

Abstract
The need for tissue-engineered constructs as replacement tissue continues to grow as the average age of the world's population increases. However, additional research is required before the efficient production of laboratory-created tissue can be realized. The multitude of parameters that affect cell growth and proliferation is particularly daunting considering that optimized conditions are likely to change as a function of growth. Thus, a generalized research platform is needed in order for quantitative studies to be conducted. In this article, an ultrasonic bioreactor is described for use in studying the response of cells to ultrasonic stimulation. The work is focused on chondrocytes with a long-term view of generating tissue-engineered articular cartilage. Aspects of ultrasound (US) that would negatively affect cells, including temperature and cavitation, are shown to be insignificant for the US protocols used and which cover a wide range of frequencies and pressure amplitudes. The bioreactor is shown to have a positive influence on several factors, including cell proliferation, viability, and gene expression of select chondrocytic markers. Most importantly, we show that a total of 138 unique proteins are differentially expressed on exposure to ultrasonic stimulation, using mass-spectroscopy coupled proteomic analyses. We anticipate that this work will serve as the basis for additional research which will elucidate many of the mechanisms associated with cell response to ultrasonic stimulation.
Introduction
Articular cartilage is irreversibly destroyed after traumatic injury or chronic illnesses such as arthritis.1,2 It is projected that by 2030, nearly 67 million adults will be afflicted with arthritis in the United States, and 25 million are projected to have activity limitations due to this disease. Since the cartilage has little capacity for self-repair, a promising alternative treatment is the transplantation of tissue-engineered cartilage.3,4 Central to a successful tissue engineering strategy to grow functional tissue equivalents is the establishment of a bioreactor or a bioprocessing unit that maintains cells seeded on biodegradable polymeric scaffolds and provides essential gas and nutrient transport between the cells and the culture media, as well as the mechanical stimuli which are necessary to promote extracellular matrix (ECM) synthesis.5–7
Bioreactors offer several advantages compared with simple tissue-flask and Petri-dish culture systems, notably the ability to provide mechanical forces influencing tissue development and to achieve better control over culture conditions.8,9 Designs of bioreactors that are currently commercially available for the cultivation of tissue-engineered constructs are mainly based on the following driving forces: hydrostatic pressure, that is, dynamic compression, hydrodynamic stress at low shear rates, that is, perfusion systems, rotating bioreactors, wavy-wall bioreactors, and conventional spinning flasks.10–16 Although ultrasonic stimulation has also been shown to influence cell growth in some cases, comprehensive studies over a wide range of parameters have not been performed.17,18
Previous studies involving ultrasound (US) have used low-intensity pulsed US (1.5 MHz, 1.0 kHz repeat, 6–40 min) to stimulate in vitro chondrocyte cultures.18–20 The effect of pulsed US stimulation was compared with rotating bioreactors over a culture period of 49 days.17 As a significant departure from such strategies, we have employed intermittent applications of low-intensity diffuse ultrasound (LIDUS) at 5.0 MHz (14 kPa) to stimulate bovine chondrocytes seeded in 3D chitosan-based matrices.21,22 Our published data provide evidence that the US stimulation regimen employed induces a marked increase in the expression of chondrogenic markers, including type-II collagen, aggrecan, and Sox-9. These results indicate that US stimulation, by itself, could enhance chondrogenic differentiation in a 3D culture.22 Results of real-time–polymerase chain reaction (RT-PCR) analysis have revealed that US stimulation increases the levels of gene expression of cell-surface integrins α5 and β1: heterodimeric adhesion receptors that regulate cell viability in response to cues from the ECM. The enhancement in abundance of mRNA transcripts on US stimulation was observed to correlate with increases in levels of protein expression of collagen type-I, collagen type-II, and integrins α5 and β1. Recently, we have shown that US stimulation of chondrocytes induced phosphorylation of focal adhesion kinase, Src, p130Crk-associated substrate (p130Cas), Crk-II, and extracellular-regulated kinase (ERK). This work strongly implicates these intracellular signaling molecules in a US-mediated signaling pathway.23 Thus, the US stimulation regimen employed has been established to modulate the proliferative capacity, biosynthetic activity, and integrin mRNA expression of articular chondrocytes maintained in 3D matrices.
To capitalize on the positive bioeffects of LIDUS and apply them to the field of cartilage tissue engineering, our laboratory has designed and developed an ultrasonic bioreactor (UBR) configuration that uses US to stimulate chondrocytes maintained in an in vitro culture (Fig. 1) over a range of US stimulations. We have chosen chondrocyte cultures as a model system, as chondrocytes have been reported to be mechano responsive and culture systems based on a variety of stimuli have been reported. US stimulation causes a variety of effects beyond cellular response that should be controlled to isolate biological effects and minimize possible experimental disturbances.24–29 Another major challenge in the postgenomic era is to decipher the spatiotemporal functions and interactions of proteins in a cell. Proteomic profiling may help identify unique markers and elucidate interconnections between different cellular signaling pathways. In this study, we will profile the global protein expression in chondrocytes subjected to US stimulation. Thus, the goals of this article are to characterize the US-induced bioeffects in the UBR, to carry-out a global proteomic profile of chondrocytes under US, and to demonstrate the culture of cell-seeded constructs in the bioreactor. Toward that end, a variety of different outcome measurement techniques are used. Since each technique has specific geometrical requirements, three different cell configurations have been used (tissue culture polystyrene [TCP] plates, coverslips, and scaffolds).
FIG. 1.

Schematic of ultrasonic bioreactor (UBR) developed at UNL. (a) The programmable stage enables the movement of the holder in x-y-z directions and allows multiple plates to be treated. A plate holder retains the six-well plates with scaffolds above the transducer array. A custom splitter allows manipulation of the ultrasound (US) signal such that all wells have identical pressure profiles. (b) K-type thermocouple coupled to a Keithley data acquisition module interfaced with a computer was employed to acquire time-dependent temperature profiles on US exposure. TCP, tissue culture polystyrene. Color images available online at www.liebertpub.com/tec
Materials and Methods
Design of the reactor
Our UBR was designed to serve as a general research platform for studying cellular response to these numerous input parameters in a well-controlled manner. The UBR consists of the following components (a schematic is shown in Fig. 1)
Incubator
All components of the UBR were designed to fit within an off-the-shelf incubator (Forma Model 3033 Steri-Cult incubator), which is used for controlling the temperature, humidity, and CO2 level. The incubator also provided a safe biological condition that minimized possible contamination of the culture plates. This design simplified our current and future studies, because it allows us to utilize standard six-well TCP plates for the cell growth. A custom insert was designed to hold nine plates in such a way to allow growth media to be changed periodically and to allow the plates to be excited consistently by the transducer array.
Transducer array
An array of six nonfocused ultrasonic transducers (Olympus V309, 5 MHz center frequency) was chosen for excitation of the cells within the TCP plates. The transducers were mounted within a fluid-filled cavity to provide consistent water coupling between the transducers and the bottom of the plates. Millipore water is used as the couplant. The transducers were excited using a USB function generator (Tektronix AFG-3021B) for control of the amplitude and frequency of excitation.
Positioning stage
The motion-control assembly consisted of two linear stages for the x-y motion (OWIS–LTM80-300-HSM; maximum horizontal movement of 30 cm) and an elevator stage for the z motion (OWIS–HVM100-30-HSM; maximum vertical movement of 3 cm). The x-y stages move the transducer array to the correct position beneath the TCP plate of interest, and the z stage raises and lowers the array during each US application cycle. These computer-controlled stages provided great flexibility with regard to experimental design—each TCP plate can be excited with a separate US profile.
Control software
Custom control software was written (MATLAB™) for control of the UBR from a laptop computer. This software allows the user to choose the application profile for each TCP plate with regard to US pressure, frequency, sonication duration, and sonication interval (i.e., number of times per day). The software then determines the application cycle for all plates for each day in order to avoid any timing conflicts. While running, a “Sonication Status” display shows either the plate currently being sonicated or a countdown for the next sonication. At the end of the testing cycle, the software generates a report that serves as a record of all parameters used for the sonications. Additional information about the bioreactor is available from the authors by request.
Temperature effects
A series of experiments (1–8 MHz; 14–60 kPa; 1–10 min of US exposure) was conducted to investigate the temperature rise induced by the US regimens employed in the bioreactor, in deionized (DI) water, cell culture medium, within a scaffold in culture medium. A K-type thermocouple coupled to a Keithley data acquisition module was used and interfaced with a computer to acquire time-dependent temperature profiles on US exposure. Typically, the TCP well was filled with 8-mL of solution, and the transducer was fixed at the plate center and in-line with the transducer. Each data point was measured in octuplicates, independently.
Noninertial cavitation
Noninertial cavitational effects of US regimens in the bioreactor were assessed through the extent of sucrose hydrolysis in dilute acid and base solutions as detailed elsewhere.30 Briefly, 5-mL of sucrose solution (10 mM in 0.1 M NaOH or 0.1 N HCl) was placed in wells of a six-well TCP plate, and exposed to US stimulation. Sucrose hydrolysis in both cases was determined by measuring the glucose content of the samples; by mixing 200 μL of each sample and 1 mL of glucose (HK) assay reagent (Sigma) and measuring the absorbance at 340 nm after an incubation at 30°C for 30 min. Glucose concentrations were calculated according to a standard curve established from standard glucose solutions. Sucrose hydrolysis in nonstimulated samples (controls) was measured in a similar manner. Sucrose hydrolysis in 1 M HCl served as the positive control. Each data point was measured in quadruplicate, independently.
Inertial cavitation
Effects from inertial cavitation (IC) were examined using electron paramagnetic resonance (EPR). 1-Hydroxy-3-methoxy carbonyl 2,2,5,5 tetramethyl pyrrolidine hydrochloride (CMH) in Krebs–Henseleit buffer with chelators at pH 7.36 was used as a spin probe for these measurements. 8-mL of degassed DI water was placed in culture wells, and 50 μL of CMH (to result in 200 μM solution) was added to the well before the indicated US exposure as detailed earlier. US sonication was provided for 8-min, 4-min, and 1 min for each experiment. At the completion of the experiment, 10–20 μL of the solution in the well was pipetted and inserted into glass tubes for measurement of the EPR signal by e-seam BRUKER (NOxygen). Nonstimulated samples and samples without CMH served as controls. Experiments with a Cup Horn Sonicator 3000 (Ultrasonic Liquid Processor; Misonix) served as the positive control. Each experiment was run in quadruplicate, independently.
Pressure profiles
The transducer array was designed to deliver a uniform pressure profile within the wells of the TCP plates and to allow a range of pressures to be studied. The pressure profiles were calibrated using a needle hydrophone (Onda HNP 400) as shown in the schematic (Fig. 1b). The first measurements were performed to determine the optimum distance between transducer face and the bottom of the TCP plate. This distance is a compromise between the spatial profile of the nonfocussed transducer and the beam spread that occurs away from it. In addition, the transducer should be close enough to a single well so as not to excite spurious waves within other wells. The hydrophone was placed in the center of a well 8 mm above the bottom plate, a distance representative of cells in a scaffold. The average pressure was measured using six different trials with a 5 MHz excitation frequency. After numerous tests, we designed our transducer array with the transducer face 23 mm below the bottom of the TCP plate. Note that this position is not unique—many configurations will likely provide an excitation profile that is sufficient for studying cellular response to US.
The spatial distribution of the pressure profile was measured in a similar way by mounting the hydrophone to a linear positioning stage. A transducer was placed below one well of a TCP plate at a specified distance. The hydrophone was placed initially at the center of the well 8 mm above the bottom of the plate (see schematic in Fig. 1b). Pressure measurements were made with an excitation of 5 MHz. The measurements were repeated at numerous positions across the well, using a step size of ∼2 mm. The measured profiles revealed that a region near the well center, ∼1.5 cm wide has a fairly constant pressure level with a slight drop outside that region. Uncertainty in these measurements is primarily associated with the diffuse interference that occurs as the US reflects within the well cavity.
Cell-seeding experiment to obtain samples for 2D-DIGE
Cell seeding and analyses
Discarded bovine shoulder joints from 6-month-old calves were obtained from a local abattoir, and chondrocytes were isolated using previously described methods.31 Freshly isolated chondrocytes were plated on six-well TCP plates at a seeding density of 2×105 cells/well and maintained in Dulbecco's modified Eagle's medium (DMEM)/F-12 (1:1) (Sigma-Aldrich) (3 mL/well) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1× antibiotic-antimyotic (Invitrogen). Plates were maintained in the CO2 incubator for a day. Media were replaced with DMEM-F12 (3 mL/well) without FBS and incubated again for a day before US exposure. When appropriate, media (DMEM-F12, w/o FBS) were replaced every 3 days.
US stimulation
Chondrocytes seeded in six-well tissue culture plates as a monolayer culture were exposed to a LIDUS signal as listed in Table 1. Cell-seeded scaffolds that are not stimulated served as controls and were handled identically to LIDUS stimulated samples, except for the US stimulation. Cell-seeded TCP plates were placed in the bioreactor (i.e., plate holders) and using the programming tool and interface, the unit was programmed to provide dedicated US stimulation to assigned plates. The US applications were equally spaced throughout each day of testing. Cell-seeded plates were stimulated, and parameters were indicated. Nonstimulated cell-seeded plates served as the control. Three identical but independent experiments were carried out at each condition.
Table 1.
List of Ultrasound Parameters Used for 2D-DIGE Analyses
| Days in culture | kPa | Applications/day | Time per application (min) |
|---|---|---|---|
| 3 | 60 | 8 | 5 |
| 3 | 60 | 16 | 5 |
| 6 | 14 | 8 | 5 |
| 6 | 60 | 8 | 5 |
2D-DIGE, two-dimensional difference gel electrophoresis.
Cell lysate preparation
On completion of the US stimulation, plates were removed from the bioreactor; media were pipetted off, washed thrice with ice-cold phosphate-buffered saline (PBS) (1×) followed by incubation in ice-cold Pierce IP lysis buffer (Thermo Scientific Pierce) supplemented with 1× Halt protease and phosphatase inhibitor cocktail (Thermo Scientific Pierce). 200 μL of IP lysis buffer with Halt inhibitor was added to each well and incubated for 15 min with periodic mixing at 4°C before combining the lysate from four wells that served as a replicate of each treatment group. Cell debris was removed, and supernatant was collected in new vials after centrifugation at 15,000 g for 10 min at 4°C. The protein concentration of each sample was determined using a QuantiPro BCA Assay kit (Sigma-Aldrich). Samples were frozen and shipped to Applied Biomics for analyses.
Two-dimensional difference gel electrophoresis
Preparation of samples
Protein samples were resuspended in 2D cell lysis buffer (30 mM Tris-HCl, pH 8.8, containing 7 M urea, 2 M thiourea, and 4% CHAPS). Protein concentration was measured using the Bio-Rad protein assay method.
CyDye labeling
For each sample, 30 μg of protein were mixed with 1.0 μL of diluted CyDye and kept in the dark on ice for 30 min. The labeling reaction was stopped by adding 1.0 μL of 10 mM Lysine to each sample and incubating in the dark on ice for an additional 15 min. The labeled samples were then mixed together. 2× 2D Sample buffer (8 M urea, 4% CHAPS, 20 mg/mL dithiothreitol (DTT), 2% pharmalytes, and trace amount of bromophenol blue), 100 ul destreak solution, and Rehydration buffer (7 M urea, 2 M thiourea, 4% CHAPS, 20 mg/mL DTT, 1% pharmalytes, and trace amount of bromophenol blue) were added to the labeling mix to bring the total volume to 250 μL. The samples were mixed well and centrifuged before loading into the strip holder.
Iso electric focusing and sodium dodecyl sulfate–polyacrylamide gel electrophoresis
After loading the labeled samples, iso electric focusing (IEF) (pH3-10 Linear) was run following the protocol provided by GE Healthcare. On finishing the IEF, the IPG strips were incubated in freshly made equilibration buffer-1 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% sodium dodecyl sulfate (SDS), trace amount of bromophenol blue, and 10 mg/mL DTT) for 15 min with gentle shaking. Then, the strips were rinsed in freshly made equilibration buffer-2 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% SDS, trace amount of bromophenol blue, and 45 mg/mL iodoacetamide) for 10 min with gentle shaking. Next, the IPG strips were rinsed in the SDS-gel running buffer before transferring onto 12% SDS-gels. The SDS-gels were run at 15°C until the dye front ran out of the gels.
Image scan and data analysis
Gel images were scanned immediately following the SDS-PAGE using Typhoon TRIO (GE Healthcare). The scanned images were then analyzed by Image Quant software (version 6.0, GE Healthcare), followed by in-gel analysis using DeCyder software version 6.5 (GE Healthcare). The fold change of the protein expression levels was obtained from in-gel DeCyder analysis.
Protein identification by mass spectrometry
Spot picking and trypsin digestion
The spots of interest were picked up by Ettan Spot Picker (GE Healthcare) based on the in-gel analysis and spot-picking design by DeCyder software. The gel spots were washed a few times, then digested in gel with modified porcine trypsin protease (Promega). The digested tryptic peptides were desalted using a Zip-tip C18 (Millipore). Peptides were eluted from the Zip-tip with 0.5 ul of matrix solution (α-cyano-4-hydroxycinnamic acid (5 mg/mL in 50% acetonitrile, 0.1% trifluoroacetic acid, and 25 mM ammonium bicarbonate) and spotted on an MALDI plate.
Mass spectrometry
MALDI-TOF MS and TOF/TOF tandem MS/MS were performed on AB SCIEX TOF/TOF™ 5800 System (AB SCIEX). MALDI-TOF mass spectra were acquired in reflectron positive ion mode, averaging 4000 laser shots per spectrum. TOF/TOF tandem MS fragmentation spectra were acquired for each sample, averaging 4000 laser shots per fragmentation spectrum on each of the 7–10 most abundant ions present in each sample (excluding trypsin autolytic peptides and other known background ions).
Database search
Both of the resulting peptide mass and the associated fragmentation spectra were submitted to GPS Explorer workstation equipped with MASCOT search engine (Matrix Science) to search the database of National Center for Biotechnology Information nonredundant (NCBInr). Searches were performed without constraining protein molecular weight or isoelectric point, with variable carbamidomethylation of cysteine and oxidation of methionine residues, and one missed cleavage also allowed in the search parameters. Candidates with either protein score CI greater than 95% were considered significant.
Scaffold preparation and cell seeding
Chitosan scaffolds were prepared by the freeze-drying and lyophilization method as detailed elsewhere (Hasanova, Noriega et al. 2011). Briefly, a 2% w/v solution of chitosan (81.7% de-acetylated, MM=276 kDa; Vanson HaloSource) in 1% acetic acid was prepared and pipetted into each well of a 24-well TCP plate (Falcon brand; Fisher), frozen at −20°C, and then lyophilized for 24–36 h. Scaffolds (5×5 mm) were punched out using a biopsy punch and neutralized with 0.25 M NaOH for 30 min, copiously rinsed with DI, and sterilized with 70% ethanol solution for 1-h; rinsed with sterile DI water; followed by sterile PBS; and then incubated for 12 h in medium (DMEM/F-12 with 10% FBS) to obtain prewetted scaffolds. Prewetted scaffold disks were seeded with bovine chondrocytes (passage-2) at a seeding density of 3×104 cells/scaffold, with six scaffolds per well in a six-well tissue culture plate. One plate with 36 scaffolds represented one test condition. Typically, 15 μL of a 2.0×106 cells/mL stock solution was pippeted on each scaffold, plates were kept in a CO2 incubator at 37°C and 95% relative humidity (RH) for 3 h, and then, 8-mL of fresh medium was added on top of the scaffolds and maintained for 20-h in the incubator. Scaffolds were transferred to a new TCP plate with 5–8 mL of fresh complete media per well, were placed in the incubator for 3 days, and were then subjected to US stimulation. Control treatments did not include US stimulation and were handled similarly to US-treated specimens. Medium was changed every alternate day. Unseeded disks were also included as controls.
Cell proliferation
Cultured chondrocytes were released from control and test scaffolds by adding 0.25% trypsin with 0.1% EDTA (ethylene diamine tetra acetic acid) followed by incubation at 37°C with 5% CO2. Medium was added to the trypsinized cells to bring the final volume to 2 mL. Cell concentration was counted using a hemocytometer. To obtain a basal value, cell counts were first determined 3 days after seeding and before the application of US stimulation. In a parallel experiment, the cell viability was also determined by (4,5-dimethyl-thiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
mRNA gene expression analysis
On completion of US exposure, cell-seeded tissue culture plates were washed with ice-cold HBSS, and incubated with 200 μL/well of Trizol reagent (Invitrogen) with periodic mixing for 5-min, and cell homogenate was collected. RNA was isolated from cell homogenate using the Qiagen RNeasy mini kit (Qiagen). The mRNA level was quantified by using quantitative RT-PCR (qRT-PCR). The qRT-PCR analysis was carried out using QuantiFast Probe RT-PCR Kit (Qiagen). Fifty nanograms of total RNA were added per 10 μL reaction vial with RT mix, RT-PCR master mix, sequence-specific primers, and Taqman probes. Sequences for all target gene primers and probes were purchased commercially from Applied Biosystmens (GAPDH was as internal control; Applied Biosystems). qRT-PCR assays were carried out in triplicate on Eppendorf's mastercycler realplex RT-PCR system (Eppendorf North America). The cycling conditions were 10 min cDNA formation by reverse transcriptase enzyme at 50°C and 5 min polymerase activation at 95°C followed by 40 cycles at 95°C for 30 s, at 55°C for 30 s, and at 72°C for 1 min. The threshold was set above the nontemplate control background and within the linear phase of target gene amplification to calculate the cycle number at which the transcript was detected.
Chondrocyte cell morphology on the samples
For scanning electron microscopy (SEM), cells seeded in scaffolds were crosslinked with 2.5% glutaraldehyde (Sigma) in PBS for 30 min, rinsed with DI water, and gradually dehydrated with a series of ethanol solutions. Hexamethyl disilazane (Fisher) was used to remove 100% ethanol. Samples were sputter coated with Au-Pd before they were examined under SEM (Hitachi, S-3000N variable pressure). A voltage of 15 kV was used to visualize the samples.
Quantification analysis
Band intensities were quantified by densitometry using ImageQuant software (v5.2; Molecular Dynamics). The values reported were normalized to unstimulated controls. For the analysis of the US stimulation effects on mRNA levels, data represent the mean and standard deviation values of 3 independent estimations.
Staining for actin
To visualize actin organization under US in the UBR, chondrocytes were seeded onto coverslips at a seeding density of 2×104 cells/coverslip, placed at the bottom of a six-well TCP plate, filled with 8-mL of media, and transferred to the plate holders in the UBR. US stimulation was applied using the programming tool/interface. Nonstimulated cells on coverslips served as controls. Cells grown on coverslips were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 2 h at room temperature followed by washing thrice with TBS. Then, coverslips were permeated with 0.1% Triton X-100 (Sigma-Aldrich) prepared in TBS for 15 min; washed thrice with TBS; and then blocked for 30 min using blocking solution (1% bovine serum albumin in TBS). Subsequently, coverslips were stained with a 1:50 dilution of Alexa-Flour594 phalloidin in blocking solution for 30 min at room temperature and rinsed extensively before mounting with aqueous mounting medium on coverslips. A confocal laser scanning microscope (Olympus FV500 Inverted Olympus IX 81) was used to obtain the images. To minimize the autofluorescence, images were converted into black and white images, and were processed using ImageJ™.
Statistical analyses
Statistical significance was evaluated using one-way analysis of variance for a comparison between the control and test groups. The values were considered statistically different when p<0.05.
Results
US bioeffects
The UBR uses US to stimulate chondrocytes maintained in in vitro culture (Fig. 1) over a range of US stimulations. Of equal importance to the direct aspects of US, our UBR should not exhibit any effects that may result from secondary bioeffects. When biological materials are exposed to US, the associated thermal and nonthermal mechanisms can impact or, in certain cases, act as the causative agent for the observed bioeffects, namely, proliferation, viability, and cell-specific processes. Since our focus is to understand the biophysical effects of the adopted US stimulation regimen, it is critical that we delineate the contribution of US-induced thermal and nonthermal mechanisms toward the observed cellular effects.
Thermal effects, which are typically associated with an increase in the bulk temperature of the medium, are usually exerted at high US intensities. In our studies, low-intensity US signals were used. The temperature increases (ΔT) caused by exposure to US stimulation regimens with amplitudes ranging from 10–60 kPa at frequencies of 5.0 to 8.5 MHz were determined in sterile water, using DMEM, within a scaffold in DMEM and within a cell-seeded scaffold. No detectable temperature rise was observed.
Nonthermal bioeffects can be grouped into two categories: inertial cavitational and noninertial-cavitational mechanisms. IC generally occurs at higher acoustic pressures, and noninertial cavitation (non-IC) occurs at lower acoustic pressures. The threshold for non-IC has been previously reported to be in the range between 6 and 8 W/cm2. We have observed intensities of 0.01–0.1 W/cm2 in the bioreactor such that no significant non-IC was expected. However, we have also assessed the non-IC effects of US by measuring the extent of sucrose hydrolysis in aqueous solutions.30 No discernible levels of hydrolysis were noted in control or solutions exposed to US, where a wide regimen of US was tested and appropriate positive controls were included.
To assess possible IC effects, a spinprobe (CMH) was added to the aqueous media at room temperature, and the solutions were sonicated over a wide range of US regimens employed in the bioreactor. The relative amount of EPR signal (an indicator of the degree of IC) was negligible and not significantly different from the EPR signal strengths obtained in control samples. However, the use of a sonic-horn (i.e., positive control) resulted in a significantly higher level of EPR signal (5.5 times higher). The ability of the US regimen to generate reactive oxygen species (ROS) was also evaluated, using the Image-iT ™ LIVE Green ROS Detection Kit, where the oxidatively stressed and nonstressed cells are reliably distinguished by fluorescence microscopy. Our results suggest that ROS was not generated by the US stimulation regimen used in this study. We conclude that any US effects in the bioreactor, apart from cellular responses, are negligible.
Characterization of the US field
The automated operation of the bioreactor is dependent on (1) the tuning of splitter channels, which in turn, controls the transducers to provide equal pressure amplitude; (2) placement of the transducers; and (3) controlled movement of the x-y-z stage. Thus, the splitter was first tuned for a 5.0 MHz, 14 kPa output for all transducers (values shown to effective in on our previous work21). The ability of the splitter to provide other inputs, uniformly, was also tested. Finally, the movement of the aquarium in x-y-z direction was controlled to avoid any spillage of water. The variation of pressure amplitude in the wells is shown in Figure 2a with regard to input voltage (Vpp). These measurements were made at points that were 8 mm above the bottom of the TCP plates and along the axis of the transducer. In our bioreactor configuration, numerous multiple reflections of the US waves are possible from the boundaries of the well and result in a diffuse field. The average radial variation of the pressure amplitude inside a well was also evaluated and is shown in Figure 2b. From these results, we estimate the usable width within each well as ∼1.5 cm, a width within which the pressure profile is uniform.
FIG. 2.

Intensity and Pressure profiles. (a) The splitter was initially tuned so that all the channels provided the same pressure amplitude (14 kPa) at low input voltage (2.5 Vpp) and an input frequency of 5 MHz. The pressure amplitudes using other input voltages from the six wells are also shown. (b) The spatial variation in average pressure amplitude was measured in the wells for a 2.5 Vpp.
While using the bioreactor, wide arrays of US regimens are possible. In this study, we have confined our analyses to a signal frequency (center) of 5 MHz so that we could establish conformity with our previously reported results and also perform comparative analyses.
US-induced proteome changes in chondrocytes
Although the effects of US on various cell types have been widely studied using DNA microarrays,32–34 the effect of US on chondrocytes has not been investigated comprehensively. We have used a proteomic approach to profile the US-induced protein expression and modifications. The protein concentration was measured and adjusted so that the same amount of each protein sample was labeled with size and charge-matched minimal fluorescent CyDye and separated on an analytical scale electrophoresis gel. A representative two-dimensional difference gel electrophoresis (2D-DIGE) image of protein lysates from US-treated chondrocytes is shown in Figure 3. About 138 protein spots were resolved and identified with high confidence (>95%). Overall, around 138 protein spots were found to be consistently up- or down-regulated by more than 1.3-fold in triplicate experiments after US treatment (5 MHz; 5-min, 14 to 60 kPa) for 3 or 6 days. We have made an initial effort to identify around 50 protein spots, encompassing a wide range of molecular weights, pI values, fold changes, and abundance. All 50 protein spots were successfully identified with high confidence by using preparative gel electrophoresis, in-gel trypsin digestion followed by tandem mass spectrometry as described earlier, and the location of each spot is labeled with a number. Proteins identified so far are listed in Table 2 and are grouped according to their primary functions.
FIG. 3.
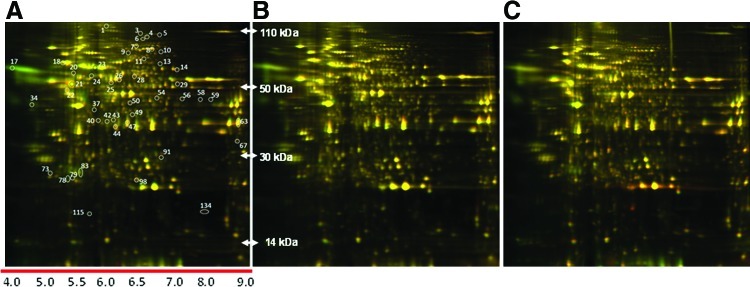
Two-dimensional difference gel electrophoresis (2D-DIGE) Global protein expression profiling. A quantitative comparative analysis of protein spots was performed using DeCyder “in-gel” or “cross-gel” analysis software. The color of a protein spot is a measure of its relative abundance. The protein expression ratios between different samples groups that were generated as an internal standard were included in all gels. Proteins that met the cut-off requirement in one or more of the comparisons presented were selected for further identification and analyses. (A) Control (day-3) versus US (60 kPa, day-3, 8-applications/day); (B) control (day-6) versus US (60 kPa, day-6, 8-applications/day) and (C) US (60 kPa, day-3, 16-applications/day) versus US (14 kPa, day-6, 8-applications/day). Color images available online at www.liebertpub.com/tec
Table 2.
Proteins Identified in Two-Dimensional Difference Gel Electrophoresis Gels
| Spot no: | Protein name | Protein Accession no. | Molecular mass kDa (theoretical) | pI (theoretical) | Panel A | Panel B | Panel C |
|---|---|---|---|---|---|---|---|
| Cytoskeletal proteins | |||||||
| 13 | Lamin A | gi|453180 | 71598.4 | 6.20 | 1.22 | −1.42 | 1.20 |
| 21 | Vimentin | gi|110347570 | 53695.1 | 5.06 | 1.59 | 1.11 | 1.13 |
| 37 | b-Actin | gi|14250401 | 40978.4 | 5.56 | −1.01 | 1.32 | −1.04 |
| 50 | ACTR1A | gi|75775168 | 41436.4 | 6.59 | 1.30 | 1.07 | −1.27 |
| Cell membrane-bound molecules | |||||||
| 1 | Sorbin and SH3 domain containing 2 isoform | gi|114597167 | 117336.7 | 8.53 | −1.00 | −2.36 | −1.05 |
| Proteins involved in matrix synthesis | |||||||
| 24 | Prolyl 4-hydroxylase subunit alpha-1 precursor | gi|115495019 | 60972 | 5.63 | −1.57 | 1.64 | 1.12 |
| 47 | Annexin | gi|74 | 38873.2 | 6.44 | −1.02 | −1.04 | −1.42 |
| Metabolic enzymes | |||||||
| 7 | Hexokinase | gi|60592784 | 102141 | 6.29 | 1.46 | −1.22 | −1.99 |
| 43 | Transaldolase | gi|164420731 | 37657.6 | 7.03 | 1.24 | 1.33 | −1.11 |
| 11 | Transketolase | gi|152941228 | 64834.1 | 6.71 | 1.43 | −1.04 | −1.52 |
| 63 | Glyceraldehyde-3-phosphate dehydrogenase | gi|77404273 | 35845.3 | 8.50 | 1.38 | −1.04 | −1.41 |
| 49 | Acetyl-CoA acetyltransferase | gi|115495669 | 41172.2 | 6.46 | 1.32 | 1.13 | −1.64 |
| 44 | l-lactate deydrogenase | gi|118572666 | 36700.2 | 6.02 | 1.10 | 1.02 | −1.21 |
| Protein synthesis and degradation | |||||||
| 25 | 26S protease regulatory subunit 4-like | gi|296222266 | 49275.7 | 5.97 | 1.35 | 1.03 | −1.29 |
| 98 | Protein DJ-1 | gi|62751849 | 20022.6 | 6.84 | 1.36 | 1.13 | −1.34 |
| 6 | Elongation factor-2 | gi|115497900 | 95307.0 | 6.41 | 1.52 | −1.03 | −1.15 |
| 8 | Glutaminyl-tRNAsynthetase | gi|77735887 | 87587.6 | 6.43 | 1.36 | −1.04 | −1.04 |
| 10 | Threonyl-tRNAsynthetase, cytoplasmic | SYTC_BOVIN | 83438.9 | 6.34 | 1.04 | −2.00 | −1.14 |
| 22 | Protein disulfide-isomerase A3 precursor | gi|148230374 | 56893.9 | 6.38 | 1.12 | 1.01 | −1.12 |
| 56 | Peptidyl-prolylcis-trans isomerase | FKB11_BOVIN | 22460.3 | 9.26 | 1.31 | 1.42 | −1.15 |
| 40 | Chain A, Crystal Structure Of DimethylarginineDimethylaminohydrolase I In Complex With S-Nitroso- | gi|109157318 | 30401.6 | 5.71 | 1.32 | 1.12 | −1.05 |
| RNA and DNA binding proteins | |||||||
| 42 | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | gi|296482303 | 35208.5 | 5.20 | 1.17 | 1.43 | −1.15 |
| 73 | Translationally controlled tumor protein | gi|62177164 | 19568.6 | 4.84 | −1.34 | 1.06 | 1.22 |
| 9 | ATP-dependent RNA helicase DDX | gi|115495959 | 82361.8 | 6.81 | 1.37 | −1.01 | −1.11 |
| 54 | COMM domain containing protein 7 | COMD7_BOVIN | 22519.6 | 5.69 | 1.30 | 1.11 | −1.03 |
| 3 | RNA polymerase II-associated protein 1 | RPAP1_BOVIN | 152762.7 | 5.91 | 1.31 | −1.39 | −1.41 |
| 17 | Chain D, Crystal Structure Of Bovine F1-C8 Sub-Complex Of Atp Synthase | gi|306991567 | 50283.3 | 4.96 | −2.46 | −1.70 | 0.79 |
| Others | |||||||
| 18 | 78 kDa glucose-regulated protein precursor (HSP-70) | gi|115495027 | 72355.5 | 5.07 | −1.33 | 1.43 | −1.12 |
| 78,83 | Apolipoprotein A-I preproprotein | gi|75832056 | 30257.9 | 5.71 | −1.73 | −1.74 | 1.10 |
| 79 | Heme-binding protein 1 | gi|115496135 | 21216.4 | 5.39 | 1.45 | 1.56 | −1.18 |
| 115 | Ferritin, heavy polypeptide | gi|154426178 | 21056.2 | 5.54 | −1.01 | 1.50 | 1.06 |
| 91 | Hemoglobin subunit alpha-I/II | HBA_BISBO | 15129.9 | 8.90 | 1.35 | 1.33 | 1.30 |
| 29 | MRS2 | gi|46362574 | 46461.3 | 5.78 | 1.45 | 1.24 | −1.29 |
| 4 | High-density lipoprotein-binding protein | gi|297473540 | 175738.3 | 9.32 | 1.04 | −2.17 | −1.21 |
| 14 | Alpha-2-HS-glycoprotein precursor | gi|27806751 | 38394.4 | 5.26 | 1.32 | −1.32 | −1.38 |
| 28 | Reticulocalbin-3 precursor | gi|114053121 | 37545.1 | 4.76 | 1.30 | −1.02 | −1.25 |
| 34 | Cathespin | gi|299522 | 37686.8 | 5.43 | −1.43 | 1.19 | 1.05 |
| Molecular chaperones | |||||||
| 26 | T-complex protein 1 subunit zeta | gi|77736031 | 57919.6 | 6.32 | 1.15 | −1.02 | −1.34 |
| 58,59 | Serpin HI precursor | gi|114051505 | 46477.2 | 9.01 | 1.50 | 1.18 | 1.18 |
| Ion Channels | |||||||
| 67 | Plasmalemmalporin | gi|437027 | 30675.6 | 8.84 | 1.37 | 1.11 | −1.48 |
Primary chondrocytes were exposed to US in the bioreactor or kept as control (no US) for 3 or 6 days. The protein lysates were subjected to 2D-DIGE, followed by silver staining and image analyses. Spots that met 1.3-fold cutoff and which appeared in nine different comparative gels were chosen for further analyses.
US, ultrasound.
Functional category of altered proteins
To gain additional insights into the biological significance and functional attributes of the differentially expressed proteins during US, the proteins were categorized according to their main biological functions collected from the UniProt protein knowledge database and PubMed. According to their main biological functions, these proteins were classified as follows: energy metabolism, RNA/DNA binding proteins/chromatin assembly, cytoskeletal, matrix synthesis, protein synthesis, degradation, and others. The challenge here is to validate the differentially expressed proteins to their respective function.
Validation of differentially expressed proteins
US-induced Erk1/2 phosphorylation
Chondrocytes that were serum deprived overnight were treated with 5.0 MHz US (14 kPa) for 3 min, and then, the cells were lysed to collect protein 15 min after the US mechanical stress. Western blotting was used to analyze the phosphorylation of Erk1/2 at threonine (T) 202/Y204 of Erk1 and T185/Y187 of Erk2 compared with total Erk1/2. US stimulation at a central frequency of 5 MHz induced transient phosphorylation of Erk1/2 (Fig. 4); with a greater level of p-ERK1/2 at 10Vpp (60 kPa) as compared with 2.5Vpp (14 kPa). The addition of ERK inhibitor (PD98059) to the medium was observed to reduce the phospho-ERK signals to baseline levels, suggesting a role for ERK-mediated signaling pathway under US.
FIG. 4.

US-induced phosphorylation of Erk at threonine 202 and tyrosine 204. Serum-deprived chondrocytes were treated with US (5 MHz/14 kPa or 5 MHz/60 kPa) for 3 min, and then, total cell lysates were collected at 15 after US treatment. Total cell lysate for control was also collected from chondrocytes that did not receive US treatment. Phosphorylation of Erk1/2 at threonine 202 and tyrosine 204, total Erk1/2, and β-actin loading control were demonstrated by Western blotting. Experiments were also conducted in the presence of ERK inhibitor, PD98059. Data are representative of three independent experiments.
Reorganization of actin under US
Confocal microscopy was used to assess the distribution and organization of the actin, where determinations made before and after US stimulation are shown in Figure 5. Figure 5A shows the actin distribution in control, nonstimulated cells (a representative frame). Several actin fiber formations can be noted in these pictures, the presence of long actin fibers that run along the length of the cell is evident, and an actin mesh that surrounds the cells is also noted. In general, most control cells exhibited the actin organization depicted in Figure 5. Figure 5B shows the distribution of actin in cells that were stimulated by US. Actin structure appeared to be nonorganized, with a punctuated membrane and cytosolic-located F-actin. A few long actin filaments were also noted along with mesh-like actin structures and long thin processes; however fewer stress fibers were observed.
FIG. 5.

Actin organization under US. Chondrocytes were seeded onto coverslips, placed on the bottom of a six-well plate, transferred to a plate holder in the UBR, and US stimulation (5 MHz; 14 kPa; 51 s; six times/day) was applied using the programming module. On completion of the stimulation, cells were fixed and stained with phalloidin and visualized under a confocal microscope. Color images available online at www.liebertpub.com/tec
A preliminary quantification of the actin cytoskeleton was done using ImageJ™, and a decrease in the total number of actin filaments was noted in US-treated samples (6.6±1.4 actin filaments) when compared with the control samples (21.0±8.0 actin filaments). In addition, the ratio of the “Feret diameter/mini Feret diameter” was noted to increase from 1.56±0.15 (for control cells) to 3.11±1.76 (US-treated cells), indicating a change in cell shape on US stimulation. This result is in accordance with the SEM images.
Cell-seeded constructs in bioreactor
Cell morphology
Time-dependent experiments were carried out on cell-seeded scaffolds in the bioreactor. Nonstimulated samples were also maintained in the bioreactor and served as controls. High cellular viabilities were noted at the indicated time periods, as shown in Figure 6. A significant difference in cellular viability (p<0.05) was noted between control and US cellular constructs at day 28. In an independent experiment, plain scaffolds (i.e., without cells; ∼50 scaffolds) were maintained in the bioreactor, and the microstructure of the scaffolds was observed to remain intact with no detectable scaffold debris.
FIG. 6.

Cellular viability and proliferation. Cultured chondrocytes were released from control and test scaffolds by adding 0.25% trypsin with 0.1% ethylene diamine tetra acetate followed by incubation at 37°C with 5% CO2. Medium was added to the trypsinized cells to bring the final volume to 2 mL (a). Cell viability was also determined by (4,5-dimethyl-thiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) assay (*p<0.05). (b) In a parallel experiment, cell concentration was counted using a hemocytometer. To obtain a basal value, cell counts were determined after seeding and before the application of US stimulation.
By design, the initial seeding density was similar for all the study groups, and data were normalized to cell counts obtained at the end of day 3 after cell seeding (2.2×105 cells/scaffold). The effect of US on the total cell number is shown in Figure 6; and total cell number of 5.5×105 cells/scaffold were obtained in the US-treated scaffolds.
SEM images of US-stimulated and control chondrocytes on scaffolds on day 10 are shown in Figure 7. The chondrocytes from the control remain spherical and nebulous. However, those subjected to US treatment show a change in morphology for which the cells show a spindle-like shape and the presence of long processes.
FIG. 7.

Scanning electron microscopy (SWM). SEM analysis of chondrocyte-seeded scaffolds stimulated by US. Scale bar is shown in the figure.
Gene expression of cartilage-specific markers
The impact of US stimulation on the mRNA expression of chondrocytic markers (collagen II, collagen I, and aggrecan) was examined by qRT-PCR (Fig. 8). Compared with the control, higher levels of gene expression were noted on US.
FIG. 8.

Gene expression. A 5.0 MHz US signal (14 kPa) was applied six times/day for 51 s/application and maintained in culture for 10 days. The mRNA levels of indicated genes were measured by quantitative real-time–polymerase chain reaction, using specific primers purchased from Applied Biosystems. The GAPDH gene was used as a loading control. Cells from seeded scaffolds that were not subjected to US stimulation served as controls. Data were normalized to the controls and are reported as the mean of three independent estimations with error bars, where an error bar represents one standard deviation (*p<0.05).
Discussion
US applications can be divided according to intensity of the US signals: (1) diagnostic US uses a frequency between 3 and 5 MHz and a low intensity (1–50 mW/cm2); (2) Disruptive US, such as those used in ultrasonic cleaning devices, uses a very low frequency (20–60 kHz) and a high intensity (8 W/cm2); and (3) therapeutic US, used in medicine and physiotherapy, uses frequencies between 1 and 3 MHz and intensities of 0.1 to 2.0 W/cm2 (SAPA).19,27,35 Our interests lie in the biological applications of low-intensity US.
Previous studies have used low-intensity pulsed US (1.5 MHz, 1.0 kHz repeat, 6–40 min) to stimulate chondrocytes.19 As a significant departure from such strategies, we have employed intermittent applications of LIDUS at 5.0 MHz (14 kPa) to stimulate bovine chondrocytes seeded in 3D chitosan-based matrices. Our ongoing research has documented the ability of US stimulation (5.0 MHz, 51-s/application) to impact the proliferative and biosynthetic activity of chondrocytes seeded in 3D matrices. The LIDUS excitation differs from previous studies, as the total application time is very long relative to the maximum travel time across the well (∼35 μs). Thus, the cells are excited in an incoherent (diffuse) manner due to the numerous multiple reflections from the boundaries of the well that the US waves experience.
While using this bioreactor, a wide array of US regimens is possible. In this study, we have confined our analyses to a signal frequency (center) of 5 MHz so that we could establish conformity with our previously reported results and also perform comparative analyses. Exposure of cells on scaffolds, coverslips, or on TCP plates in the UBR yielded results that are very comparable to previously reported trends and values21 in which the US stimulation was provided manually and from the top of the TCP plate. This provides validity to our pressure amplitude measurements using both configurations.
ERK1 and ERK2 are often referred to together as ERK1/2, and when activated in response to extracellular signals, undergo phosphorylation. They then regulate proliferation, differentiation, cell-cycle processes, and survival, as well as many other cell processes.36–38 Activated ERKs can translocate to the nucleus, where they phosphorylate and regulate various transcription factors, ultimately leading to changes in gene expression.39,40 Our results suggest that the cellular effects of US on chondrocytes are mediated by the ERK1/2 pathway, a topic which will be investigated in detail in our future studies.
Our UBR is designed to capitalize on the positive bioeffects of LIDUS and to apply them to the field of tissue engineering. In conclusion, we have shown that the US-assisted bioreactor can serve as a useful platform for dissecting molecular and biochemical events. However, our ongoing and future efforts will involve the testing of the US bioreactor under a variety of experimental conditions. Further, the 2D protein reference map of chondrocytes will facilitate future studies on chondrocyte functions and differentiation in response to US stimulation, will help us elucidate connections between broad cellular pathways/molecules, and complement traditional biochemical analyses.
Acknowledgments
This work was supported, in part, by the American Recovery and Reinvestment Act of 2009 research grant 1R21RR024437-01A1 from the Department of Health and Human Services. The expert technical assistance of Mr. Leonard Akert and Mr. Paul Marxhausen is truly appreciated. The authors thank Dr. Mark Zimmermann (University of Nebraska, Medical Center, Omaha, NE) for the use of the EPR spectrometer.
Disclosure Statement
No competing financial interests exist.
References
- 1.McGinty J.B. Articular cartilage regeneration: a matter of time and creativity. Am J Knee Surg. 2000;13:7. [PubMed] [Google Scholar]
- 2.Grana W.A. Healing of articular cartilage: a review. Am JKnee Surg. 2000;13:29. [PubMed] [Google Scholar]
- 3.Getgood A. Brooks R. Fortier L. Rushton N. Articular cartilage tissue engineering: today's research, tomorrow's practice? J Bone Joint Surg Br Vol. 2009;91:565. doi: 10.1302/0301-620X.91B5.21832. [DOI] [PubMed] [Google Scholar]
- 4.Temenoff J.S. Mikos A.G. Review: tissue engineering for regeneration of articular cartilage. Biomaterials. 2000;21:431. doi: 10.1016/s0142-9612(99)00213-6. [DOI] [PubMed] [Google Scholar]
- 5.Concaro S. Gustavson F. Gatenholm P. Bioreactors for tissue engineering of cartilage. Adv Biochem Eng Biotechnol. 2009;112:125. doi: 10.1007/978-3-540-69357-4_6. [DOI] [PubMed] [Google Scholar]
- 6.Danisovic L. Varga I. Zamborsky R. Bohmer D. The tissue engineering of articular cartilage: cells, scaffolds and stimulating factors. Exp Biol Med (Maywood) 2012;237:10. doi: 10.1258/ebm.2011.011229. [DOI] [PubMed] [Google Scholar]
- 7.Portner R. Goepfert C. Wiegandt K. Janssen R. Ilinich E. Paetzold H., et al. Technical strategies to improve tissue engineering of cartilage-carrier-constructs. Adv Biochem Eng Biotechnol. 2009;112:145. doi: 10.1007/978-3-540-69357-4_7. [DOI] [PubMed] [Google Scholar]
- 8.Schulz R.M. Bader A. Cartilage tissue engineering and bioreactor systems for the cultivation and stimulation of chondrocytes. Eur Biophys J. 2007;36:539. doi: 10.1007/s00249-007-0139-1. [DOI] [PubMed] [Google Scholar]
- 9.Wescoe K.E. Schugar R.C. Chu C.R. Deasy B.M. The role of the biochemical and biophysical environment in chondrogenic stem cell differentiation assays and cartilage tissue engineering. Cell Biochem Biophys. 2008;52:85. doi: 10.1007/s12013-008-9029-0. [DOI] [PubMed] [Google Scholar]
- 10.Bilgen B. Uygun K. Bueno E.M. Sucosky P. Barabino G.A. Tissue growth modeling in a wavy-walled bioreactor. Tissue Eng Part A. 2009;15:761. doi: 10.1089/ten.tea.2008.0078. [DOI] [PubMed] [Google Scholar]
- 11.Bonassar L.J. Grodzinsky A.J. Frank E.H. Davila S.G. Bhaktav N.R. Trippel S.B. The effect of dynamic compression on the response of articular cartilage to insulin-like growth factor I. J Orthop Res. 2001;19:11. doi: 10.1016/S0736-0266(00)00004-8. [DOI] [PubMed] [Google Scholar]
- 12.Buschmann M.D. Gluzband Y.D. Grodzinsky A.J. Hunzinker E.B. Mechanical compression modulates matrix biosynthesis in chondrocyte/agarose culture. J Cell Sci. 1995;108:1497. doi: 10.1242/jcs.108.4.1497. [DOI] [PubMed] [Google Scholar]
- 13.Carver S.E. Heath C.A. Influence of intermittent pressure, fluid flow, and mixing on the regenerative properties of articular chondrocytes. Biotechnol Bioeng. 1999;65:274. [PubMed] [Google Scholar]
- 14.Davisson T. Kunig S. Chen A. Sah R. Ratcliffe A. Static and dynamic compression modulate matrix metabolism in tissue engineered cartilage. J Orthop Res. 2002;20:842. doi: 10.1016/S0736-0266(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 15.Lee D.A. Martin I. Bioreactor culture techniques for cartilage-tissue engineering. Methods Mol Biol. 2004;238:159. doi: 10.1385/1-59259-428-x:159. [DOI] [PubMed] [Google Scholar]
- 16.Anton F. Suck K. Diederichs S. Behr L. Hitzmann B. van Griensven M., et al. Design and characterization of a rotating bed system bioreactor for tissue engineering applications. Biotechnol Prog. 2008;24:140. doi: 10.1021/bp070241b. [DOI] [PubMed] [Google Scholar]
- 17.Hsu S.-H. Kuo C.-C. Whu S.W. Lin C.-H. Tsai C.-L. The effect of ultrasound stimulation versus bioreactors on neocartilage formation in tissue engineering scaffolds seeded with human chondrocytes in vitro. Biomol Eng. 2006;23:259. doi: 10.1016/j.bioeng.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 18.Lee H.J. Choi B.H. Min B.-H. Son Y.S. Park S.R. Low-intensity ultrasound stimulation enhances chondrogenic differentiation in alginate culture of mesenchymal stem cells. Artif Organs. 2006;30:707. doi: 10.1111/j.1525-1594.2006.00288.x. [DOI] [PubMed] [Google Scholar]
- 19.Parvizi J. Wu C. Lewallwen D.G. Greenleaf J.F. Bolander M.E. Low-intensity ultrasound stimulates proteoglycan synthesis in rat chondrocytes by increasing aggrecan gene expression. J Orthop Res. 1999;17:488. doi: 10.1002/jor.1100170405. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi R. Ryo A. Komitsu N.N. Mikuni-Takagaki Y. Fukui A. Takagi Y., et al. Low-intensity pulsed ultrasound activates the phosphatidylinositol 3 kinase/Akt pathway and stimulates the growth of chondrocytes in three-dimensional cultures: a basic science study. Arthritis Res Therapy. 2008;10:R77. doi: 10.1186/ar2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noriega S.E. Mammedov T. Turner J.A. Subramanian A. Intermittent applications of continuous ultrasound on the viability, proliferation, morphology and matrix production of chondrocytes in 3D matrices. Tissue Eng. 2007;13:611. doi: 10.1089/ten.2006.0130. [DOI] [PubMed] [Google Scholar]
- 22.Hasanova G.I. Noriega S.E. Mamedov T.G. Thakurta S.G. Turner J.A. Subramanian A. The effect of ultrasound stimulation on the gene and protein expression of chondrocytes seeded in chitosan scaffolds. J Tissue Eng Regen Med. 2011;5:815. doi: 10.1002/term.384. [DOI] [PubMed] [Google Scholar]
- 23.Whitney N.P. Lamb A.C. Louw T.M. Subramanian A. Integrin-mediated mechanotransduction pathway of low-intensity continuous ultrasound in human chondrocytes. Ultrasound Med Biol. 2012;38:1734. doi: 10.1016/j.ultrasmedbio.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J. Nyborg W.L. Ultrasound, cavitation bubbles and their interaction with cells. Adv Drug Deliv Rev. 2008;60:1103. doi: 10.1016/j.addr.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Kimmel E. Cavitation bioeffects. Crit Rev Biomed Eng. 2006;34:105. doi: 10.1615/critrevbiomedeng.v34.i2.10. [DOI] [PubMed] [Google Scholar]
- 26.Krasovitski B. Frenkel V. Shoham S. Kimmel E. Intramembrane cavitation as a unifying mechanism for ultrasound-induced bioeffects. Proc Natl Acad Sci U S A. 2011;108:3258. doi: 10.1073/pnas.1015771108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merritt C.R.B. Kremkau F.W. Hobbins J.C. Diagnostic ultrasound: bioeffects and safety. Ultrasound Obstet Gynecol. 1992;2:366. doi: 10.1046/j.1469-0705.1992.02050366.x. [DOI] [PubMed] [Google Scholar]
- 28.Dalecki D. Mechanical bioeffects of ultrasound. Annu Rev Biomed Eng. 2004;6:229. doi: 10.1146/annurev.bioeng.6.040803.140126. [DOI] [PubMed] [Google Scholar]
- 29.Hallow D.M. School of Chemical and Biomolecular Engineering, Georgia Institute of Technology; Georgia: 2006. Measurement and correlation of acoustic cavitation with cellular and tissue bioeffects [Ph.D. dissertation] [DOI] [PubMed] [Google Scholar]
- 30.Buldakov M.A. Hassan M.A. Zhao Q.L. Feril L.B., Jr. Kudo N. Kondo T., et al. Influence of changing pulse repetition frequency on chemical and biological effects induced by low-intensity ultrasound in vitro. Ultrason Sonochem. 2009;16:392. doi: 10.1016/j.ultsonch.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Noriega S.E. Subramanian A. Chitosan as cell culture scaffold material: consequences of scaffold pretreatment on chondrocyte proliferation, attachment and cytoskeletal organization. Int J Carbohydr Chem. 2011 doi: 10.1155/2011/809743. Article ID. 809743, 13 pages. [DOI] [Google Scholar]
- 32.Tabuchi Y. Kondo T. Ogawa R. Mori H. DNA microarray analyses of genes elicited by ultrasound in human U937 cells. Biochem Biophys Res Commun. 2002;290:498. doi: 10.1006/bbrc.2001.6203. [DOI] [PubMed] [Google Scholar]
- 33.Sironen R.K. Karjalainen H.M. Elo M.A. Kaarniranta K. Torronen K. Takigawa M., et al. cDNA array reveals mechanosensitive genes in chondrocytic cells under hydrostatic pressure. Biochim Biophys Acta. 2002;1591:45. doi: 10.1016/s0167-4889(02)00247-1. [DOI] [PubMed] [Google Scholar]
- 34.Myokai F. Oyama M. Nishimura F. Ohira T. Yamamoto T. Arai H., et al. Unique genes induced by mechanical stress in periodontal ligament cells. J Periodont Res. 2003;38:255. doi: 10.1034/j.1600-0765.2003.00602.x. [DOI] [PubMed] [Google Scholar]
- 35.ter Haar G. Therapeutic ultrasound. Eur J Ultrasound. 1999;9:3. doi: 10.1016/s0929-8266(99)00013-0. [DOI] [PubMed] [Google Scholar]
- 36.Junttila M.R. Li S.P. Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 37.Meloche S. Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 38.Rubinfeld H. Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- 39.Zuber J. Tchernitsa O.I. Hinzmann B. Schmitz A.C. Grips M. Hellriegel M., et al. A genome-wide survey of RAS transformation targets. Nat Genet. 2000;24:144. doi: 10.1038/72799. [DOI] [PubMed] [Google Scholar]
- 40.Schulze A. Nicke B. Warne P.H. Tomlinson S. Downward J. The transcriptional response to Raf activation is almost completely dependent on mitogen-activated protein kinase kinase activity and shows a major autocrine component. Mol Biol Cell. 2004;15:3450. doi: 10.1091/mbc.E03-11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]