCT-disease (20-21). Dexametasone reversed the MCT-disease in rats and improved survival, through mechanisms that involve reduction of IL-6-expressing inflammatory cells, restoration of pulmonary BMPR2 and reduced proliferation of vascular smooth muscle cells (22).
The objectives of this study were to evaluate the effects of Sildenafil treatment during the early stages of the MCT-disease in the Wistar rats, using a morphopathological assessment of the pulmonary parenchyma.
We hypothesized that Sildenafil administration from day 1 following MCT injection will prevent/reduce both pulmonary vascular remodeling and acute inflammation. ❑
METHODS AND MATERIALS
Study design
Thirty-six female Wistar rats (mean age of 120 days; mean weight of 196 g), kept in a temperature-controlled room, with a daily constant 14:10 h light/dark ratio, were randomly divided into 3 groups:
-
-
saline-injected control group (C-G; n=6);
-
-
MCT-injected group (MCT-G; n=15);
-
-
MCT-injected and sildenafil-treated group from day 1 to day 28 (oral administration of 1 ml of Sil solution, every 12 h; total of 2 mg/kg/day) (SIL-G; n=15).
Each animal had ad libitum access to rat chow and water.
MCT-exposed rats received on day 1 a single subcutaneous inter-scapular injection of MCT – 60 mg/kg.
Sacrifices were made in each group on day 14 (C-G n=3; MCT-G n=9; SIL-G n=9) and on day 28 (C-G n=3; MCT-G n=6; SIL-G n=6), using an intra-peritoneal injection of 50 mg/kg sodium thiopental.
The experiments performed during our study respected the Romanian legislation regarding laboratory animal testing.
Solutions protocols
For the MCT solution, 1 g of monocrotaline (Sigma-Aldrich) was dissolved in 5.2 ml of 1 N
HCl, to which 30 ml of sterile water was then added. The pH was adjusted to 7.0 using 1 N
NaOH, after which sterile water was added for a final 50 ml total volume of MCT solution.
The sildenafil solution was obtained using Sildenafil tablets of 50 mg (Viagra®). One half tablet (25 mg of sildenafil) was crushed into thin powder, which was then dissolved into 125 ml of sterile water. A watery solution of Sildenafil 0.2 mg/ml was obtained.
Morphological analysis. Immunohistochemistry
Both lungs were harvested from each rat and received an intra-tracheal low-pressure formol injection, in order to keep the airways opened and to avoid baro-trauma. Afterwards, the lungs were preserved in the same formol solution.
Sections of 10 μ m thick paraffin-embedded peripheral parenchyma (one sample for each lung) were prepared and stained with hematoxylin and eosin.
Immunohistochemistry using actine monoclonal antibody as a smooth muscle cell marker was also performed.
The slides were evaluated by light microscopy and the types of lesions, the extent of inflammation and the degree of vascular remodeling were assessed by a researcher blinded to the treatment groups.
Each slide suffered a blinded qualitative and quantitative analysis.
Pulmonary arterial morphometry
The hematoxylin-eosin stained slides and the actine-marked immunohistochemistry slides were computer-scanned using the MOTIC 2000 microscope. The images were acquired using the Motic Image Plus 2.0 software.
Two slides/animal (one/lung) were analyzed. In medium, ten randomly chosen resistance arteries (diameter < 500 μ m) were evaluated per slide. The following measurements were performed: internal diameter (ID measuring the luminal diameter) and external diameter (ED measuring the total diameter of the vessel), using the GSA ImageAnalyser v3.8.1 morphometry software. The hypertrophy index (HI; the wall/vessel thickness %) was defined as: (ED –ID) / ED %.
HI = (ED – ID) / ED %
Inflammatory index (Iix)
The Iix defines the percentage of tissue occupied space for each pulmonary slide and was measured using the GSA ImageAnalyser v3.8.1 morphometry software. It is a new measurable inflammatory marker which we propose for objective evaluation of the degree of inflammation and overall pulmonary remodeling.
In order to determine the Iix we measured the tissue surface area (TSA) and the complete section area (CSA) for each slide.
Iix = TSA/CSA %
We consider the Iix to be influenced:
-
-
in the acute phase (day 14) by the degree of edema (vascular, perivascular, bronchial, peribronchial, alveolar septae) and by the amount of the alveolar infiltrates
-
-
in the chronic phase (day 28) by the degree of vascular remodeling and pulmonary fibrosis (edema significantly diminished)
Data analysis
The resulting data are expressed as mean ± standard deviation. Data were analyzed using the Kolmogorov-Smirnov normality test follo wed by the one-way Anova test and Tukey’s multiple comparison text. Differences were considered significant for p <0.05. Analyses were performed using Excel 2007, GraphPad Prism v4.03 and InStat v3.05. ❑
RESULTS
Typical pulmonary lesions present both in spontaneously deceased and in sacrificed MCT-inoculated rats show that we have successfully realized the monocrotaline experimental disease in the Wistar rat.
The control group showed no significant abnormalities, except for a minimum degree of emphysema – a disruption of the alveolar walls due to the preparation technique (Figure 1). Furthermore, a small degree of emphysema appears equally in all three groups, thus not significantly influencing the conclusions of our study (Figures 2, 3 and 4).
Figure 1. Photograph from saline-injected control group (C-G). Normally looking parenchyma with small degree “emphysema” (alveolar wall disruption) – secondary to the preparation technique (intra-tracheal formol injection). Hematoxylin-eosin stain.

Figure 2. Photograph from MCT-injected group (MCT-G) – day 14. Peri-vascular edema, diffuse lympho-monocitary infiltrates; mild thickening of intimal and medial layers of small arteries secondary to edema and endothelium hypertrophy. Hematoxylin-eosin stain.
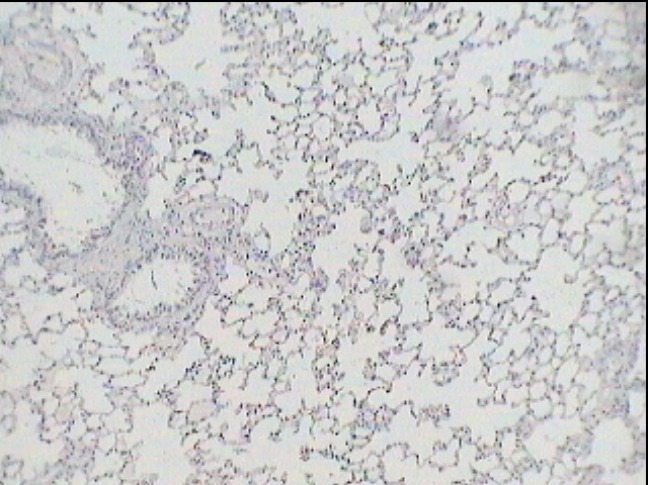
Figure 3. Photograph from MCT-injected group (MCT-G) – day 14. Vascular wall thickening and severe inflammatory infiltrate. Monoclonal Anti-Actine Antibody Stain.
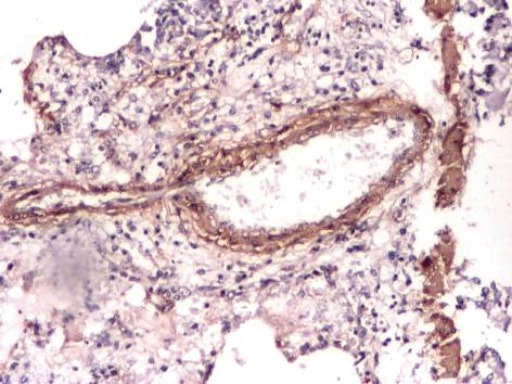
Figure 4. Photograph from MCT-injected sildenafil-treated group (SIL-G) – day 14. Near to normal looking parenchyma with minimal pari-vascular inflammatory infiltrate, without edema or vascular wall thickening. Monoclonal Anti-Actine Antibody Stain.
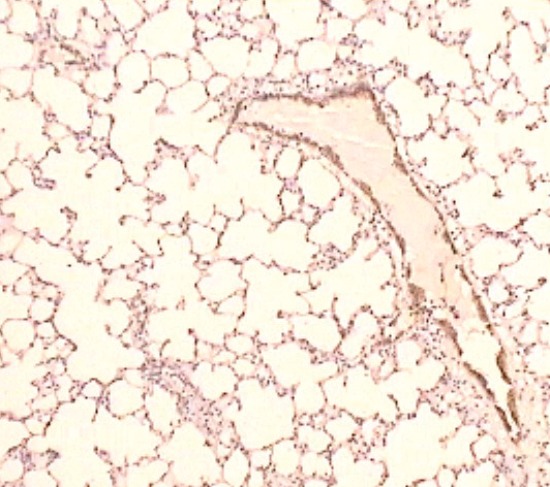
MCT exposure leads to the development of a series of modifications involving both small arteries and small airways. An acute pneumonia-like process – with vascular, peri-vascular, alveolar and peri-bronchial lympho-monocitary infiltrates and edema, appears during the first 14 days (Figures 2, 3 and 4). Between day 14 and 28, the inflammatory lesions regress, the small pulmonary vessels suffer extensive vascular remodeling – with intimal and medial hypertrophy of the muscular arteries and arterioles, and interstitial fibrosis develops. All these modifications observed in our MCT-exposed rats represent typical findings in the monocrotaline-induced experimental disease (23-25).
Sildenafil improves survival during the first 14 days in the MCT-induced disease in the Wistar rat (Figure 5).
Figure 5. Survival rate during the first 14 days in MCT-G and SIL-G.

A total of 9 rats died during the first 14 days following MCT: 4 in the MCT-G and 1 in the SIL-G, while the rest of them were sacrificed respecting the protocol at day 14 (C-G n=3; MCT-G n=3; SIL-G n=6) and day 28 (C-G n=3; MCT-G n=6; SIL-G n=6). There was no spontaneous death in the control group. Two deaths occurred in both MCT and SIL groups between day 14 and 28.
Sildenafil significantly reduces inflammation in the MCT-induced disease in the Wistar rat.
The qualitative comparison analysis shows the presence of the same types of lesions in both MCT-exposed groups (MCT-G in Table 1; SIL-G in Table 2). However, the extension degree and severity of inflammatory lesions are strikingly lower in the sildenafil-treated group. The Control group showed no significant pulmonary lesions (Figure 1).
Table 1.
Morpho-pathological findings in the MCT-exposed group
| Slide no | Edema | Atelectasis | Alveolar macrophage | Lymphoplasmocitary infiltrates | BALT* | Alveolar hemorrhages | Hypertro-phic vessels |
|---|---|---|---|---|---|---|---|
| 56 | ++ | - | - | Moderate | + | + | + |
| 57 | ++ | - | - | Moderate | + | + | + |
| 151 | +++ | - | - | Moderate | + | + | + |
| 237 | ++ | - | - | Moderate | + | + | + |
| 238 | ++ | - | - | Moderate | + | + | + |
| 166 | +++ | - | - | Moderate | + | + | + |
Table 2.
Morpho-pathological findings in the MCT-exposed sildenafil-treated group
| Slide no | Edema | Atelectasis | Alveolar macrophage | Lymphoplasmocitary infiltrates | BALT* | Alveolar hemorrhages | Hypertro-phic vessels |
|---|---|---|---|---|---|---|---|
| 29 | - | - | - | Minimal | + | - | - |
| 30 | - | - | - | Minimal | + | - | - |
| 33 | + | - | - | Mild | + | - | + |
| 34 | + | - | - | Mild | + | - | + |
| 209 | ++ | - | - | Moderate | + | + | + |
| 210 | ++ | - | - | Moderate | + | - | + |
| 297 | - | - | - | Minimal | + | - | - |
| 298 | - | - | - | Minimal | + | - | - |
The Iix comparison (Table 3) showed a significant reduction of 6.038 % (p <0.05) in the sildenafil treated group (Iix SIL-G = 52.94 ± 6.605 %) in comparison with the MCT group (Iix MCT-G = 58.97 ± 7.832 %) (Figure 6; Figure 7).
Table 3.
Inflammatory index measurements in the 3 groups. Sildenafil significantly reduces the Iix in the MCT exposed rats (Figures 6 and 7).
| MCT | SIL | Control | |
|---|---|---|---|
| Mean Iix % | 58,97 | 52,94 | 46,33 |
| Std. Deviation % | 7,832 | 6,605 | 8,913 |
| Std. Error | 1,846 | 1,377 | 2,573 |
| Tukey's Multiple Comparison Test | Mean Diff. | P value | 95% CI of diff |
|---|---|---|---|
| MCT vs SIL | 6,038 | P < 0.05 | 0.2647 to 11.81 |
Figure 6. Inflammatory index in the SIL-G significantly lower than in the MCT-G.

Figure 7. Inflammatory index (Iix) measurement.Rats sacrificed at day 14: A from the Control Group, B from the MCT-exposed not-treated group and C from the MCT-exposed Sildenafil-treated group. The digitally acquired pulmonary slides (original images: A2; B2; C2) are analyzed using the morphometry software which uses color codes (images A1; B1; C1) in order to define and measure the tissue surface area – TSA (black area) and the complete surface area – CSA (yellow + black area). The values are numerically expressed in μm2 and the software automatically calculates the Iix = TSA/CSA %, as shown in the red box.

Sildenafil prevents the pulmonary arterial remodeling in the MCT-induced disease in the Wistar rat (Figure 8).
Figure 8. Hypertrophy index in SIL-G significantly lower than in MCT-G.

The saline-injected control group has a mean HI of 24.84 ± 6.572, significantly lower in comparison to the MCT-exposed groups. Sildenafil significantly decreases by Δ = 7.306 % (p<0.001) the HI in the MCT-exposed rats (HI MCT-G = 50.38 ± 11.44 %; HI SIL-G = 43.07 ± 11.11 %) (Table 4). Sildenafil prevents and reduces the degree of thickening and hyperplasia of muscular pulmonary arteries and arterioles, thus diminishing the pulmonary arterial remodeling. ❑
Table 4.
Hypertrophy index measurements in the 3 groups. Sildenafil significantly reduces the HI in the MCT exposed rats (Figure 8).
| MCT-G | SIL-G | Control | |
|---|---|---|---|
| Mean HI % | 50,38 | 43,07 | 24,84 |
| Std. Deviation | 11,44 | 11,11 | 6,572 |
| Std. Error | 0,8254 | 0,6985 | 0,7005 |
| Tukey's Multiple Comparison Test | Mean Diff. | P value | 95% CI of diff |
|---|---|---|---|
| MCT vs SIL | 7,306 | P < 0.001 | 4.897 to 9.715 |
DISCUSSIONS
The MCT-induced disease is a PAH inflammatory model. It has an initial acute inflammatory phase, which reaches its maximum during the first 14 days, primarily involving the endothelium of small pulmonary arteries, with progressive perivascular, alveolar and peribronchial extension – when most severe, taking the form of an acute diffuse pneumonitis (pneumonia-like process). The second phase of the disease evolves between day 14 and 21, with regression of the inflammation and development of pulmonary artery remodeling – with muscularization of the small pulmonary arteries and arterioles, associated with thickening and hyperplasia of the intimal and medial layers. At day 28 – end-phase of the disease, the inflammatory lesions are almost completely gone and PAH with right ventricular hypertrophy develop (23-25).
The complete pathogenesis of the MCT-induced disease leading to PAH in the Wistar rat remains unknown.
MCT is metabolized in the liver to Ehrlich reactive (E+) metabolites, the monocrotaline pyrroles (MCTP). These MCTP are entirely responsible for the pulmonary toxic effect. Without the MCT activation by the liver, no pulmonary disease develops. The pulmonary endothelial cell is considered to be the main target for the MCT intoxication. This hypothesis is supported by the circulatory proximity of the liver to the lung endothelium, evidence of increased thymidine uptake and decreased 5-hydroxytryptamine clearance by endothelial cells, and extra-vascular leakage of large macromolecules. Furthermore, specific pulmonary endothelial cell proteins have been identified as binding targets for MCTP: galectin-1, protein-disulfide isomerase, probable protein-disulfide isomerase, b- or gcytoplasmic actin, and cytoskeletal tropomyosin, affecting the integrity of the endothelial layer.
Previous studies on the MCT model have shown the efficacy of early (before day 14) anti-inflammatory or immunosuppressive therapy for preventing the pulmonary arterial remodeling (22,26-31).
Sildenafil has been proven efficient in the MCT model during the pulmonary remodeling process (after day 14) and in the late-stage disease, after PAH development (day 21-28) (6-9), beneficial effects up until now thought to be secondary only to vasodilatation and pulmonary vascular proliferation reduction.
Our study offers a new insight into the understanding of the interaction between sildenafil and the MCT-induced disease, by showing that sildenafil has also the ability, when administered early, to reduce inflammation and improve survival – with a very probable causal effect. Thus, besides the vasodilator and anti-proliferative effects, sildenafil also manifests a clinically significant anti-inflammatory effect in the MCT-disease. This finding is concordant with previous experimental observations regarding the potential anti-inflammatory effect of sildenafil (32,33) and has direct clinical consequences.
Although most idiopathic PAH patients are considered to be non-responsive to anti-inflammatory therapies, significant clinical improvement has been observed in those with concomitant inflammatory diseases (PAH associated with scleroderma (34) or connective-tissue disease [35[) who received immunosuppressive / anti-inflammatory therapy. Fur th er more, the greatest benefit occurs in those with earlystage disease and less severe PAH (36), suggesting that anti-inflammatory therapy is probably more efficient on “active” proliferative lesions rather than on late “fixated” vascular lesions (22). Thus, patients suffering from inflammatory diseases at risk for developing in time PAH, could benefit from sildenafil treatment during the early sub-clinical phases of the pulmonary disease.
The main limitations of our study are the absence of an MCT-exposed sildenafil-treated starting day 14 group and also the lack of hemodynamic evaluation. ❑
CONCLUSIONS
Early administration of Sildenafil starting day 1 following MCT exposure 1) improves survival during the first 14 days, 2) significantly reduces inflammation during the acute phase of the disease and 3) prevents/reduces the disease-specific pulmonary arterial remodeling.
We have shown that early administration of sildenafil reduces the degree of inflammation, prevents and diminishes the pulmonary vascular remodeling and improves survival in the monocrotaline-induced disease in the Wistar rat, thus offering new perspectives for the use of sildenafil in human PAH associated with inflammatory diseases. Sildenafil remains interesting as a therapeutic approach for other acute pulmonary interstitial inflammation.
References
- 1.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 2.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 3.Pietra GG, Capron F, Humbert M, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Morrell NW, Voelkel NF, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 5.Reda E, Hunter C, Gregory B, et al. Decreased Exhaled Nitric Oxide in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2005;172:352–357. doi: 10.1164/rccm.200412-1684OC. [DOI] [PubMed] [Google Scholar]
- 6.Schermuly RT, Kreisselmeier KP, Ghofrani HA, et al. Chronic sildenafil treatment inhibits monocrotalineinduced pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;169:39–45. doi: 10.1164/rccm.200302-282OC. [DOI] [PubMed] [Google Scholar]
- 7.Clozel M, Hess P, Rey M, et al. Bosentan, sildenafil, and their combination in the monocrotaline model of pulmonary hypertension in rats. Exp Biol Med. [PubMed] [Google Scholar]
- 8.Yen CH, Leu S, Lin YC, et al. Sildenafil limits monocrotaline-induced pulmonary hypertension in rats through suppression of pulmonary vascular remodeling. . J Cardiovasc Pharmacol. 2010;55:574–84. doi: 10.1097/FJC.0b013e3181d9f5f4. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Yang L, Shen J, et al. The antiproliferative effect of sildenafil on pulmonary artery smooth muscle cells is mediated via upregulation of mitogen-activated protein kinase phosphatase-1 and degradation of extracellular signal-regulated kinase 1/2 phosphorylation. Anesth Analg. 2007;105:1034–4. doi: 10.1213/01.ane.0000278736.81133.26. [DOI] [PubMed] [Google Scholar]
- 10.Bhatia S, Frantz RP, Severson CJ, et al. Immediate and longterm hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clin Proc. 2003;78:1207–1213. doi: 10.4065/78.10.1207. [DOI] [PubMed] [Google Scholar]
- 11.Michelakis ED, Tymchak W, Noga M, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation. 2003;108:2066–2069. doi: 10.1161/01.CIR.0000099502.17776.C2. [DOI] [PubMed] [Google Scholar]
- 12.Ghofrani HA, Schermuly RT, Rose F, et al. Sildenafil for long-term treatment of nonoperable chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. 2003;167:1139–1141. doi: 10.1164/rccm.200210-1157BC. [DOI] [PubMed] [Google Scholar]
- 13.Galie N, Ghofrani HA, Torbicki A, et al. The Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group - Sildenafil citrate therapy for pulmonary arterial hypertension. New Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 14.Badesch DB, Hill NS, Burgess G, et al. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol. 2007;34:2417–2422. [PubMed] [Google Scholar]
- 15.Dorfmuller P, Perros F, Humbert M, et al. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 16.Dorfmuller P, Zarka V, Durand-Gasselin I, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:534–9. doi: 10.1164/ajrccm.165.4.2012112. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez O, Marcos E, Humbert M, et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2007;176:1041–7. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 18.Balabanian K, Foussat A, Dorfmuller P, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:1419–25. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- 19.Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin- 6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 20.Bhargava A, Kumar A, Yuan N, et al. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis. 1999;1:126–32. [PubMed] [Google Scholar]
- 21.Gillespie MN, Goldblum SE, Cohen DA, et al. Interleukin 1 bioactivity in the lungs of rats with monocrotalineinduced pulmonary hypertension. Proc Soc Exp Biol Med. 1988;187:26–32. doi: 10.3181/00379727-187-42632. [DOI] [PubMed] [Google Scholar]
- 22.Price L, Montani D, Tcherakian C, et al. Dexamethasone reverses monocrotaline- induced pulmonary arterial hypertension in rats. ERJ Express. doi: 10.1183/09031936.00028310. [DOI] [PubMed] [Google Scholar]
- 23.Alexandru B, Bogdan MA. Monocrotaline induced pulmonary hypertension in animal models. Pneumologia. 2001;50:85–9. [PubMed] [Google Scholar]
- 24.Meyrick B, Gamble W, Reid L. Development of Crotalaria pulmonary hypertension: hemodynamic and structural study. Am J Physiol. 1980;239:H692–702. doi: 10.1152/ajpheart.1980.239.5.H692. [DOI] [PubMed] [Google Scholar]
- 25.Wilson DW, Segall HJ, Pan LC, et al. Progressive inflammatory and structural changes in the pulmonary vasculature of monocrotaline-treated rats. Microvasc Res. 1989;38:57–80. doi: 10.1016/0026-2862(89)90017-4. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura T, Faul JL, Berry GJ, et al. 40-O-(2-hydroxyethyl)-rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2001;163:498–502. doi: 10.1164/ajrccm.163.2.2006093. [DOI] [PubMed] [Google Scholar]
- 27.Bonnet S, Rochefort G, Sutendra G, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104:11418–23. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki C, Takahashi M, Morimoto H, et al. Mycophenolate mofetil attenuates pulmonary arterial hypertension in rats. Biochem Biophys Res Commun. 2006;349:781–8. doi: 10.1016/j.bbrc.2006.08.109. [DOI] [PubMed] [Google Scholar]
- 29.Lu J, Shimpo H, Shimamoto A, et al. Specific inhibition of p38 mitogenactivated protein kinase with FR167653 attenuates vascular proliferation in monocrotaline-induced pulmonary hypertension in rats. J Thorac Cardiovasc Surg. 2004;128:850–9. doi: 10.1016/j.jtcvs.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Voelkel NF, Tuder RM, Bridges J, et al. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. 1994;11:664–7. doi: 10.1165/ajrcmb.11.6.7946395. [DOI] [PubMed] [Google Scholar]
- 31.Kimura H, Kasahara Y, Kurosu K, et al. Alleviation of monocrotalineinduced pulmonary hypertension by antibodies to monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Lab Invest. 1998;78:571–81. [PubMed] [Google Scholar]
- 32.Wang T, Liu Y, Chen L, et al. Effect of sildenafil on acrolein-induced airway inflammation and mucus production in rats. ERJ. 2009;33:1122–1132. doi: 10.1183/09031936.00055908. [DOI] [PubMed] [Google Scholar]
- 33.Sawatzky DA, Megson IL, Rossi AG. Sildenafil offers protection against NSAID-induced gastric injury. Br J Pharmacol. 2005;146:477–478. doi: 10.1038/sj.bjp.0706362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karmochkine M, Wechsler B, Godeau P, et al. Improvement of severe pulmonary hypertension in a patient with SLE. Ann Rheum Dis. 1996;55:561–2. doi: 10.1136/ard.55.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez O, Humbert M, Sitbon O, et al. Treatment of pulmonary hypertension secondary to connective tissue diseases. Thorax. 1999;54:273–7. doi: 10.1136/thx.54.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jais X, Launay D, Yaici A, et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum. 2008;58:521–31. doi: 10.1002/art.23303. [DOI] [PubMed] [Google Scholar]