

Abstract
Significance: Despite advances made in the treatment of cancer, a significant number of patients succumb to this disease every year. Hence, there is a great need to develop new anticancer agents. Recent Advances: Emerging data show that malignant cells have a greater requirement for iron than normal cells do and that proteins involved in iron import, export, and storage may be altered in cancer cells. Therefore, strategies to perturb these iron-dependent steps in malignant cells hold promise for the treatment of cancer. Recent studies show that gallium compounds and metal-thiosemicarbazone complexes inhibit tumor cell growth by targeting iron homeostasis, including iron-dependent ribonucleotide reductase. Chemical similarities of gallium(III) with iron(III) enable the former to mimic the latter and interpose itself in critical iron-dependent steps in cellular proliferation. Newer gallium compounds have emerged with additional mechanisms of action. In clinical trials, the first-generation-compound gallium nitrate has exhibited activity against bladder cancer and non-Hodgkin's lymphoma, while the thiosemicarbazone Triapine® has demonstrated activity against other tumors. Critical Issues: Novel gallium compounds with greater cytotoxicity and a broader spectrum of antineoplastic activity than gallium nitrate should continue to be developed. Future Directions: The antineoplastic activity and toxicity of the existing novel gallium compounds and thiosemicarbazone-metal complexes should be tested in animal tumor models and advanced to Phase I and II clinical trials. Future research should identify biologic markers that predict tumor sensitivity to gallium compounds. This will help direct gallium-based therapy to cancer patients who are most likely to benefit from it. Antioxid. Redox Signal. 00, 000–000.
Inhibition of Tumor Cell Growth with Gallium Compounds–Iron Mimicry and Beyond
Introduction
Iron, iron proteins, and tumor growth. The role of iron in cell viability and proliferation is well known; it has been reviewed elsewhere in this forum and will not be reiterated here. Coupled with advances in our knowledge of iron metabolism has been an increasing appreciation that certain malignant cells have a far greater requirement for iron than normal cells do (126). This importance of iron in tumor cell growth is exemplified by observations which show that the expression of transferrin (Tf) receptors on lymphoma, breast cancer, and bladder cancer cells is increased relative to normal cells and that elevated levels of this receptor correlate with adverse clinical outcomes (62, 86, 120). These changes in the level of Tf receptors may also be associated with alterations in the expression of ferritin, the iron-storage protein in cells (136). The relevance of iron metabolism in cancer was recently underscored by Pinnix et al., who reported on gene signatures of iron-related proteins in biopsies from patients with breast cancer and showed that breast cancer cells displayed a decrease in ferroportin, a protein involved in the efflux of iron from cells (94, 104, 126). It is known that cell-surface ferroportin is down-regulated by hepcidin in the circulation (68). These investigators demonstrated that increased serum levels of hepcidin and decreased ferroportin in breast cancers were associated with a more aggressive clinical behavior (increased metastases and decreased survival) when compared with breast cancers lacking these features (94, 104). The significance of the changes in these proteins in breast cancer is that an elevation in Tf receptor expression along with a diminution of ferroportin would favor an increase in intracellular iron needed to support the aggressive biological behavior of this tumor. Iron-dependent proteins known to be altered in breast cancer are summarized in Figure 1.
FIG. 1.

Iron proteins in breast cancer cells. Under physiologic conditions, iron is bound to transferrin (Tf) in the circulation and is incorporated into cells by transferrin receptor1 (TfR1)-mediated endocytosis of Tf-Fe complexes. The binding site of the wild-type hemochromatosis protein (wt HFE) partially overlaps with the Tf-binding site on TfR1 and can, thus, competitively inhibit Tf binding to its receptor. This regulatory effect of HFE on Tf-Fe-TfR binding is lost with the HFE C282Y mutation, as the latter is degraded within the cell and no longer associates with the TfR to interfere with its binding to Tf-Fe. The Tf-Fe-TfR complex translocates from the cell surface to an intracellular acidic endosome, where Fe(III) dissociates from Tf and is reduced to Fe(II) by STEAP3 (six-membrane epithelial antigen of the prostate 3) (not shown). Fe(II) exits the endosome through divalent metal transporter1 (DMT1, not shown) to a labile iron “pool.” From here, iron trafficks to different compartments (mitochondria, ribonucleotide reductase [RR], and others). Excess iron is stored in ferritin. Iron exits from the cell through cell membrane-based ferroportin. Ferroportin levels can be lowered by hepcidin, which binds to it and translocates it to the lysosome for degradation. Cytoplasmic iron regulatory proteins (IRPs) function as sensors of cellular iron status and regulate the synthesis of Tf receptors, ferritin, and ferroportin at the mRNA translational level by interactions with iron response elements (IREs) present in the untranslated regions of their respective mRNAs. Iron proteins known to be altered in breast cancer are marked with an asterisk (*) and include an increase in TfR and ferritin as well as a reduction in ferroportin levels. In addition, the C282Y HFE mutation may be associated with an increased risk of breast cancer development. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Targeting tumor iron homeostasis with gallium compounds—multiple sites for gallium's action
Perturbation of cellular iron transport and uptake by gallium compounds
Given the increased iron requirements of certain cancer cells, it is not surprising that chelators which bind iron and limit its availability to cells may inhibit malignant cell growth. However, the blockade of cellular iron utilization may also be achieved through other strategies. Studies have shown that iron homeostasis in cancer cells can be perturbed by gallium compounds, leading to an arrest in cell proliferation and cell death. The growth-inhibitory effects of gallium can be enhanced by decreasing cellular iron through the exposure of cells to iron chelators before incubation with gallium and can be diminished by elevating intracellular iron content with soluble iron salts or Tf-iron (90). These interactions of gallium with iron metabolism are possible, because gallium(III) shares certain properties with iron(III) with regard to its ionic radius and its binding to Tf with high avidity in the circulation. It is primarily incorporated into cells via Tf receptor1-mediated endocytosis of Tf-gallium complexes (38, 87). Early insights into the process of gallium uptake by cells were provided by the studies of Harris and Sephton, who showed that the cellular uptake of 67Ga citrate (used for tumor imaging in humans) could be enhanced in vitro by Tf (66), a finding that was confirmed by others (38, 87). Further studies demonstrated that the cytotoxicity of gallium in malignant cell lines in vitro could be enhanced by Tf and could be reversed by iron salts (30, 108). The steps in cellular iron metabolism that are targeted by gallium are summarized in Figure 2.
FIG. 2.

Interaction of gallium with cellular iron metabolism. The potential sites of gallium's interaction with cellular iron metabolism are identified in the bordered boxes. Membrane transport: TfR mediated. In the circulation, gallium(III) is transported and bound to Tf as Tf-Ga and is incorporated into TfR1-expressing cells by TfR1-mediated endocytosis of Tf-Ga complexes. Tf-Ga inhibits cellular iron incorporation by interfering with TfR-mediated uptake of Tf-Fe and the release of Fe from Tf within the endosome. Non-TfR mediated. Low-molecular-weight gallium and iron chelates in the circulation may also be taken up by certain cells through a shared Tf-independent pathway. In this pathway, gallium may enhance cellular iron uptake and vice versa. In cells, gallium can be detected in a low-molecular-weight pool. TfR1 and ferritin mRNA translation: Consistent with the induction of cellular iron deprivation, cells exposed to gallium display an increase in TfR1 mRNA and protein. This results from an increase in IRP-IRE mRNA interaction that leads to increased TfR mRNA translation. RR: Gallium blocks iron incorporation into the R2 subunit and may itself be incorporated into this subunit, rendering it inactive. Gallium also blocks RR enzyme activity more directly through competitive inhibition of substrate binding. This could result from the formation of Ga-ADP/CDP complexes that compete with (Fe)-ADP/CDP binding to the enzyme. The inhibition of RR activity contributes to gallium-induced cell death. Iron-dependent mitochondrial function: Gallium may perturb mitochondrial function by action on the numerous iron-containing proteins present in the citric acid cycle and the electron transport chain (shown in Fig. 4). Upstream from the mitochondrion (mito), gallium may activate proapoptotic Bax, which translocates to the mitochondria, produces a loss of mitochondrial membrane potential, and the release of cytochrome c and apoptogenic factors to the cytoplasm. This triggers the activation of effector caspases-3 and -7, leading to apoptotic cell death. ADP, adenosine diphosphate; CDP, cytidine diphosphate.
Evidence of the role of Tf receptor1 in gallium uptake was demonstrated in an animal model bearing implanted melanoma cells; here, the tumor uptake of 67Ga was inhibited by a monoclonal antibody to the Tf receptor (16). The cellular uptake of 67Ga-Tf may also be modulated by the expression of the wild-type (wt) or mutant hemochromatosis (HFE) gene. HFE protein associates with newly synthesized Tf receptor1 in the cell and, along with beta-2 microglobulin, trafficks to the cell surface. Since Tf and HFE share overlapping binding sites on the Tf receptor, the association with HFE may modify Tf receptor1-mediated uptake of Tf-iron, especially when Tf levels are low (113). With the HFE C282Y mutation (seen in hereditary hemochromatosis), however, HFE protein is degraded within the cell, and the HFE regulation of Tf-iron uptake is lost (112). In HeLa cells engineered to express the wt HFE gene under the control of tetracycline (tet-off system), the overexpression of wt HFE was shown to significantly decrease 67Ga uptake by cells (32). These observations raise the intriguing question as to whether tumors in cancer patients with HFE mutations will express mutant HFE and whether these tumors will differ in their response to treatment with gallium compounds when compared with individuals with wt HFE. Given the high frequency of HFE mutations in the general population (93), this is an important question to be addressed in the clinic.
The concentration of gallium in the circulation appears to be important for its speciation in vivo. Bernstein has suggested that at a concentration of approximately 50 μM gallium, greater than 99.9% of gallium in the circulation exists at Tf-gallium; at gallium concentrations of 50 μM, ∼95% of gallium is Tf bound. When gallium concentrations exceed Tf saturation, however, the fraction of gallium bound to Tf decreases, and gallium exists primarily as gallate [Ga(OH)−4] in the circulation (10). Although the Tf-transferrin receptor pathway appears to be the primary mechanism of gallium uptake by cells, it is clear that certain cells may also incorporate gallium via a Tf-independent mechanism (38). Interestingly, this Tf-independent gallium uptake pathway is similar to that used by Tf-independent iron (29). However, while Tf-gallium inhibits the cellular uptake of Tf-iron, Tf-independent gallium actually enhances the uptake of Tf-independent iron in HL60 cells in vitro (29). The relevance of Tf-independent gallium uptake to the cytotoxicity of gallium is not clear. Possible explanations include the possibility that this pathway may enable cells to incorporate gallium when their endogenous expression of Tf receptors is low. Alternatively, Tf-independent uptake may enable cells to acquire iron when Tf receptor-mediated uptake of iron is blocked by gallium.
Although there is no known physiologic role for gallium in humans, gallium's binding to Tf in the circulation enables it to home in on Tf receptor-bearing cells and complete with Tf-iron for binding to its receptor. This is possible in vivo, because, under physiologic conditions, approximately one-third of Tf in the blood is occupied by iron leaving the remainder available to bind gallium and circulate as Tf-gallium. This mimicry of iron by gallium allows it to be initially handled by cells as though it were iron. Unlike iron (III), however, gallium(III) is not reduced to gallium(II) and, thus, does not directly participate in redox reactions in the cell. Nonetheless, the consequence of gallium incorporation into cells is the disruption of iron homeostasis at various levels. In human leukemic HL60 cells, Tf-gallium inhibits the cellular uptake of Tf-iron and blocks the dissociation of iron from Tf within an acidic endosome (30). As a result, gallium's action at this level leads to the generation of cellular iron deprivation; this is evidenced by a decrease in intracellular ferritin and an up-regulation of Tf receptor mRNA and protein (30, 65). Other studies have also confirmed the interaction of gallium with iron metabolism. In murine erythroleukemia cells, Tf-gallium inhibits the induction of hemoglobin production; however, this block can be reversed by the co-incubation of cells with Tf-iron or iron pyridoxal isonicotinoyl hydrazone, a compound that delivers iron directly to reticulocytes for heme synthesis (37). Consistent with in vitro studies, gallium administered to patients binds exclusively to Tf in the circulation (3, 129) and patients being treated with gallium nitrate may develop microcytic anemia that is characterized by an elevated erythrocyte protoporphyrin level, a marker of tissue iron deficiency (116).
Effect of gallium on ribonucleotide reductase
Cellular iron requirements for DNA synthesis are related to the iron-dependent activity of ribonucleotide reductase (RR), the enzyme that is responsible for the reduction of ribonucleoside diphosphates to deoxyribonucleoside diphosphates (Fig. 3). The latter are converted to deoxyribonucleotides, the substrate for DNA polymerase (44, 124). Since the activity of RR is rate limiting for DNA synthesis, this enzyme holds a critical position in cell division. Human RR consists of two heterodimeric subunits termed R1 and R2 that are located on chromosomes 11 and 2, respectively (52, 139). The R1 subunit contains substrate- and effector-binding sites, while the R2 subunit contains a dinuclear iron center and a tyrosyl free radical that can be detected by electron paramagnetic resonance (EPR) spectroscopy (44, 60). The activity of the R2 subunit and the amplitude of the tyrosyl radical EPR signal increase as cells enter the S-phase of the cell cycle (51, 54). Destruction of the tyrosyl radical or interference with iron incorporation in the R2 subunit results in a loss of RR activity and an arrest in DNA synthesis. In proliferating cells, the R2 subunit has a half-life of 3 h, indicating that it should be continuously regenerated to maintain DNA synthesis (54). Hence, a steady supply of intracellular iron to the R2 subunit is needed for RR activity (99).
FIG. 3.

Ribonucleotide reductase (RR). RR is responsible for the reduction of ribonucleoside diphosphates (NDPs) to deoxyribonucleoside diphosphates (dNDPs). The enzyme is a heterodimer that consists of an R1 and R2 subunit. The R2 subunit contains a dinuclear iron center and a tyrosyl-free radical (*Tyr) that is detected by EPR spectroscopy to produce the signal shown in the figure. The amplitude of the EPR signal is closely correlated with enzyme activity and is dependent on the presence of iron in the R2 subunit. EPR, electron paramagnetic resonance.
Given the importance of iron in DNA synthesis, it is not surprising that one consequence of gallium-induced cellular iron deprivation is an inhibition of RR (25). Specifically, the incubation of intact leukemic cell lines with Tf-gallium or gallium nitrate results in a decrease in the activity of RR as evidenced by a loss of the R2 subunit tyrosyl radical EPR signal, a decrease in the levels of dNTPs, and a block in overall enzymatic activity (25). A consideration in such analysis is that under normal conditions, the magnitude of the R2 EPR signal increases as cells transition from G1 to S phase of the cell cycle; hence, any gallium-induced changes in cell cycle distribution could also impact the level of the EPR signal in cells. However, in these studies, gallium-treated and control cells were shown to display a similar cell cycle distribution at the time of EPR analysis, indicating that the diminution in the tyrosyl radical EPR signal in gallium-treated cells was not due to cell cycle arrest in the G1 phase (25). That this was secondary to the inhibition of iron utilization was revealed by the observation that the co-incubation of gallium-treated cells with hemin as an iron source abrogated the gallium-induced decrease in the R2 subunit tyrosyl radical signal (25). The gallium-induced loss of the R2 tyrosyl radical EPR signal results from an interference of iron incorporation into R2 and not a decrease in R2 protein. Other studies showed that the EPR signal in cytoplasmic extracts from gallium-treated cells could be regenerated within 10 min after the addition of iron salts to the cell extract (96).
Although an interruption in the intracellular trafficking of iron to the R2 subunit by gallium may be sufficient to inhibit RR activity, this does not appear to be the sole mechanism at play. Gallium nitrate directly inhibits RR (adenosine diphosphate [ADP] and cytidine diphosphate [CDP] reductase) activity when directly added to a cell-free assay (26). Interestingly, the mechanism of inhibition appears to be due to the competitive inhibition of substrate interactions with the enzyme.
Further insights into the interaction of gallium with iron homeostasis and RR have been obtained through studies by the use of cell lines that are resistant to growth inhibition by gallium nitrate. These cells were developed through continuous exposure of the parental gallium-sensitive cell line to incremental concentrations of gallium nitrate. HL60 cells with acquired resistance to gallium nitrate display a decrease in Tf receptor 1 expression along with a diminished uptake of iron and gallium relative to gallium-sensitive cells (45). Moreover, gallium resistance leads to changes in the intracellular trafficking and distribution of iron and gallium (45). With regard to RR, in the absence of gallium, the amplitude of the tyrosyl radical EPR signal is similar in both gallium-sensitive and gallium-resistant HL60 cells; however, this EPR signal is suppressed by gallium in gallium-sensitive cells but not in gallium-resistant cells (40). In contrast, in human leukemic T-lymphoma CCRF-CEM cells with acquired resistance to growth inhibition by gallium, gallium-resistant cells display a decrease in iron and gallium uptake and a decrease in their basal activity of RR (31, 34). Hence, it would appear that although gallium inhibits RR, the development of tumor cell resistance to gallium nitrate does not uniformly involve compensatory changes in RR expression and activity.
Overall, the inhibition of RR activity by gallium appears to be due to a combination of an indirect (iron deprivation) and a direct action of the metal on this enzyme. However, several questions regarding the latter mechanism remain unanswered: Is gallium incorporated into the R2 subunit in place of iron? Although it appears reasonable that this could occur, gallium substitution for iron in R2 protein has never been demonstrated. Could the competitive inhibition of RR by gallium suggested in the enzyme assay analysis result from the formation of gallium-nucleoside complexes (Ga-CDP, Ga-ADP) that compete with substrate (ADP or CDP) binding to the enzyme? Gallium binding to nucleosides has been demonstrated in other studies (91), thus providing support for the latter mechanism.
Effect of gallium on iron-dependent mitochondrial function
Recent studies have suggested that the disruption of mitochondrial function is central to gallium-induced cell death in lymphoma cells. The incubation of lymphoma cells with gallium compounds results in a loss of mitochondrial membrane potential, release of cytochrome c to the cytoplasm followed by the activation of effector caspase-3, and apoptosis (35). In some cells, cell death induction by gallium nitrate appears to involve the activation of proapoptotic Bax, a protein that translocates from the cytoplasm to the mitochondria to initiate a change in mitochondria membrane permeability and the release of cytochrome c (35). In contrast, gallium maltolate appears to disrupt mitochondrial membrane permeability without Bax activation, thereby suggesting that gallium may act directly on mitochondria (Chitambar and Wereley, unpublished observations). This mechanism of gallium's action may be explained by the fact that several enzymes of the citric acid cycle and electron transport chain contain iron-sulfur clusters that are essential for mitochondrial function (Fig. 4). Given the ability of gallium to interact with metal-binding sites and to interfere with iron incorporation into certain proteins, it is reasonable to speculate that gallium may disrupt mitochondrial function by targeting iron-sulfur cluster-containing proteins in this organelle. In this regard, gallium nitrate was shown to block gene translation of mitochondrial aconitase, an iron-sulfur cluster-containing enzyme, and inhibit aconitase enzymatic activity in prostate cancer cells (127). Other studies also lend support for an action of gallium on the mitochondria. CCRF-CEM cells incubated with gallium maltolate for 1–4 h displayed a decrease in their reduced glutathione (GSH)/oxidized glutathione (GSSG) ratio and an increase in intracellular reactive oxygen species (ROS). The latter was blocked by mitoquinone, a mitochondria-targeted antioxidant, thus suggesting that intracellular ROS induced by gallium originated in the mitochondria and could possibly be the result of gallium-induced inhibition of mitochondrial function (28). Clearly, further studies are needed to advance our understanding of gallium's action on the mitochondrion.
FIG. 4.

Potential sites of action for gallium in the mitochondria. The iron-sulfur cluster (Fe-S) proteins in the citric acid cycle and mitochondrial complexes are potential targets for the cytotoxic action of gallium compounds.
Action of gallium beyond iron metabolism
While the cellular gallium and iron uptake occurs through similar pathways and both metals can be found in the lysosome (13, 144), more recent studies indicate that gallium and iron differ in other aspects of their intracellular trafficking (45). In HL60 cells, most of the iron incorporated is retained by the cell and is stored in ferritin. In contrast, a significant amount of gallium that enters cells by Tf receptor-mediated uptake cycles back out of the cells, and only a small amount of gallium associates with ferritin; the majority intracellular gallium can be detected in a labile cytoplasmic pool (39, 45). Hence, while iron homeostasis is the primary target for gallium compounds, its action on other cellular process may also contribute to its cytotoxicity.
The noniron targets for gallium are somewhat diverse. Early studies showed that gallium could inhibit DNA polymerases and tyrosine phosphatase (9, 131), but this effect could not be correlated with an inhibition of cell growth and, hence, the significance to cytotoxicity is unclear. On the other hand, gallium has been shown to inhibit Mg-dependent ATPase and tubulin polymerase; both these actions could contribute its growth-inhibitory effects (4, 100). More recently, novel gallium complexes with pyridine and phenolate ligands agents have been described (21, 117). These gallium complexes inhibit proteasome function and block malignant cell proliferation in vitro and in a rodent tumor model bearing prostate cancer xenografts (21). This mechanism of the action of gallium is important, as proteasome inhibitor drugs have emerged as clinically important drugs for the treatment of multiple myeloma, lymphoma, and other malignancies (20, 76).
Studies utilizing human CCRF-CEM lymphoma cells with acquired resistance to growth inhibition by gallium nitrate have yielded further information about the action of gallium on iron-independent cellular processes. A comparison of metal metabolism-related genes in gallium-resistant and gallium-sensitive cells revealed that the resistant cells had a higher level of expression of the metallothionein-2A and zinc transporter-1 genes relative to gallium-sensitive cells (138). Hence, it appears that the action of gallium in cells is not limited to iron metabolism but includes zinc homeostasis. In these studies, it was shown that a zinc-induced increase in metallothionein in gallium-sensitive CCRF-CEM cells produced a partial reduction in the cytotoxicity of gallium nitrate; this protection was lost when metallothionein levels returned to baseline (138). Furthermore, the growth-inhibitory effects of gallium nitrate in a panel of lymphoma cell lines were shown to correlate with their endogenous levels of metallothionein; here, gallium's cytotoxicity was lower in cells with the highest expression of metallothionein (138). While these observations suggest an association of metallothionein with lymphoma cell sensitivity to gallium nitrate, it is unclear whether overexpression of metallothionein per se is sufficient to confer tumor cell resistance to gallium nitrate. Under physiologic conditions, metallothionein plays a central role in zinc metabolism and also sequesters toxic divalent metals such as cadmium to protect against their toxicity (46, 77). However, since metallothionein is not involved in iron metabolism, it would not be expected to play a role in the cellular handling of gallium and, thus, a gallium-induced up-regulation of metallothionein would not be expected. Recent studies, though, provide at least a partial explanation for this observation. CCRF-CEM cells incubated with gallium nitrate display an increase in ROS and a decrease in the ratio of GSH/GSSG within 4 h of gallium exposure; this is followed by an increase in the expression of metallothionein and heme oxygenase-1 (137). The gallium-induced up-regulation of heme oxygenase-1 gene expression involves activation of the transcription factor Nrf-2, while metallothionein gene up-regulation involves a shift in intracellular zinc and the activation of metal transcription factor-1. Both these events appear to be triggered by ROS, as they can be abrograted by the antioxidant N-acetyl cysteine. This suggests that the up-regulation of metallothionein and heme oxygenase-1 by gallium nitrate results from gallium-induced oxidant stress in cells (137). While the mechanism by which gallium generates ROS in cells remains to be determined, additional experiments show that this increase in ROS can be blocked by mitoquinone, a mitochondria-targeted antioxidant (28). The latter finding supports a central position of the mitochondria in gallium's mechanisms of action.
Gallium-induced cell death is determined by the balance between cytoprotective and cytotoxic events
Based on our current understanding of the mechanisms of action of gallium compounds, a model can be proposed in which the initial response of a cell to gallium is to elevate proteins such as metallothionein and heme oxygenase-1 that provide a level of protection to the cell. These cytoprotective responses to gallium may differ among cells and may play a role in determining the clinical antineoplastic activity of gallium compounds in various tumors. However, with continued exposure of cells to sufficiently high concentrations of gallium, these cytoprotective responses are overwhelmed and cell death eventually follows (Fig. 5). Further investigation will undoubtedly unravel other processes involved in tipping the balance between life and death in cells exposed to gallium compounds.
FIG. 5.
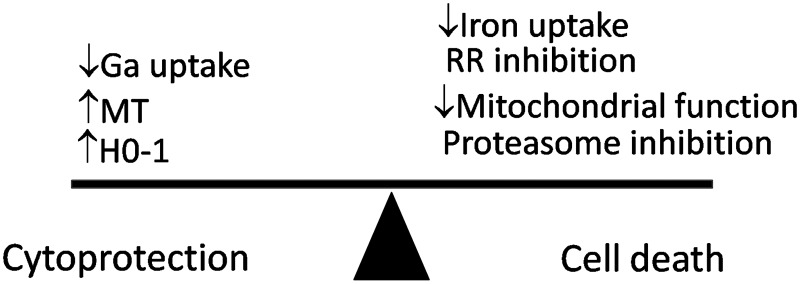
Cytoprotection and cell death with gallium compounds. Events that confer cytoprotection or cell death by gallium compounds are summarized. The exposure of cells to gallium nitrate produces an early increase in cellular oxidative stress that leads to an increase in metallothionein-2A (MT2A) and hemoxygenase-1 (HO-1) as a cytoprotective reaction. The negative effect of gallium on cellular iron balance, RR activity, mitochondrial function, and other processes tilts the balance toward cytotoxicity. Cell death ensues when the cytoprotective responses are overcome. It is possible that cytoprotective responses to gallium may vary in different cells and may, thus, contribute to differences in cell sensitivity to gallium compounds.
Gallium compounds in the clinic
Gallium nitrate
As a first-generation therapeutic gallium compound, gallium nitrate (Fig. 6A) has been extensively evaluated for its toxicity and anticancer activity in preclinical and clinical studies (22, 57). The clinical development of gallium nitrate followed the discovery that 67Ga citrate, when injected into tumor-bearing animals, accumulated in malignant tissues to a greater extent than in normal tissues. The latter observation led to the development of the gallium-67 scan for the detection of certain tumors in humans and prompted further investigation into the antineoplastic activity of nonradioactive gallium and other metal salts (1, 69). These studies showed gallium nitrate to have significant antitumor activity in tumor-bearing rodents (1). Following toxicology studies in animals, gallium nitrate was advanced to the status of an experimental drug (NSC 15200) by the National Cancer Institute for further evaluation of toxicity and efficacy in Phase 1 and Phase 2 clinical trials.
FIG. 6.

Chemical structures of gallium compounds currently in use in the clinic. The chemical structures of (A) gallium nitrate, (B) gallium maltolate, and (C) Tris(8quinolato)gallium(III) (KP46) are shown.
Several Phase 2 clinical trials have consistently demonstrated gallium nitrate to have antineoplastic activity in bladder cancer and non-Hodgkin's lymphoma (23, 49, 122). In an early Phase 1–2 clinical trial, the efficacy of intravenous gallium nitrate infusion was evaluated in patients with bladder cancer that had relapsed after treatment with conventional chemotherapy; partial responses to treatment lasting 4–8 months were seen in 4 of 23 patients (115). These encouraging results led to further clinical trials in which gallium nitrate was combined with vinblastine and ifosfamide and administered to patients with relapsed urothelial malignancies; 44%–67% of the patients receiving this combination therapy responded to treatment (48, 50).
The efficacy of gallium nitrate in the treatment of non-Hodgkin's lymphoma that has progressed or relapsed following conventional therapy has been demonstrated in at least five different clinical trials [reviewed in ( 23, 107, 122)]. These studies have demonstrated responses in 30%–43% of patients treated with gallium nitrate by continuous intravenous infusion administered over 5–7 days. Interestingly, some subtypes of lymphoma, such as diffuse large B-cell lymphoma and mantle cell lymphoma, were found to be more responsive to gallium nitrate than other histologic subtypes (107).
The basis for the preferential sensitivity of certain lymphoma subtypes to gallium nitrate is not understood and is an important area for future investigation. In this regard, the potential ability of metallothionein to modulate lymphoma cell sensitivity to gallium nitrate may be relevant to the clinic. An examination of metallothionein in biopsies of lymphomatous tissue from patients showed that its expression varied in different histological types of non-Hodgkin's lymphoma (138). Other studies showed a correlation between metallothionein expression and prognosis in non-Hodgkin's lymphoma (106). Thus, the identification of biologic markers that may predict tumor sensitivity to gallium compounds would be a major advance in the field, as it may allow for the selective use of these agents to treat patients with lymphomas that are more likely to respond. Of equal importance, it would also help in identifying patients who are unlikely to benefit from gallium compounds and, thus, spare them the toxicities of unnecessary treatment.
Apart from its antineoplastic activity, gallium nitrate has important action on bone metabolism. In early clinical trials which evaluated the antineoplastic activity of gallium nitrate, it was noted that this drug could block pathologic bone resorption and lower blood calcium levels (132, 133). The mechanisms of action of gallium on bone metabolism appear to be independent of its interaction with iron and iron proteins (15). Several clinical studies confirmed a benefit of gallium nitrate in pathologic bone disease, and the drug was approved by the Food and Drug Administration for the treatment of cancer-associated hypercalcemia (88, 125).
Although gallium nitrate has proved its efficacy in the treatment of lymphoma, bladder cancer, and hypercalcemia, its bioavailability when administered by the oral route is low. This necessitates that it be administered to patients by continuous intravenous infusion over 5–7 days to achieve maximum efficacy with a low level of toxicity (133). Recent efforts by the manufacturer of gallium nitrate (Genta Incorporated) have focused on the development of an oral tablet formulation of gallium nitrate, termed G4544, which employs a proprietary drug delivery system (98). Pharmacokinetic studies of G4544 in healthy human volunteers have shown that a single oral dose of 30–150 mg produces plasma gallium levels as high as 485 ng/ml, which is similar to those reported with intravenous gallium nitrate (98).
Gallium maltolate
Gallium maltolate, tris(3-hydroxy-2methyl-4H-pyran-4-onato)gallium (Fig. 6B) is an orally administered gallium in clinical development; it consists of three maltolate ligands that are bidentately bound to a central gallium atom in a propeller-like arrangement (11). In vitro, gallium maltolate induces apoptosis in hepatoma and lymphoma cell lines at lower concentrations than gallium nitrate (28, 41). Pharmacokinetic studies in humans show that a single oral dose of 100–500 mg gallium maltolate (equivalent to 15.7–78.4 mg gallium) produces maximum serum gallium levels of 0.115 and 0.569 μg/ml, respectively (11). After its oral administration, virtually all of the gallium present in the circulation is bound to Tf with very little of it being unbound (3). The clinical antitumor activity of oral gallium maltolate has recently been demonstrated. Bernstein et al. reported on a patient with advanced hepatocellular carcinoma who displayed a significant reduction in the size of his hepatic tumor with improvement in symptoms after treatment with 1500 mg/day of oral gallium maltolate for 4 weeks (12).
Tris(8-quinolonato)gallium(III)
Tris(8-quinolonato)gallium(III) (KP46) (Fig. 6C) is a gallium complex with an organic ligand 8-quinolinol that is involved in clinical trials as an oral gallium compound (114). This agent has undergone extensive preclinical evaluation in a variety of solid tumor cells, including melanoma, ovary, breast, colon, and lung cancer cell lines, where its IC50 was found to be lower than that reported for gallium nitrate and ranged from 0.85–10.4 μM (128). Its tissue distribution and pharmacology has been studied in detail in mice (42). In patients, KP46 is reported to be well tolerated at doses ranging from 30–480 mg/m2 daily for 14 days, and appreciable dose-limiting toxicities were not encountered (43, 70). Responses to treatment in patients with renal cancer have been noted, a finding that warrants further investigation (43, 70).
Drug synergy between gallium and other chemotherapeutic agents
An important strategy in the treatment of cancer is to use different drugs in combination with the goal of enhancing antitumor activity through action on multiple cellular targets. In clinical trials, gallium nitrate has been used in combination with etoposide, mitoguazone, and hydroxyurea, thus demonstrating that it can be safely administered to patients in combination chemotherapy protocols (36, 134). A further assessment of its potential synergism with other chemotherapeutic drugs in lymphoma and other cell lines in vitro has been made using strict pharmacologic criteria. These studies showed that gallium nitrate acts synergistically to inhibit cell growth when it is combined with hydroxyurea, fludarabine, interferon-alpha, gemcitabine, paclitaxel, or bortezomib (25, 27, 33, 67, 90). Since these drugs are currently in use for the treatment of cancer, it appears logical to combine them with gallium compounds for further study in the clinic.
Gallium compounds in preclinical development
Pyridine and phenolate ligand complexes of gallium
A number of interesting gallium compounds are in various stages of preclinical development. Pyridine and phenolate ligand complexes of gallium have been shown to possess antitumor activity in a prostate cancer xenograft implanted in a rodent tumor model (21). These compounds appear to induce cell death through the inhibition of proteasome function in cells and, thus, have a primary mechanism of action that is different from that of other gallium compounds (21, 117). The proteasome has emerged as an important therapeutic target in the treatment of lymphoma and multiple myeloma (20, 56); thus, the inhibition of proteasome function by these gallium complexes is highly relevant to the clinical application of gallium.
Gallium complexes with thiosemicarbazones
The synthesis of different 2-acetylpyridine thiosemicarbazones-gallium complexes as potential therapeutic agents was first reported by Kratz et al. (85). Enyedy et al. compared the formation of high stability bis-liganded complexes of Triapine® [3-aminopyridine-2-carboxylaldehyde thiosemicarbazone, 3-AP (Fig. 7)] with gallium and iron and showed that at physiologic pH, Triapine formed complexes with both metals (53). Another study comparing the cytotoxicity of gallium versus iron complexes of Triapine in 41M (ovarian carcinoma) and SK BR 3 (mammary carcinoma) cell lines demonstrated that the cytotoxicity of this thiosemicarbazone was enhanced by gallium but weakened by iron (81). The gallium(III) complexes were found to be 210–3300-fold more cytotoxicity in 41M cells and 150–4000-fold more cytotoxic in SK-BR-3 cells than the corresponding iron complexes, respectively (81). Popovic-Bijelic et al. have provided some insight into the mechanisms involved in the enhanced cytotoxicity of the Triapine-gallium complex with regard to action on the R2 subunit of RR (105). Although Triapine-gallium was more potent than Triapine in inhibiting the growth of 41M cells in culture, it displayed similar kinetics as metal-free Triapine in inhibiting the tyrosyl radical of R2 protein in solution (105). An explanation for this is that in an aqueous solution at pH 7.6, gallium dissociates from Triapine and the latter, in turn, is available to chelate iron which is released from the R2 subunit (105). These observations suggest that the enhanced cytotoxicity of the Triapine-gallium complex after its uptake by cells results from the superimposed actions of gallium hydroxides or other gallium complexes, free Triapine, and subsequent complexes formed with Triapine. The antineoplastic activity and interactions of Triapine with other metals are discussed further in the next section of this review.
FIG. 7.

Chemical structures of various thiosemicarbazones.
Other gallium complexes
Gallium compounds other than those previously discussed have been reported to have anti-proliferative activity against malignant cell lines. These include gallium complexes with azole, thiolato, and pyridoxal isonicotinoyl hydrazone ligands (7, 24, 58, 59, 74, 75, 78, 92, 111, 145). However, these gallium complexes have only been examined in tissue culture cell lines in vitro, and a confirmation of their antineoplastic activity in vivo remains to be demonstrated.
Inhibition of Tumor Growth by the Iron Chelator Triapine: A Critical Analysis of Its Action on RR and Interaction with Metals
Introduction
Triapine®, T [3-aminopyridine-2-carboxylaldehyde thiosemicarbazone, 3-AP (Fig. 7) is one of the many derivatives from the parent ligand 2-formyl pyridine monothiosemicarbazone, L. Thiosemicarbazones and their anti-tumor activity have been recently reviewed (140) and further reviewed elsewhere in this issue. One of the intents of this review is to add new insights for both Triapine and gallium-Triapine interactions in cells and to interpret them, but citations are used as examples and do not include all references. The reader is referred to these recent reviews, which contain more comprehensive references (140, 141, 143) and references therein.
Triapine inhibits RR
The antineoplastic activity of thiosemicarbazones is generally attributed to an inhibition of RR, an enzyme that converts ribonucleosides to deoxyribonucleosides (Fig. 3). An obvious mechanism is that (i) iron from the di-ferric site in the R2 subunit is removed by forming an iron complex with the thiosemicarbazone or (ii) iron is not available to form the di-ferric site. A thorough study of the inhibition of RR showed that Triapine, T, plus RR formed Fe(III)T2+, but only 2.5 μM from 30 μM RR (105), and the tyrosyl radical was partially lost with T, but completely lost with FeT2+. With the addition of dithiolthreitol (DTT), a reducing agent, and Triapine or the Triapine metal complex, the tyrosyl radical is completely lost. With Triapine alone, 7.9 μM of Fe(II)T2 was formed, indicating the removal of Fe from RR. Some of the formation of FeT2 on the addition of T is probably the result of spontaneous loss of Fe from RR. Considering that RR has a reserve capacity, it seems possible that enough Fe is still bound to RR to provide enough activity for cells to grow, but the activity is inhibited as indicated by the loss of the tyrosyl radical. The tyrosyl radical from RR is not lost with low concentrations of Triapine and Triapine metal complexes [0.125 μM], except for FeT2 with DTT, suggesting that preformed FeT2 is active and less FeT2 is formed with a low concentration of T. In the next section, it is suggested that the iron which forms FeT2 is readily available from iron bound to Tf, Fe-Tf, competing with RR as a source of Fe for T. It is concluded that a more effective mechanism for the inhibition of RR is the generation of ROS with a catalytic amount of FeT2.
Inhibition of RR is not the only cellular interaction for Triapine
It was shown that an Fe-thiosemicarbazone complex plus ascorbate generates ROS and damages RR (118), but Fe-thiosemicarbazone plus ascorbate would damage a lot of enzymes. The Gräslund group has argued that Fe(II)T2 is an efficient inhibitor of mouse R2, where catalytic amounts are needed for loss of the tyrosyl radical and where bound cysteines from RR act as the reducing agent (105). The role of FeT2 is discussed further in the next section.
An alternative hypothesis is that thiosemicarbazones deprivation of iron leads to an alteration in cell cycle control and leads to G1/S phase cell cycle arrest (140). RR increases in the S phase and decreases in other phases; so, a reduction in RR could be due to a phase change. Clearly, in the future, it needs to be determined whether cells are arrested in the S phase when chelators are added.
While the depletion of iron through the formation of an iron complex with a dipyridyl thiosemicarbazone, 25 μM di-2-pyridyl-ketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT) (Fig. 7) is highly effective at up-regulating NDRG-1, a growth and metastasis suppressor gene, the IC50 is 0.1 μM (140). The low IC50 suggests that other mechanisms are involved in cell death. How FeT2 is involved still needs to be considered. The up-regulation of NDRG-1 is an important goal, but it still needs to be determined that the up-regulation of NDRG-1 is a major mechanism for the anti-tumor activity of Triapine. Nevertheless, the study with Dp44mT has opened the door for the up-regulation of a growth and metastasis suppressor on the addition of thiosemicarbazones.
Triapine or Cu-Triapine inhibits iron uptake and increases the concentration of Tf receptor (19, 97, 110, 135). It is assumed that ROS are generated after the formation of either FeT2 or CuT, and ROS cause the inhibition. Difficult experiments in which, for example, free radical quenchers such as spin traps are taken up in the same compartments as FeT2 might measure the activity of ROS.
Derivatives of isatin-β-thiosemicarbazones (Fig. 7) were evaluated for multidrug-resistant cells expressing P-glycoprotein (63). Isatin-β-thiosemicarbazones were shown to kill P-glycoprotein expressing cells more than nonresistant cells. The strategy is to kill P-glycoprotein expressing cells, which correlates with multidrug resistance and poor patient prognosis (63). While certain isatin-β-thiosemicarbazones do not appear to inhibit P-glycoprotein, the activity of the thiosemicarbazone correlates with the expression of P-glycoprotein. From a bio-inorganic perspective, it seems that a step toward understanding the mechanism is to understand whether iron or copper isatin-β-thiosemicarbazones are involved in the mechanism. Studies with other thiosemicarbazones including Triapine need to be compared with isatin-β-thiosemicarbazone, where a parallel mechanism might exist.
Is iron-Triapine the active antitumor complex?
“Hold me tight” hypothesis
Administration of the precursor to Triapine, 2-formyl pyridine thiosemicarbazone (L) resulted in dark green urine that is often attributed to the iron complex (47, 121). We have given the cupric complex of this precursor to rats and obtained green urine, the color of the cupric complex, which was confirmed by its EPR spectrum. Fe(II)L2 is blue and EPR silent, so blue and yellow giving green could also contribute to the color in urine. One conclusion is that metal complexes of Triapine remain intact through excretion.
The binding constant for FeT2 is very high, as Triapine can compete with DFO for iron, although the equilibrium occurs slowly (140). Thiosemicarbazone derivatives were effective for mobilizing intercellular 59Fe (109). If the binding constant for the second Triapine ligand is greater than or about equal to the binding constant for the first Triapine ligand in the formation of FeT2, then FeT2, and not FeT, is the stable complex formed. Using the parent ligand, 2-formyl pyridine monothiosemicarbazone, L, the overall formation constant, logKFeFeL2 for Fe(II)L2 is 23.0 and logKFeFeL2+ is 26.5 for Fe(III)L2+ (102). In this example, KFeLFeL2>KFeFeL and there is no evidence for the presence of FeL in solution. The active form is most likely to be Fe(III)L2+ and Fe(II)L2. Once formed, FeL2+ reacts very slowly in ligand substitution reactions (102). That FeL2 is stable in a complex biological medium is referred to as the “hold me tight” hypothesis.
It is argued that there is an equilibrium between Fe(II)T2 and Fe(III)T2+ in cells. A reaction of H2O2 with Fe(II)T2, the Fenton reaction, gives a very reactive, but not very specific, hydroxyl radical, •OH. What is the reducing agent for reducing Fe(III)T2+ to Fe(II)T2? If the reducing agent is thiol, then thiols from cysteine should be considered, because the concentration of cysteine amino acids in proteins is greater than the concentration of GSH (8). When the local environment of cysteine amino acids decreases the pKa value through, for example, the effects of nearby positively charged amino acids, the thiolate form is a more reactive reducing agent [(72) and references 55 and 56 therein]. Used as a model for protein thiols, cysteine can reduce Fe(III)T2+ to Fe(II)T2 (95).
“Kiss and run” hypothesis from the Richardson group (71)
Strong support for the hypothesis that Triapine and/or FeT2 is localized in the mitochondria is that Triapine treatment of human lung A549 cells (with T, presumably FeT2 was formed, see sections below) caused an almost complete oxidation of Trx2 and its dependent peroxiredoxin (Prx3), but there was no effect on Trx1 redox status (95). The positive charge for Fe(III)T2+ may also favor transport to mitochondria. If Triapine picks up iron from Tf, then it is possible that after endocytosis, FeT2 is transported directly to the mitochondrion, that is, the “kiss and run” hypothesis (71).
Fe-thiosemicarbazone, for example Fe-Triapine, sometimes exerts little effect on the cytotoxicity (83), but in at least one case for bis(1-formylisoquinoline thiosemicarbazone), the iron complex was 40 times more active than the ligand against mouse ascites sarcoma 180 (2). If Fe(III)T2+ is added to cells instead of T, Fe may not be picked up from Tf and may not be as well localized in the endosome. Therefore, a straightforward pathway to mitochondria is not as obvious.
Several derivatives of 2-formyl pyridine monothiosemicarbazone, L, were found to “possess considerable Fe chelation efficacy and reduced liver, spleen and Tf Fe levels in Fe-overloaded mice” (121). We have shown that the addition of 2-formyl pyridine monothiosemicarbazone, L, and ascorbate to Fe-Tf gives FeL2+ with oxygen present (102). This is suggestive that a source of iron for thiosemicarbazones is Fe-Tf and that the formation of FeL2+ occurs during endocytosis, where FeL2+ may be readily reduced. Thiosemicarbazone chelators can directly remove iron from Fe-Tf (109), but the addition of ascorbate better mimics the reaction in the endosome. These chelators also inhibit the cellular uptake of Fe from Fe-Tf. One thought is that as the concentration of Fe-thiosemicarbazone complexes in the endosomes increases, ROS are generated that damage the TfR. An increase in Fe-Tf signal is observed in peripheral blood lymphocytes from patients with refractory solid tumors treated with Triapine (79, 80). One explanation is that the uptake of Fe-Tf is inhibited by the damage to TfR, resulting in more oxidized Fe-Tf in peripheral blood lymphocytes.
Triapine free radical hypothesis
A third hypothesis for the activity of iron-thiosemicarbazone complexes is that a free radical from the ligand is formed, leading to free radical chemistry that leads to an attack on cellular targets (82). These investigators found that the redox potential for the ligand is very low in physiological systems, that is, −400 mv. Without free radicals being generated by Triapine itself, the formation of a metal-Triapine complex is the next option to generate ROS.
Fe-Triapine, FeT2, is redox active
Fe-thiosemicarbazone complexes (and Cu-thiosemicarbazone complexes) are redox active, and they affect cellular anti-oxidant systems. Thiosemicarbazones significantly elevated oxidized trimeric thioredoxin levels to 213% of the control and decreased thioredoxin reductase, TrxR, activity to 65% (142). Consistent with a decrease in TrxR, the glutathione/oxidized-glutathione ratio and the activity of glutaredoxin that requires glutathione as a reductant were reduced. It is suggested that thiosemicarbazones could have effects on major thiol-containing systems. These effects are dependent on the formation of an Fe-thiosemicarbazone complex. As stated earlier for the Fe complex with 5-hydroxy-2-formylpyridine thiosemicarbazone (47), Fe-thiosemicarbazones are sometimes more active RR inhibitors when added to cells as an iron or copper complex than as the ligand alone (19, 55, 105, 118). The treatment of A549 cells (human alveolar carcinoma epithelial cells) resulted in the complete oxidation of Trx2, supporting a mechanism involving thiols (95). Even more effectively, a pronounced oxidation of Prx3 occurred. In contrast to Trx2, the redox state of Trx1 was not altered suggesting little effect on RR in the cytosol, while mitochondria are greatly affected (95). One hypothesis is that Fe-Triapine is partitioned into the mitochondria or Triapine enters the mitochondria and forms Fe-Triapine. Here, Fe-Triapine possibly generates predominately two electron transfer of electrons or two one electron transfers to form peroxides, including H2O2, which greatly affects peroxiredoxin3 before other sites such as thioredoxin and has a minimal effect on thiols in the cytosol, resulting in little inhibition of RR until the concentration of Triapine increases to where it affects all thiols in both mitochondria and cytosol. The localization of Triapine in the mitochondria is consistent with the “kiss and run” hypothesis discussed earlier.
Copper Triapine, combination of effects from released copper
Lessons from bis(thiosemicarbazones)
The anti-tumor properties of bis thiosemicarbazones and especially their copper complexes (Fig. 8) have been recognized since the 1960s (101, 103). While toxicity has prevented their use in the clinic, less toxic derivatives of the original bis thiosemicarbazones became viable radiopharmaceuticals as copper complexes to measure blood flow (61). Later, copper complexes of bis thiosemicarbazones, including CuATSM, copper(II)-diacetyl-bis(N4-methylthiosemicarbazone) (Fig. 8), were used to target hypoxia (130). High lipophilicity and low redox potential are properties of copper(II)-ATSM that support hypoxic regions of cells. Due to the low redox potential of CuATSM, NADH-cytochrome b5 reductase and NADPH-cytochrome P450 reductase in the microsomes reduce a large fraction of CuATSM. In contrast, the redox potentials of Cu-Triapine and Fe-Triapine are not low and could also be reduced in mitochondria, but it is useful to understand and compare how thiosemicarbazone complexes are reduced in cells. The mechanism for labeling hypoxic cells involves reduction to Cu(I)ATSM followed by release of Cu(I), whereby Cu(I) is irreversibly trapped in a hypoxic region of the cell. These studies with CuATSM by analogy provide an insight into the mechanism for Triapine and vice versa. For example, after unloading Cu(I), does ATSM pick up another metal, and if ATSM picks up iron, what are the consequences of FeATSM? Similarly, if Cu(I) is unloaded from Cu-Triapine, is Fe-Triapine formed? Finally, is Cu(I) incorporated into copper metabolism or does Cu(I) contribute to other problems such as Alzheimers disease?
FIG. 8.

Chemical structures for cupric complexes of bis-thiosemicarbazones.
CuPTSM2 (Fig. 8) is rapidly cleared from the brain, while CuPTSM (Fig. 8) is retained to at least 2 h postinjection (14). Myocardial uptake was highest for the PTSM complex. Only a purified mitochondrial fraction reduced CuPTSM to give Cu(I) (14), as found for CuKTS (17). Is it feasible that much of the mono- and bis-thiosemicarbazone complexes, including Cu-Triapine and Fe-Triapine, are localized and reduced in the mitochondria? Others found that a cytosplasmic component is responsible for the reduction of CuPTSM [(130) and references therein]. In addition, there is little selectivity for a subcellular compartment, and Cu(I) enters copper metabolism pathways (130).
Cytotoxicity of CuT
The cytotoxicity of copper complexes of L, and its derivatives, T, Dp44mT, and so on (Fig. 7), range from about 50 nM to μM concentrations (73, 102) and their refs therein. For a cupric complex bound to a single chelator, Dp44mT, IC50 is 230 nM at 24 h and <10 nM at 72 h (73). Often, the cupric complexes are very toxic. One hypothesis is that CuT at low concentrations generates ROS through the reduction of Cu(II)T by thiols in the cells to Cu(I)T or even to a more stable Cu(I) complex followed by the oxidation of Cu(I) to Cu(II)T by oxygen and hydrogen peroxide. This continues until oxygen is depleted, thiols are oxidized, or Cu(I) is sequestered by metallothionein and chaparones (84). It may be that metallothionein and chaparones for Cu(I) are the most influential factors in stopping the generation of ROS from Cu. The mechanism for cytoxicity of CuT at nanomolar concentrations initially involves the generation of ROS through CuT. However, cuprous ion is removed over time. One theme of this review is that thiosemicarbazones, such as Triapine, form an iron complex, FeT2. Thus, over extended periods of time, that is, 72 h, the effect of both the preformed CuT after uptake and FeT2 at a later stage combine to increase cytotoxicity.
An exception to the combined effect of Cu- and Fe-thiosemicarbazones is when the Fe-thiosemicarbazone complex inhibits antiproliferation activity. For example, the Fe-thiosemicarbazone complex, where the thiosemicarbazone is 2-pyridinecarbaldehyde N,N-bis(2-pyridinylmethyl) thiosemicarbazone (Fig. 7), inhibits antiproliferation activity as Fe(2+) with HL60 cells and as Fe(3+) with PC3 cells (64). The Cu-thiosemicarbazone complex enhances activity almost fivefold. For example, the IC50 values are 0.85 μM for the thiosemicarbazone, 0.2 μM for the Cu(2+)-thiosemicarbazone, 0.7 μM for the Fe(3+)-thiosemicarbazone, and >3 μM for the Fe(2+)-thiosemicarbazone (64). One complication from the bio-inorganic standpoint is that 2 molar equivalents of metal were added. A clean formation of metal complex requires 1 molar equivalent of Cu and 0.5 molar equivalents of Fe. With regard to the addition of iron as hemin, Tf, Fe(2+), or Fe(3+), it is not clear what complexes are formed extracellularly or intracellularly. For example, hemoglobin, as well as the Fe in heme, has a strong binding site for Cu(2+) that will compete with thiosemicarbazone for Cu(2+) (5, 6). It appears that Cu-thiosemicarbazone is taken up by cells, generates ROS for an unspecified time, then hypothetically, the Cu is removed as Cu(1+), possibly Fe-thiosemicarbazone complexes are formed, which appear to be less toxic than the Cu-thiosemicarbazone. A twist to this hypothesis is that the reduced toxicity of Fe-thiosemicarbazone may be more effective than the too toxic nanomolar toxicity of the Cu-thiosemicarbazone or that preformed iron-thiosemicarbazone complexes are not as effective as iron-thiosemicarbazone complexes formed in the cell as previously discussed. However, mice tolerated both the thiosemicarbazone and the Cu-thiosemicarbazone (64). The uptake of Cu-thiosemicarbazone complexes is less complicated in that Cu(II)-thiosemicarbazone complexes have no charge if the counter-ion has a negative charge, which facilitates uptake. Nevertheless, studies determining the contrast with cell lines, cell cycle analysis, and apoptosis and necrosis discrimination as done for this thiosemicarbazone (64) need to be performed with more thiosemicarbazones. The nanomolar concentrations in this study suggest that the targets are low in concentration, suggesting that the metal-thiosemicarbazone complex acts as a catalyst. The authors of a recent study emphasizing the basis of the mechanism for Cu-thiosemicarbazones report that Cu-Dp44mT localizes in lysosomes (89). First, Dp44mT is retained in lysosomes at pH 5, because the ligand is protonated. A Cu-Dp44mT complex is the active species, resulting in the generation of ROS and apoptosis. While details for metal metabolism in lysosomes (and mitochondria) are not known, this is clearly a unique perspective, for which the role of iron and copper complexes needs to be unraveled.
Insights
Given that K2, the binding constant for the second thiosemicarbazone ligand bound, is greater than K1, the binding constant for the first ligand, 2-formylpyridine monothiosemicarbazone, the parent compound, for thiosemicarbazone derivatives where K2>K1, the major complex is FeT2 with little FeT or FeTX, where X is a ligand from the cell. This favors the intact FeT2 complex in biological media.
At low concentrations, about 100 nM or less, thiosemicarbazone forms a stable iron complex, FeT2. FeT2, acting as a catalyst, generates ROS and damages DNA. ROS are generated until either oxygen is consumed or thiols, including cysteine amino acids, are oxidized. The consequences of DNA damage are monitored by biomarkers, that is, cyclins, cdks, altered expression of NDRG1, RR activating protein, and so on and/or changes in the cell cycle. Damage at sites other than DNA occurs, but this damage only partially inhibits enzyme activity. Whether the damage is similar in different cells or cells from different individuals needs to be determined. This model is taken from studies on Fe-bleomycin, where double-strand scission is considered the mechanism of action. Since it is difficult to study metal complexes at nanomolar concentrations, what has just been cited is considered a hypothesis.
At intermediate concentrations of about 10 μM, thiosemicarbazone forms FeT2, acts as a catalyst, decreases tyrosyl radical in RR, inhibits iron uptake, causes mitochondrial thiol redox stress, and so on. A case can be made for the accumulation of FeT2+ in mitochondria, which enhances the concentration in mitochondria while decreasing the concentration in the cytosol.
At high concentrations, thiosemicarbazone, in addition to the effects at lower concentrations for iron complexes, results in iron deprivation.
Challenges and Opportunities
Despite many advances in the treatment of malignancy, it is estimated that 577,190 individuals in the United States will die from cancer in 2012 (119). Hence, there is a great need to develop new antineoplastic drugs to combat this disease. In an ideal situation, an anticancer agent should be sufficiently selective so that it kills malignant cells while sparing normal cells. In reality, however, antineoplastic drugs are not highly selective and may, thus, act on both normal and malignant cells. Nonetheless, treatment can be successful, because biologic differences that exist between normal and cancer cells may render the latter cells more susceptible to a particular drug. As an example, our understanding of differences in signaling and metabolic pathways between cancer and normal cells has sparked interest in developing therapeutic agents that act preferentially in these pathways (18, 123). There is an increasing body of evidence that cancer cells require greater amounts of iron and display changes in iron proteins, which give them a proliferative advantage compared with normal cells. These differences, therefore, provide an opportunity to exploit iron-dependent tumor growth with drugs that disrupt cellular iron homeostasis such as novel gallium compounds, metal-thiosemicarbazone complexes, and iron chelators. This is an area of cancer therapeutics that is underdeveloped and warrants further research. Agents that appear promising in vitro should be evaluated for their antineoplastic activity and toxicity in animal studies and moved forward to clinical trials. These studies should also explore strategies to reduce the toxicity of these compounds to normal cells so that they can be safely administered to patients. In this regard, pharmacologic agents that could provide protection to normal cells without diminishing the cytotoxicity to cancer cells would be of great interest. Other challenges in this field include the need to understand the basis of tumor cell resistance to gallium, thiosemicarbazones, and iron chelators, and to identify biological markers that can predict sensitivity or resistance to treatment with these agents in the clinic. The association between Tf receptor expression and aggressive tumor behavior in lymphoma and other cancers has been described (62, 86, 120). However, whether iron and iron proteins in lymphoma cells in patients predict tumor response to gallium compounds is not known and needs to be studied in the clinic. Knowledge of biologic markers of gallium sensitivity would be highly relevant to the design of clinical trials and the selection of patients who are likely to respond to treatment.
Conclusions
Exciting advances in our understanding of the importance of tumor iron metabolism have provided an impetus for the development of a group of anticancer drugs that target iron homeostasis in malignant cells. Gallium has a unique mechanism of action in that it can interpose itself into proteins and processes in lieu of iron and disrupt critical iron-dependent steps in cell function. However, gallium's activity is not limited to its functioning as an iron mimetic; it may also interfere with other aspects of cell function. Thus, gallium's mechanisms of action include iron targeting and noniron targeting, both of which act in concert to enhance its potency as an antineoplastic agent. The next generation of gallium compounds appears to have a wider range of antitumor activity than gallium nitrate and, importantly, they inhibit the growth of malignant cells that are resistant to gallium nitrate. As a result of their unique mechanisms of action, drugs that perturb tumor iron homeostasis do not share cross-resistance to conventional chemotherapeutic agents and are, thus, an important addition to the therapeutic armamentarium against cancer. The gallium complexes with different ligands represent an important advance in the development of therapeutic gallium-based drugs. Further investigation into their antineoplastic activity and toxicity in tumor-bearing animal models needs to be vigorously pursued to determine whether these agents should be advanced to clinical trials.
Abbreviations Used
- ADP
adenosine diphosphate
- ATSM
diacetyl-bis(N4-methylthiosemicarbazone)
- CDP
cytidine diphosphate
- Dp44mT
di-2-pyridyl-ketone-4,4-dimethyl-3-thiosemicarbazone
- DTT
dithiolthreitol
- EPR
electron paramagnetic resonance
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- KTS
3-ethoxy-2-oxobutraldehyde bis(thiosemicarbazone)
- L
2-formyl pyridine monothiosemicarbazone
- NSC689534
2-pyridinecarbaldehyde N,N-bis(2-pyridinemethyl)thiosemicarbazone
- NSC73306
1-isatin-4-(4′-methoxyphenyl)-3-thiosemicarbazone
- PTSM
pyruvaldehyde bis(N4-methylthiosemicarbazone)
- PTSM2
pyruvaldehyde bis(N4-dimethylthiosemicarbazone)
- ROS
reactive oxygen species
- RR
ribonucleotide reductase
- T
Triapine® [3-aminopyridine-2-carboxylaldehyde thiosemicarbazone, 3-AP]
- Tf
transferrin
- wt
wild-type
Acknowledgments
Research on gallium was supported by USPHS Grant 1095818 and an award from the Medical College of Wisconsin Cancer Center to CRC.
References
- 1.Adamson RH. Canellos GP. Sieber SM. Studies on the antitumor activity of gallium nitrate (NSC-l5200) and other group IIIa metal salts. Cancer Chemother Rep. 1975;59:599–610. [PubMed] [Google Scholar]
- 2.Agrawal KC. Booth BA. Moore EC. Sartorelli AC. Antitumor effects of transition metal chelates of 1-formylisoquinoline thiosemicarbazone. Proc Am Assoc Cancer Res. 1974;15:1974. [Google Scholar]
- 3.Allamneni KP. Burns RB. Gray DJ. Valone FH. Bucalo LR. Sreedharan SP. Gallium maltolate treatment results in transferrin-bound gallium in patient serum. Proc Am Assoc Cancer Res. 2004;45:230. (abstract 1013) [Google Scholar]
- 4.Anghileri LJ. Robert J. Radiogallium as a probe for magnesium-binding sites. Magnes-Bull. 1982;2:197–200. [Google Scholar]
- 5.Antholine WE. Taketa F. Effects of 2-formylpyridine monothiosemicarbazonato copper II on red cell components. J Inorg Biochem. 1984;20:69–78. doi: 10.1016/0162-0134(84)80007-0. [DOI] [PubMed] [Google Scholar]
- 6.Antholine WE. Taketa F. Wang JT. Manoharan PT. Rifkind JM. Interaction between bound cupric ion and spin-labeled cysteine beta-93 in human and horse hemoglobins. J Inorg Biochem. 1985;25:95–108. doi: 10.1016/0162-0134(85)80018-0. [DOI] [PubMed] [Google Scholar]
- 7.Arion VB. Jakupec MA. Galanski M. Unfried P. Keppler BK. Synthesis, structure, spectroscopic and in vitro antitumour studies of a novel gallium(III) complex with 2-acetylpyridine 4N-dimethylthiosemicarbazone. J Inorg Biochem. 2002;91:298–305. doi: 10.1016/s0162-0134(02)00419-1. [DOI] [PubMed] [Google Scholar]
- 8.Beer SM. Taylor ER. Brown SE. Dahm CC. Costa NJ. Runswick MJ. Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant defense. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 9.Berggren MM. Burns LA. Abraham RT. Powis G. Inhibition of protein tyrosine phosphatase by the antitumor agent gallium nitrate. Cancer Res. 1993;53:1862–1866. [PubMed] [Google Scholar]
- 10.Bernstein LR. Mechanisms of therapeutic activity for gallium. Pharmacol Rev. 1998;50:665–682. [PubMed] [Google Scholar]
- 11.Bernstein LR. Tanner T. Godfrey C. Noll B. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Metal-Based Drugs. 2000;7:33–47. doi: 10.1155/MBD.2000.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein LR. van der Hoeven JJM. Boer RO. Hepatocellular detection by gallium scan and subsequent treatment by gallium maltolate: rationale and case study. Anit-cancer Agents Med Chem. 2011;11:585–590. doi: 10.2174/187152011796011046. [DOI] [PubMed] [Google Scholar]
- 13.Berry JP. Poupon MF. Galle S. Escaig F. Role of lysosomes in gallium concentration by mammalian tissues. Biol Cell. 1984;51:43–51. doi: 10.1111/j.1768-322x.1984.tb00282.x. [DOI] [PubMed] [Google Scholar]
- 14.Blower PJ. Lewis JS. Zweit J. Copper radionuclides and radiopharmaceuticals in nuclear medicine. Nucl Med Biol. 1996;23:957–980. doi: 10.1016/s0969-8051(96)00130-8. [DOI] [PubMed] [Google Scholar]
- 15.Bockman R. The effects of gallium nitrate on bone resorption. Semin Oncol. 2003;30:5–12. doi: 10.1016/s0093-7754(03)00170-2. [DOI] [PubMed] [Google Scholar]
- 16.Chan SM. Hoffer PB. Duray P. Inhibition of Gallium-67 uptake in melanoma by an anti-human transferrin receptor monoclonal antibody. J Nucl Med. 1987;28:1303–1307. [PubMed] [Google Scholar]
- 17.Chan-Stier CH. Minkel D. Petering DH. Reactions of bis(thiosemicarbazonato) copper(II) complexes with tumor cells and mitochondria. Bioinorg Chem. 1976;6:203–217. doi: 10.1016/s0006-3061(00)80227-6. [DOI] [PubMed] [Google Scholar]
- 18.Chapuis N. Tamburini J. Green AS. Willems L. Bardet V. Park S. Lacombe C. Mayeux P. Bouscary D. Perspectives on inhibiting mTOR as a future treatment strategy for hematological malignancies. Leukemia. 2010;24:1686–1699. doi: 10.1038/leu.2010.170. [DOI] [PubMed] [Google Scholar]
- 19.Chaston TB. Lovejoy DB. Watts RN. Richardson DR. Examination of the antiproliferative activity of iron chelators: multiple cellular targets and the different mechanism of action of triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin Cancer Res. 2003;9:402–414. [PubMed] [Google Scholar]
- 20.Chauhan D. Hideshima T. Mitsiades C. Richardson P. Anderson KC. Proteasome inhibitor therapy in multiple myeloma. Mol Cancer Ther. 2005;4:686–692. doi: 10.1158/1535-7163.MCT-04-0338. [DOI] [PubMed] [Google Scholar]
- 21.Chen D. Frezza M. Shakya R. Cui QC. Milacic V. Verani CN. Dou QP. Inhibition of the proteasome activity by gallium(III) complexes contributes to their anti prostate tumor effects. Cancer Res. 2007;67:9258–9265. doi: 10.1158/0008-5472.CAN-07-1813. [DOI] [PubMed] [Google Scholar]
- 22.Chitambar CR. Gallium compounds as antineoplastic agents. Curr Opin Oncol. 2004;16:547–552. doi: 10.1097/01.cco.0000142071.22226.d2. [DOI] [PubMed] [Google Scholar]
- 23.Chitambar CR. Gallium nitrate for the treatment of non-Hodgkin's lymphoma. Expert Opin Investig Drugs. 2004;13:531–541. doi: 10.1517/13543784.13.5.531. [DOI] [PubMed] [Google Scholar]
- 24.Chitambar CR. Boon P. Wereley JP. Evaluation of transferrin and gallium-pyridoxal isonicotinoyl hydrazone as potential therapeutic agents to overcome lymphoid leukemic cell resistance to gallium nitrate. Clin Cancer Res. 1996;2:1009–1015. [PubMed] [Google Scholar]
- 25.Chitambar CR. Matthaeus WG. Antholine WE. Graff K. O'Brien WJ. Inhibition of leukemic HL60 cell growth by transferrin-gallium: effects on ribonucleotide reductase and demonstration of drug synergy with hydroxyurea. Blood. 1988;72:1930–1936. [PubMed] [Google Scholar]
- 26.Chitambar CR. Narasimhan J. Guy J. Sem DS. O'Brien WJ. Inhibition of ribonucleotide reductase by gallium in murine leukemic L1210 cells. Cancer Res. 1991;51:6199–6201. [PubMed] [Google Scholar]
- 27.Chitambar CR. Purpi DP. A novel gallium compound synergistically enhances bortezomib-induced apoptosis in mantle cell lymphoma cells. Leuk Res. 2010;34:950–953. doi: 10.1016/j.leukres.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chitambar CR. Purpi DP. Woodliff J. Yang M. Wereley JP. Development of gallium compounds for treatment of lymphoma: gallium maltolate, a novel hydroxypyrone gallium compound induces apoptosis and circumvents lymphoma cell resistance to gallium nitrate. J Pharmacol Exp Ther. 2007;322:1228–1236. doi: 10.1124/jpet.107.126342. [DOI] [PubMed] [Google Scholar]
- 29.Chitambar CR. Sax D. Regulatory effects of gallium on transferrin-independent iron uptake by human leukemic HL60 cells. Blood. 1992;80:505–511. [PubMed] [Google Scholar]
- 30.Chitambar CR. Seligman PA. Effects of different transferrin forms on transferrin receptor expression, iron uptake and cellular proliferation of human leukemic HL60 cells: mechanisms responsible for the specific cytotoxicity of transferrin-gallium. J Clin Invest. 1986;78:1538–1546. doi: 10.1172/JCI112746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chitambar CR. Wereley JP. Transferrin receptor-dependent and -independent iron transport in gallium-resistant human lymphoid leukemic cells. Blood. 1998;91:4686–4693. [PubMed] [Google Scholar]
- 32.Chitambar CR. Wereley JP. Expression of the hemochromatosis (HFE) gene modulates the cellular uptake of 67Ga. J Nucl Med. 2003;44:943–946. [PubMed] [Google Scholar]
- 33.Chitambar CR. Wereley JP. Haq RU. Synergistic inhibition of T-lymphoblastic leukemic CCRF-CEM cell growth by gallium and recombinant human α-interferon through action on cellular iron uptake. Cancer Res. 1994;54:3224–3228. [PubMed] [Google Scholar]
- 34.Chitambar CR. Wereley JP. Heiman T. Antholine WE. O'Brien WJ. Cellular adaptation to down-regulated iron transport into lymphoid leukaemic cells: effects on the expression of the gene for ribonucleotide reductase. Biochem J. 2000;345:681–685. [PMC free article] [PubMed] [Google Scholar]
- 35.Chitambar CR. Wereley JP. Matsuyama S. Gallium-induced cell death in lymphoma: role of transferrin receptor cycling, involvement of Bax and the mitochondria, and effects of proteasome inhibition. Mol Cancer Ther. 2006;5:2834–2843. doi: 10.1158/1535-7163.MCT-06-0285. [DOI] [PubMed] [Google Scholar]
- 36.Chitambar CR. Zahir SA. Ritch PS. Anderson T. Evaluation of continuous-infusion gallium nitrate and hydroxyurea in combination for the treatment of refractory non-Hodgkin's lymphoma. Am J Clin Oncol. 1997;20:173–178. doi: 10.1097/00000421-199704000-00015. [DOI] [PubMed] [Google Scholar]
- 37.Chitambar CR. Zivkovic Z. Inhibition of hemoglobin production by transferrin-gallium. Blood. 1987;69:144–149. [PubMed] [Google Scholar]
- 38.Chitambar CR. Zivkovic Z. Uptake of gallium-67 by human leukemic cells: demonstration of transferrin receptor-dependent and transferrin-independent mechanisms. Cancer Res. 1987;47:3929–3934. [PubMed] [Google Scholar]
- 39.Chitambar CR. Zivkovic-Gilgenbach Z. Role of the acidic receptosome in the uptake and retention of 67Ga by human leukemic HL60 cells. Cancer Res. 1990;50:1484–1487. [PubMed] [Google Scholar]
- 40.Chitambar CR. Zivkovic-Gilgenbach Z. Narasimhan J. Antholine WE. Development of drug resistance to gallium nitrate through modulation of cellular iron uptake. Cancer Res. 1990;50:4468–4472. [PubMed] [Google Scholar]
- 41.Chua MS. Bernstein LR. Li R. So SK. Gallium maltolate is a promising chemotherapeutic agent for the treatment of hepatocellular carcinoma. Anticancer Res. 2006;26:1739–1743. [PubMed] [Google Scholar]
- 42.Collery P. Domingo JL. Keppler BK. Preclinical toxicology and tissue gallium distribution of a novel antitumor gallium compound: tris (8-quinolinolato) gallium (III) Anticancer Res. 1996;16:687–692. [PubMed] [Google Scholar]
- 43.Collery P. Jakupec MA. Kynast B. Keppler BK. Preclinical and early clinical developemnt of the oral Gallium complex KP46 (FFC11) In: Alpoim MC, editor; Morais PV, editor; Santos MA, editor; Cristovao L, editor; Centeno JA, editor; Collery P, editor. Metal Ions in Biology and Medicine. Paris: John Libbey Eurotext; 2006. pp. 521–524. [Google Scholar]
- 44.Cory JG. Role of ribonucleotide reductase in cell division. In: Cory JG, editor; Cory AH, editor. Inhibitors of Ribonucleoside Diphosphate Reductase Activity. New York: Pergamon Press; 1989. pp. 1–16. [Google Scholar]
- 45.Davies NP. Rahmanto YS. Chitambar CR. Richardson DR. Resistance to the antineoplastic agent gallium nitrate results in marked alterations in intracellular iron and gallium trafficking: identification of novel intermediates. J Pharmacol Exp Ther. 2006;317:153–162. doi: 10.1124/jpet.105.099044. [DOI] [PubMed] [Google Scholar]
- 46.Davis SR. Cousins RJ. Metallothionein expression in animals: a physiological perspective on function. J Nutr. 2000;130:1085–1088. doi: 10.1093/jn/130.5.1085. [DOI] [PubMed] [Google Scholar]
- 47.DeConti RC. Toftness BR. Agrawal KC. Tomchick R. Mead JA. Bertino JR. Sartorelli AC. Creasey WA. Clinical and pharmacological studies with 5-hydroxy-2-formylpyridine thiosemicarbazone. Cancer Res. 1972;32:1455–1462. [PubMed] [Google Scholar]
- 48.Dreicer R. Propert KJ. Roth BJ. Einhorn LH. Loehrer PJ. Vinblastine, Ifosfamide, and Gallium Nitrate—an active new regimen in patients with advanced carcinoma of the urothelium. Cancer. 1997;79:110–114. [PubMed] [Google Scholar]
- 49.Einhorn L. Gallium nitrate in the treatment of bladder cancer. Semin Oncol. 2003;30:34–41. doi: 10.1016/s0093-7754(03)00174-x. [DOI] [PubMed] [Google Scholar]
- 50.Einhorn LH. Roth BJ. Ansari R. Dreicer R. Gonin R. Loehrer PJ. Phase II trial of vinblastine, ifosfamide, and gallium combination chemotherapy in metastatic urothelial carcinoma. J Clin Oncol. 1994;12:2271–2276. doi: 10.1200/JCO.1994.12.11.2271. [DOI] [PubMed] [Google Scholar]
- 51.Engstrom Y. Eriksson S. Jildevik I. Skog S. Thelander L. Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–9116. [PubMed] [Google Scholar]
- 52.Engstrom Y. Francke U. Assignment of the structural gene for subunit M1 of human ribonucleotide reductase to the short arm of chromosome 11. Exp Cell Res. 1985;158:477–483. doi: 10.1016/0014-4827(85)90470-7. [DOI] [PubMed] [Google Scholar]
- 53.Enyedy EA. Primik MF. Kowol CR. Arion VB. Kiss T. Keppler BK. Interaction of Triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): a comparative solution equilibrium study. Dalton Trans. 2011;40:5895–5905. doi: 10.1039/c0dt01835j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eriksson S. Gräslund A. Skog S. Thelander L. Tribukait B. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J Biol Chem. 1984;259:11695–11700. [PubMed] [Google Scholar]
- 55.Finch RA. Liu MC. Cory AH. Cory JG. Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): an inhibitor of ribonucleotide reductase with antineoplastic activity. Adv Enzyme Regul. 1999;39:3–12. doi: 10.1016/s0065-2571(98)00017-x. [DOI] [PubMed] [Google Scholar]
- 56.Fisher RI. Bernstein SH. Kahl BS. Djulbegovic B. Robertson MJ. de VS. Epner E. Krishnan A. Leonard JP. Lonial S. Stadtmauer EA. O'Connor OA. Shi H. Boral AL. Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 57.Foster BJ. Clagett-Carr K. Hoth D. Leyland-Jones B. Gallium nitrate: the second metal with clinical activity. Cancer Treat Rep. 1988;70:1311–1319. [PubMed] [Google Scholar]
- 58.Gallego B. Kaluderovic MR. Kommera H. Hey-Hawkins E. Paschke R. Kaluderovic GN. Novel gallium(III) complexes containing phthaloyl derivatives of neutral amino acids with apoptotic activity in cancer cells. J Organomet Chem. 2009;694:2191–2197. [Google Scholar]
- 59.Gallego B. Kaluderovic MR. Kommera H. Paschke R. Hey-Hawkins E. Remmerbach TW. Kaluderovic GN. Gomez-Ruiz S. Cytotoxicity, apoptosis and study of the DNA-binding properties of bi- and tetranuclear gallium(III) complexes with heterocyclic thiolato ligands. Invest New Drugs. 2011;29:932–944. doi: 10.1007/s10637-010-9449-8. [DOI] [PubMed] [Google Scholar]
- 60.Gräslund A. Sahlin M. Sjöberg B-M. The tyrosyl free radical in ribonucleotide reductase. Environ Health Perspect. 1985;64:139–149. doi: 10.1289/ehp.64-1568609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Green MA. Mathias CJ. Welch MJ. McGuire AH. Perry D. Fernandez-Rubio F. Perlmutter JS. Raichle ME. Bergmann SR. Copper-62-labeled pyruvaldehyde bis(N4-methylthiosemicarbazonato)copper(II): synthesis and evaluation as a positron emission tomography tracer for cerebral and myocardial perfusion. J Nucl Med. 1990;31:1989–1996. [PubMed] [Google Scholar]
- 62.Habashy HO. Powe DG. Staka CM. Rakha EA. Ball G. Green AR. Aleskandarany M. Paish EC. Douglas MR. Nicholson RI. Ellis IO. Gee JM. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat. 2010;119:283–293. doi: 10.1007/s10549-009-0345-x. [DOI] [PubMed] [Google Scholar]
- 63.Hall MD. Brimacombe KR. Varonka MS. Pluchino KM. Monda JK. Li J. Walsh MJ. Boxer MB. Warren TH. Fales HM. Gottesman MM. Synthesis and structure-activity evaluation of isatin-beta-thiosemicarbazones with improved selective activity toward multidrug-resistant cells expressing P-glycoprotein. J Med Chem. 2011;54:5878–5889. doi: 10.1021/jm2006047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hancock CN. Stockwin LH. Han B. Divelbiss RD. Jun JH. Malhotra SV. Hollingshead MG. Newton DL. A copper chelate of thiosemicarbazone NSC 689534 induces oxidative/ER stress and inhibits tumor growth in vitro and in vivo. Free Radic Biol Med. 2011;50:110–121. doi: 10.1016/j.freeradbiomed.2010.10.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haq RU. Chitambar CR. Modulation of transferrin receptor mRNA by transferrin-gallium in human myeloid HL60 cells and lymphoid CCRF-CEM cells. Biochem J. 1993;294:873–877. doi: 10.1042/bj2940873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harris AW. Sephton RG. Transferrin promotion of 67Ga and 59Fe uptake by cultured mouse myeloma cells. Cancer Res. 1977;37:3634–3638. [PubMed] [Google Scholar]
- 67.Hata Y. Sandler A. Loehrer PJ. Sledge GW., Jr. Weber G. Synergism of taxol and gallium nitrate in human breast carcinoma cells: schedule dependency. Oncol Res. 1994;6:19–24. [PubMed] [Google Scholar]
- 68.Hentze MW. Muckenthaler MU. Galy B. Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 69.Hoffer P. Status of gallium-67 in tumor detection. J Nucl Med. 1980;21:394–398. [PubMed] [Google Scholar]
- 70.Hofheinz RD. Dittrich C. Jakupec MA. Drescher A. Jaehde U. Gneist M. Graf von KN. Keppler BK. Hochhaus A. Early results from a phase I study on orally administered tris(8-quinolinolato)gallium(III) (FFC11, KP46) in patients with solid tumors—a CESAR study (Central European Society for Anticancer Drug Research—EWIV) Int J Clin Pharmacol Ther. 2005;43:590–591. doi: 10.5414/cpp43590. [DOI] [PubMed] [Google Scholar]
- 71.Huang ML. Lane DJ. Richardson DR. Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid Redox Signal. 2011;15:3003–3019. doi: 10.1089/ars.2011.3921. [DOI] [PubMed] [Google Scholar]
- 72.Hurd TR. Requejo R. Filipovska A. Brown S. Prime TA. Robinson AJ. Fearnley IM. Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jansson PJ. Sharpe PC. Bernhardt PV. Richardson DR. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: identification of pronounced redox activity and characterization of their antitumor activity. J Med Chem. 2010;53:5759–5769. doi: 10.1021/jm100561b. [DOI] [PubMed] [Google Scholar]
- 74.Kaluderovic MR. Gomez-Ruiz S. Gallego B. Hey-Hawkins E. Paschke R. Kaluderovic GN. Anticancer activity of dinuclear gallium(III) carboxylate complexes. Eur J Med Chem. 2010;45:519–525. doi: 10.1016/j.ejmech.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 75.Kaluderovic MR. Kaluderovic GN. Gomez-Ruiz S. Paschke R. Hemprich A. Kuhling J. Remmerbach TW. Organogallium(III) complexes as apoptosis promoting anticancer agents for head and neck squamous cell carcinoma (HNSCC) cell lines. J Inorg Biochem. 2011;105:164–170. doi: 10.1016/j.jinorgbio.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 76.Kane RC. Dagher R. Farrell A. Ko CW. Sridhara R. Justice R. Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 77.Klaassen CD. Liu J. Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol. 1999;39:267–294. doi: 10.1146/annurev.pharmtox.39.1.267. [DOI] [PubMed] [Google Scholar]
- 78.Knorr GM. Chitambar CR. Gallium-pyridoxal isonicotinoyl hydrazone (Ga-PIH), a novel cytotoxic gallium complex. A comparative study with gallium nitrate. Anticancer Res. 1998;18:1733–1738. [PubMed] [Google Scholar]
- 79.Kolesar JM. Sachidanandam K. Schelman WR. Eickhoff J. Holen KD. Traynor AM. Alberti DB. Thomas JP. Chitambar CR. Wilding G. Antholine WE. Cytotoxic Evaluation of 3-Aminopyridine-2-Carboxaldehyde Thiosemicarbazone, 3-AP, in Peripheral Blood Lymphocytes of Patients with Refractory Solid Tumors using Electron Paramagnetic Resonance. Exp Ther Med. 2011;2:119–123. doi: 10.3892/etm.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kolesar JM. Schelman WR. Geiger PG. Holen KD. Traynor AM. Alberti DB. Thomas JP. Chitambar CR. Wilding G. Antholine WE. Electron paramagnetic resonance study of peripheral blood mononuclear cells from patients with refractory solid tumors treated with Triapine. J Inorg Biochem. 2008;102:693–698. doi: 10.1016/j.jinorgbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kowol CR. Berger R. Eichinger R. Roller A. Jakupec MA. Schmidt PP. Arion VB. Keppler BK. Gallium(III) and iron(III) complexes of alpha-N-heterocyclic thiosemicarbazones: synthesis, characterization, cytotoxicity, and interaction with ribonucleotide reductase. J Med Chem. 2007;50:1254–1265. doi: 10.1021/jm0612618. [DOI] [PubMed] [Google Scholar]
- 82.Kowol CR. Reisner E. Chiorescu I. Arion VB. Galanski M. Deubel DV. Keppler BK. An electrochemical study of antineoplastic gallium, iron and ruthenium complexes with redox noninnocent alpha-N-heterocyclic chalcogensemicarbazones. Inorg Chem. 2008;47:11032–11047. doi: 10.1021/ic8013249. [DOI] [PubMed] [Google Scholar]
- 83.Kowol CR. Trondl R. Heffeter P. Arion VB. Jakupec MA. Roller A. Galanski M. Berger W. Keppler BK. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J Med Chem. 2009;52:5032–5043. doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- 84.Kraker A. Krezoski S. Schneider J. Minkel D. Petering DH. Reaction of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazonato) Cu(II) with Ehrlich cells. Binding of copper to metallothionein and its relationship to zinc metabolism and cell proliferation. J Biol Chem. 1985;260:13710–13718. [PubMed] [Google Scholar]
- 85.Kratz F. Nuber B. Weis J. Keppler BK. Synthesis and characterization of potential antitumor and antiviral gallium(III) complexes of a-(N)-heterocyclic thiosemicarbazones. Synth React Inorg Met-Org Chem. 1991;21:1601–1615. [Google Scholar]
- 86.Kvaloy S. Langholm R. Kaalhus O. Michaelsen T. Funderud S. Foss AA. Godal T. Transferrin receptor and B-lymphoblast antigen—their relationship to DNA synthesis, histology and survival in B-cell lymphomas. Int J Cancer. 1984;33:173–177. doi: 10.1002/ijc.2910330204. [DOI] [PubMed] [Google Scholar]
- 87.Larson SM. Rasey JS. Allen DR. Nelson NJ. Grunbaum Z. Harp GD. Williams DL. Common pathway for tumor cell uptake of Gallium-67 and Iron-59 via a transferrin receptor. J Natl Cancer Inst. 1980;64:41–53. [PubMed] [Google Scholar]
- 88.Leyland-Jones B. Treatment of cancer-related hypercalcemia: the role of gallium nitrate. Semin Oncol. 2003;30:13–19. doi: 10.1016/s0093-7754(03)00171-4. [DOI] [PubMed] [Google Scholar]
- 89.Lovejoy DB. Jansson PJ. Brunk UT. Wong J. Ponka P. Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011;71:5871–5880. doi: 10.1158/0008-5472.CAN-11-1218. [DOI] [PubMed] [Google Scholar]
- 90.Lundberg JL. Chitambar CR. Interaction of gallium nitrate with fludarabine and iron chelators: effects on the proliferation of human leukemic HL60 cells. Cancer Res. 1990;50:6466–6470. [PubMed] [Google Scholar]
- 91.Marzilli LG. de Castro B. Caradonna JP. Stewart RC. Van Vuuren CP. Nucleoside complexing. A Raman and 13C NMR spectroscopic study of the binding of hard and soft metal species. J Am Chem Soc. 1980;102:916–924. [Google Scholar]
- 92.Mendes IC. Soares MA. Dos Santos RG. Pinheiro C. Beraldo H. Gallium(III) complexes of 2-pyridineformamide thiosemicarbazones: cytotoxic activity against malignant glioblastoma. Eur J Med Chem. 2009;44:1870–1877. doi: 10.1016/j.ejmech.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 93.Merryweather-Clarke AT. Pointon JJ. Shearman JD. Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet. 1997;34:275–278. doi: 10.1136/jmg.34.4.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller LD. Coffman LG. Chou JW. Black MA. Bergh J. D'Agostino R., Jr. Torti SV. Torti FM. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71:6728–6737. doi: 10.1158/0008-5472.CAN-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Myers JM. Antholine WE. Zielonka J. Myers CR. The iron-chelating drug triapine causes pronounced mitochondrial thiol redox stress. Toxicol Lett. 2011;201:130–136. doi: 10.1016/j.toxlet.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 96.Narasimhan J. Antholine WE. Chitambar CR. Effect of gallium on the tyrosyl radical of the iron-dependent M2 subunit of ribonucleotide reductase. Biochem Pharmacol. 1992;44:2403–2408. doi: 10.1016/0006-2952(92)90686-d. [DOI] [PubMed] [Google Scholar]
- 97.Narasimhan J. Antholine WE. Chitambar CR. Petering DH. Inhibition of iron uptake in HL60 cells by 2-formylpyridine monothiosemicarbazonato Cu(II) Arch Biochem Biophys. 1991;289:393–398. doi: 10.1016/0003-9861(91)90429-m. [DOI] [PubMed] [Google Scholar]
- 98.Novick SC. Julian TN. Majuru S. Mangelus M. Brown BD. Mehta B. Warrell RP. Initial phase I clinical and pharmacokinetic assessment of G4544, an oral gallium-containing compound. J Clin Oncol. 2008;26:8592. [Google Scholar]
- 99.Nyholm S. Mann GJ. Johansson AG. Bergeron RJ. Gräslund A. Thelander L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J Biol Chem. 1993;268:26200–26205. [PubMed] [Google Scholar]
- 100.Perchellet EM. Ladesich JB. Collery P. Perchellet JP. Microtubule-disrupting effects of gallium chloride in vitro. Anticancer Drugs. 1999;10:477–488. doi: 10.1097/00001813-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 101.Petering DH. Physicochemical properties of the antitumor agent, 3 ethoxy 2 oxobutyraldehyde bis(thiosemicarbazonato) copper(II) Bioinorg Chem. 1972;1:255–271. [Google Scholar]
- 102.Petering DH. Antholine WE. Saryan LA. Metal complexes as antitumor agents. In: Ottenbrite RM, editor; Butler GB, editor. Anticancer and Interferon Agents. New York and Basel: Marcel Dekker, Inc.; 1984. pp. 203–246. [Google Scholar]
- 103.Petering HG. Buskirk HH. Underwood GE. The anti-tumor activity of 2-keto-3ethoxybutyraldehyde bis(thiosemicarbazone) and related compounds. Cancer Res. 1964;24:367–372. [PubMed] [Google Scholar]
- 104.Pinnix ZK. Miller LD. Wang W. D'Agostino R., Jr. Kute T. Willingham MC. Hatcher H. Tesfay L. Sui G. Di X. Torti SV. Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43–56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Popovic-Bijelic A. Kowol CR. Lind ME. Luo J. Himo F. Enyedy EA. Arion VB. Gräslund A. Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): a combined experimental and theoretical study. J Inorg Biochem. 2011;105:1422–1431. doi: 10.1016/j.jinorgbio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Poulsen CB. Borup R. Borregaard N. Nielsen FC. Moller MB. Ralfkiaer E. Prognostic significance of metallothionein in B-cell lymphomas. Blood. 2006;108:3514–3519. doi: 10.1182/blood-2006-04-015305. [DOI] [PubMed] [Google Scholar]
- 107.Pro B. Bociek RG. Chitambar CR. Gregory SA. Leonard JP. Smith S. Novick S. Phase 2 multicenter trial of gallium nitrate in patients with advanced non-Hodgkin's lymphoma (NHL) Blood. 2004;104:682A. [Google Scholar]
- 108.Rasey JS. Nelson NJ. Larson SM. Tumor cell toxicity of stable gallium nitrate: enhancement by transferrin and protection by iron. Eur J Cancer Clin Oncol. 1982;l8:66l–668. doi: 10.1016/0277-5379(82)90212-7. [DOI] [PubMed] [Google Scholar]
- 109.Richardson DR. Kalinowski DS. Richardson V. Sharpe PC. Lovejoy DB. Islam M. Bernhardt PV. 2-Acetylpyridine thiosemicarbazones are potent iron chelators and antiproliferative agents: redox activity, iron complexation and characterization of their antitumor activity. J Med Chem. 2009;52:1459–1470. doi: 10.1021/jm801585u. [DOI] [PubMed] [Google Scholar]
- 110.Richardson DR. Sharpe PC. Lovejoy DB. Senaratne D. Kalinowski DS. Islam M. Bernhardt PV. Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J Med Chem. 2006;49:6510–6521. doi: 10.1021/jm0606342. [DOI] [PubMed] [Google Scholar]
- 111.Richardson DR. Tran EH. Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995;86:4295–4306. [PubMed] [Google Scholar]
- 112.Roy CN. Enns CA. Iron homeostasis: new tales from the crypt. Blood. 2000;96:4020–4027. [PubMed] [Google Scholar]
- 113.Roy CN. Penny DM. Feder JN. Enns CA. The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J Biol Chem. 1999;274:9022–9028. doi: 10.1074/jbc.274.13.9022. [DOI] [PubMed] [Google Scholar]
- 114.Rudnev AV. Foteeva LS. Kowol C. Berger R. Jakupec MA. Arion VB. Timerbaev AR. Keppler BK. Preclinical characterization of anticancer gallium(III) complexes: solubility, stability, lipophilicity and binding to serum proteins. J Inorg Biochem. 2006;100:1819–1826. doi: 10.1016/j.jinorgbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 115.Seidman AD. Scher HI. Heinemann MH. Bajorin DF. Sternberg CN. Dershaw DD. Silverberg M. Bosl GJ. Continuous infusion gallium nitrate for patients with advanced refractory urothelial tumors. Cancer. 1991;68:2561–2565. doi: 10.1002/1097-0142(19911215)68:12<2561::aid-cncr2820681205>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 116.Seligman PA. Moran PL. Schleicher RB. Crawford ED. Treatment with gallium nitrate: evidence for interference with iron metabolism in vivo. Am J Hematol. 1992;41:232–240. doi: 10.1002/ajh.2830410403. [DOI] [PubMed] [Google Scholar]
- 117.Shakya R. Peng F. Liu J. Heeg MJ. Verani CN. Synthesis, structure, and anticancer activity of gallium(III) complexes with asymmetric tridentate ligands: growth inhibition and apoptosis induction of cisplatin-resistant neuroblastoma cells. Inorg Chem. 2006;45:6263–6268. doi: 10.1021/ic060106g. [DOI] [PubMed] [Google Scholar]
- 118.Shao J. Zhou B. Di Bilio AJ. Zhu L. Wang T. Qi C. Shih J. Yen Y. A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 119.Siegel R. Naishadham D. Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 120.Smith NW. Strutton GM. Walsh MD. Wright GR. Seymour GJ. Lavin MF. Gardiner RA. Transferrin receptor expression in primary superficial human bladder tumours identifies patients who develop recurrences. Br J Urol. 1990;65:339–344. doi: 10.1111/j.1464-410x.1990.tb14752.x. [DOI] [PubMed] [Google Scholar]
- 121.Spingarn NE. Sartorelli AC. Synthesis and evaluation of the thiosemicarbazone, dithiocarbazonate, and 2′-pyrazinylhydrazone of pyrazinecarboxaldehyde as agents for the treatment of iron overload. J Med Chem. 1979;22:1314–1316. doi: 10.1021/jm00197a007. [DOI] [PubMed] [Google Scholar]
- 122.Straus DJ. Gallium nitrate in the treatment of lymphoma. Semin Oncol. 2003;30:25–33. doi: 10.1016/s0093-7754(03)00173-8. [DOI] [PubMed] [Google Scholar]
- 123.Tennant DA. Duran RV. Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 124.Thelander L. Reichard P. Reduction of ribonucleotides. Ann Rev Biochem. 1979;48:133–158. doi: 10.1146/annurev.bi.48.070179.001025. [DOI] [PubMed] [Google Scholar]
- 125.Todd PA. Fitton A. Gallium nitrate: a review of its pharmacological properties and therapeutic potential in cancer-related hypercalcemia. Drugs. 1991;42:261–273. doi: 10.2165/00003495-199142020-00007. [DOI] [PubMed] [Google Scholar]
- 126.Torti SV. Torti FM. Ironing out cancer. Cancer Res. 2011;71:1511–1514. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tsui K-H. Feng T-H. Cheng P-L. Juang H-H. Gallium deregulates mitochondrial aconitase gene in human prostate carcinoma cells. Androl Update. 2007;1:304–312. [Google Scholar]
- 128.Valiahdi SM. Jakupec MA. Marculescu R. Keppler BK. Tris(8-quinolinolato)gallium(III) Exerts Strong Antiproliferative Effects in Melanoma Cells. Paris: Libbey Eurotext; 2006. pp. 282–286. [Google Scholar]
- 129.Vallabhajosula SR. Harwig JF. Siemsen JK. Wolf W. Radiogallium localization in tumors:Blood binding and transport and the role of transferrin. J Nucl Med. 1980;21:650–656. [PubMed] [Google Scholar]
- 130.Vavere AL. Lewis JS. Cu-ATSM: a radiopharmaceutical for the PET imaging of hypoxia. Dalton Trans. 2007:4893–4902. doi: 10.1039/b705989b. [DOI] [PubMed] [Google Scholar]
- 131.Waalkes TP. Sanders K. Smith RG. Adamson RH. DNA polymerases of Walker 256 carcinosarcoma. Cancer Res. 1974;34:385–391. [PubMed] [Google Scholar]
- 132.Warrell RP., Jr. Bockman RS. Coonley CJ. Isaacs M. Staszewski H. Gallium nitrate inhibits calcium resorption from bone and is effective treatment for cancer-related hypercalcemia. J Clin Invest. 1984;73:1487–1490. doi: 10.1172/JCI111353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Warrell RP., Jr. Coonley CJ. Straus DJ. Young CW. Treatment of patients with advanced malignant lymphoma using gallium nitrate administered as a seven-day continuous infusion. Cancer. 1983;51:1982–1987. doi: 10.1002/1097-0142(19830601)51:11<1982::aid-cncr2820511104>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 134.Warrell RP., Jr. Danieu L. Coonley CJ. Atkins C. Salvage chemotherapy of advanced lymphoma with investigational drugs: mitoguazone, gallium nitrate and etoposide. Cancer Treat Rep. 1987;71:47–51. [PubMed] [Google Scholar]
- 135.Whitnall M. Howard J. Ponka P. Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang DC. Wang F. Elliott RL. Head JF. Expression of transferrin receptor and ferritin H-chain mRNA are associated with clinical and histopathological prognostic indicators in breast cancer. Anticancer Res. 2001;21:541–549. [PubMed] [Google Scholar]
- 137.Yang M. Chitambar CR. Role of oxidative stress in the induction of metallothionein-2A and heme oxygenase-1 gene expression by the antineoplastic agent gallium nitrate in human lymphoma cells. Free Radic Biol Med. 2008;45:763–772. doi: 10.1016/j.freeradbiomed.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang M. Kroft SH. Chitambar CR. Gene expression analysis of gallium-resistant and gallium-sensitive lymphoma cells reveals a role for metal-responsive transcription factor-1, metallothionein-2A, and zinc transporter-1 in modulating the antineoplastic activity of gallium nitrate. Mol Cancer Ther. 2007;6:633–643. doi: 10.1158/1535-7163.MCT-06-0557. [DOI] [PubMed] [Google Scholar]
- 139.Yang-Feng TL. Barton DE. Thelander L. Lewis WH. Srinivasan PR. Francke U. Ribonucleotide reductase M2 subunit sequences mapped to four different chromosomal sites in humans and mice: functional locus identified by its amplification in hydroxyurea-resistant cell lines. Genomics. 1987;1:77–86. doi: 10.1016/0888-7543(87)90108-x. [DOI] [PubMed] [Google Scholar]
- 140.Yu Y. Kalinowski DS. Kovacevic Z. Siafakas AR. Jansson PJ. Stefani C. Lovejoy DB. Sharpe PC. Bernhardt PV. Richardson DR. Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. J Med Chem. 2009;52:5271–5294. doi: 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- 141.Yu Y. Kovacevic Z. Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–1994. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- 142.Yu Y. Suryo RY. Hawkins CL. Richardson DR. The potent and novel thiosemicarbazone chelators di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone and 2-benzoylpyridine-4,4-dimethyl-3-thiosemicarbazone affect crucial thiol systems required for ribonucleotide reductase activity. Mol Pharmacol. 2011;79:921–931. doi: 10.1124/mol.111.071324. [DOI] [PubMed] [Google Scholar]
- 143.Yu Y. Wong J. Lovejoy DB. Kalinowski DS. Richardson DR. Chelators at the cancer coalface: desferrioxamine to Triapine and beyond. Clin Cancer Res. 2006;12:6876–6883. doi: 10.1158/1078-0432.CCR-06-1954. [DOI] [PubMed] [Google Scholar]
- 144.Yu Z. Persson HL. Eaton JW. Brunk UT. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med. 2003;34:1243–1252. doi: 10.1016/s0891-5849(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 145.Zanias S. Papaefstathiou GS. Raptopoulou CP. Papazisis KT. Vala V. Zambouli D. Kortsaris AH. Kyriakidis DA. Zafiropoulos TF. Synthesis, structure, and antiproliferative activity of three gallium(III) azole complexes. Bioinorg Chem Appl. 2010;2010:168030. doi: 10.1155/2010/168030. [DOI] [PMC free article] [PubMed] [Google Scholar]