

Abstract
Numerous inflammatory conditions are associated with elevated YKL‐40 expression by infiltrating macrophages. Thus, we were surprised to observe minimal macrophage and abundant astrocyte expression of YKL‐40 in neuroinflammatory conditions. The aims of the current study were to better delineate this discrepancy, characterize the factors that regulate YKL‐40 expression in macrophages and astrocytes and study whether YKL‐40 expression correlates with cell morphology and/or activation state. In vitro, macrophages expressed high levels of YKL‐40 that was induced by classical activation and inhibited by alternative activation. Cytokines released from macrophages induced YKL‐40 transcription in astrocytes that was accompanied by morphological changes and altered astrocytic motility. Because coculturing of astrocytes and macrophages did not reverse this in vitro expression pattern, additional components of the in vivo central nervous system (CNS) milieu must be required to suppress macrophage and induce astrocyte expression of YKL‐40.
Keywords: astrocytes, macrophages, neuroinflammation, YKL‐40
INTRODUCTION
YKL‐40 [chitinase 3‐like protein 1 (CHI3L1), human cartilage glycoprotein 39 (HC‐gp39), gp38k, chondrex, breast regression protein 39 (BRP‐39)] is a member of the glycosyl hydrolase family 18; however, it does not possess hydrolase activity (19). YKL‐40 is expressed and released by chondrocytes, synovial cells, neutrophils and macrophages during cellular differentiation 19, 24, 45. YKL‐40 is upregulated in inflamed tissues of ulcerative colitis, Crohn's disease, rheumatoid arthritis, osteoarthritis, asthma, chronic obstructive pulmonary disease and liver cirrhosis, as well as solid cancers 7, 8, 9, 15, 17, 19, 28, 31, 41, 43. It has been shown by us and others that differentiated macrophages express YKL‐40 in vitro 3, 27, 45. In vivo, peritumoral macrophages in small cell lung cancer (21) and macrophages in atherosclerotic plaques (38) and in a murine asthma model (30) express YKL‐40.
The physiological role and specific cell surface receptor are not known; however, YKL‐40 has been hypothesized to be involved in tissue remodeling during inflammation. In a recent study, BRP‐39 knockout mice showed a blunted immune response to allergic sensitization [eg, decreased accumulation of dendritic cells in the lungs, decreased immunoglobulin E (IgE) production, increased percentage of apoptotic T cells and macrophages in the bronchoalveolar lavage] accompanied by reduced peribronchial fibrosis and collagen content (30).
Our recent studies showed significant elevation of YKL‐40 in the cerebrospinal fluid (CSF) of a variety of acute [eg, lentiviral encephalitis, brain infarction and traumatic brain injury (TBI)] and chronic neuroinflammatory conditions [eg, multiple sclerosis (MS)]3, 4, 5. CSF YKL‐40 levels were higher in patients who eventually died following TBI than in patients who survived. YKL‐40 levels significantly correlated with CSF levels of inflammatory cytokines like IL‐1β and TNFα, as well as the inflammatory marker C‐reactive protein. Controlled cortical impact in rats showed that YKL‐40 transcription was primarily associated with reactive astrocytes in pericontusional cortex (5). Time course analysis showed that YKL‐40 transcription in astrocytes began 1 day after injury, remained elevated for several days and then declined by day 12 and was coincident with IL‐1β expression. Taken together, these findings demonstrated that YKL‐40 was induced in astrocytes during acute neuroinflammation and was temporally related to inflammatory mediator expression. It was not clear why in vivo YKL‐40 transcription in the brain was less evident in activated tissue macrophages/microglia. The current in vitro study was carried out to identify mediators of YKL‐40 transcription in astrocytes and macrophages and to begin assessing functional changes associated with YKL‐40 expression in those cells.
MATERIALS AND METHODS
Materials
IL‐2, IL‐6, IL‐12, IL‐13, IL‐17 and IL‐18 were purchased from Cell Sciences (Canton, MA, USA). IFNγ, IL‐1β, IL‐1 receptor antagonist, TNFα and recombinant basic fibroblast growth factor (bFGF) were purchased from R&D Systems (Minneapolis, MN, USA). The NF‐κB inhibitors Bay 11‐7082 and IKKγ NEMO binding domain were purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA) and AnaSpec (Fremont, CA, USA), respectively. sIL‐6 receptor was purchased from PeproTech (Rocky Hill, NJ, USA). Purified YKL‐40 was purchased from Quidel Corporation (San Diego, CA, USA). IL‐4 and all other chemicals were purchased from Sigma‐Aldrich (St. Louis. MO, USA).
Human specimens
Paraffin sections from a wide variety of human neuroinflammatory conditions were used for immunohistochemical and in situ hybridization (ISH) analysis of YKL‐40 expression. As a control for expression during gliosis in the absence of neuroinflammation, cases of the spongiform encephalopathy Creutzfeldt–Jakob disease (CJD) were assessed. For illustration purposes, a case of acute bacterial meningitis, progressive multifocal leukoencephalopathy (PML), posterior reversible encephalopathy syndrome (PRES) and CJD are shown. All human studies were approved by our institutional review board.
Immunofluorescence
Glial fibrillary acidic protein (GFAP) staining was performed using polyclonal rabbit antihuman GFAP (1:500; DAKO, Carpinteria, CA, USA). CD68 staining was performed using monoclonal mouse antihuman CD68 (1:100; DAKO). Iba‐1 staining was performed using polyclonal rabbit anti‐Iba‐1 (1:500; Wako, Richmond, VA, USA). YKL‐40 staining was performed using goat antihuman CHI3L1 (1:100; R&D Systems) followed by tyramine signal amplification as described previously (55). All primary antibodies were followed by goat anti‐rabbit/mouse Cy3 or goat anti‐rabbit/mouse Dylight 488 antibody (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA, USA).
ISH
Antisense YKL‐40 DNA templates containing the T7 promoter were generated by polymerase chain reaction (PCR) using primers corresponding to base pairs 19–241 from the pUC57 vector (GenScript, Piscataway, NJ, USA) containing the full length YKL‐40 cDNA. 35S‐labeled RNA probes were generated using MAXIscript in vitro transcription kit (Ambion, Austin, TX, USA). For ISH, cell cultures were fixed and then incubated in hybridization buffer (1X HYB buffer, 0.6 M NaCl, 10% dextran, 50 µg/mL tRNA, 0.1 M DTT) containing radiolabeled YKL‐40 probe (50 000 cpm/µL) at 50°C overnight. The following day, the cell cultures were washed and processed for immunofluorescence staining if required. Cell cultures were dipped in emulsion (Eastman Kodak company, Rochester, NY, USA) and exposed for 10 days at 4°C and then ISH signal was developed using D19 (Sigma‐Aldrich) and fixed by Rapid fix (Sigma‐Aldrich).
Primary human macrophage cultures
Macrophage cultures were obtained from human peripheral blood mononuclear cells isolated from HIV and hepatitis B seronegative buffy coats obtained from the Central Blood Bank (Pittsburgh, PA, USA) using Cellgro lymphocyte separation medium (Mediatech Inc., Herndon, VA, USA). Macrophages were differentiated in growth medium (AIM‐V, Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) in the presence of macrophage colony‐stimulating factor (M‐CSF) (10 ng/mL) for 3 days. At day 4 in culture, the medium was replaced with medium without M‐CSF. More than 95% of the cells were macrophages that stained for CD68.
YKL‐40 and TNFα enzyme‐linked immunosorbent assay (ELISA)
YKL‐40 or TNFα levels in the culture growth medium were determined, in duplicate, using the MicroVue YKL‐40 ELISA kit (Quidel Corporation) or human TNFα enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer's protocol. Absorbance was measured using a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Western blot analysis
Cells were lysed with CelLytic M (Sigma‐Aldrich) lysis buffer. Samples were separated on 12% Novex Tris‐glycine gels (Invitrogen), transferred to polyvinylidene difluoride membranes (Bio‐Rad Laboratories, Hercules, CA, USA), blocked with 5% milk or bovine serum albumin (Fisher Scientific, Pittsburgh, PA, USA) in phosphate‐buffered saline (PBS) containing Tween 20 and then incubated with either antihuman CHI3L1 (goat polyclonal, 1:50; R&D Systems) or anti‐β‐actin (mouse monoclonal, 1:5000; Abcam, Cambridge, MA, USA). Membranes were washed and incubated with species‐specific secondary antibodies tagged with horseradish peroxidase. A signal was visualized using SuperSignal West Pico chemiluminescent substrate system (Pierce, Rockford, IL, USA).
Luminex analysis
Human macrophages cultures from three donors were cultured as mentioned before. The release of inflammatory analytes into the growth medium over time was quantified with a Luminex 100 (Luminex Corporation, Austin, TX, USA) using microsphere‐based multiplexing technology. The human 27‐Plex kit from Bio‐Rad was composed of analyte‐specific components for the simultaneous measurement of the following human cytokines (IL‐1β, IL‐1Ra, IL‐2, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, IL‐9, IL‐10, IL‐12(p70), IL‐13, IL‐15, IL‐17, Eotaxin, bFGF, granulocyte colony‐stimulating factor (G‐CSF), granulocyte macrophage colony‐stimulating factor (GM‐CSF), IFNγ, CXCL10, CCL2, CCL3, CCL4, platelet‐derived growth factor‐BB (PDGF‐BB), CCL5, TNFα and vascular endothelial growth factor (VEGF).
YKL‐40 and IL‐1β real‐time PCR (RT‐PCR)
RNA was extracted from the cells using the QIAGEN RNeasy (Valencia, CA, USA) RNA extraction protocol. cDNA was generated using the RETROscript kit two step protocol (Ambion). YKL‐40 and IL‐1β transcription were measured using TaqMan Gene Expression Assays (Applied Biosystems, Carlsbad, CA, USA) with 2X TaqMan Gene Expression Master Mix (Applied Biosystems) and analyzed on an Applied Biosystems StepOne RT‐PCR machine.
Flow cytometry analysis
Primary human macrophage cultures were prepared as described previously. Cells were plated at a concentration of 19.7 × 106 cells per 75‐cm2 flask in Dulbecco's modified Eagle medium (DMEM) with 10% FBS. On days 4, 7, 11 in culture, media was either replaced or cells were harvested using 5 mL of Accutase (eBioscience, San Diego, CA, USA) at 37°C for approximately 45 minutes. During incubation with Accutase, flasks were frequently tapped to promote detachment. Approximately 12 h prior to harvesting, GolgiPlug (BD Biosciences, San Jose, CA, USA) was added to culture media.
Staining of macrophages for flow cytometry analysis
For surface antibodies, the following fluorochrome‐conjugated antibodies were used: CD3‐PerCP (BD Biosciences) and CD11b‐APC (eBioscience) and CD11c‐fluorescein isothiocyanate (FITC), HLA‐DR‐PerCP, CD14‐PerCP, CD16‐PerCP, CD206‐phycoerythrin (PE) (Biolegend, San Diego, CA, USA). Macrophages were stained as described previously 1, 14. Briefly, cells were washed and resuspended in PBS with 4% FCS and incubated for 20 minutes at 4°C followed by incubation with a cocktail of monoclonal antibodies for 30 minutes at 4°C. After washing, cells were prepared for intracellular staining using the BD Cytofix/Cytoperm (BD Biosciences) fixation and permeabilization solution according to the manufacturer's recommendations. For intracellular antibodies, the following fluorochrome‐conjugated antibodies were used: CD68‐FITC (eBioscience), YKL‐40 biotin‐conjugated (see following) and IL1‐β‐FITC, TNFα‐APC and IL12 p40/p70‐APC (Biolegend). Following the final wash, cells were fixed with 2% paraformaldehyde for analysis.
Intracellular YKL‐40 staining
A goat antihuman CHI3L1 (YKL‐40) polyclonal antibody (R&D Systems) was biotinylated using the One‐Step Antibody Biotinylation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) per manufacturer's recommendations. The staining protocol was as described previously with the exception of an additional incubation with anti‐biotin‐PE or anti‐biotin‐FITC antibodies (Miltenyi Biotec) following the intracellular staining step. Staining of YKL‐40‐expressing cells was validated by the following methods (Supporting Information Figure S1): +/− GolgiStop, +/− intracellular staining, anti‐biotin antibody only, dilution of the YKL‐40 antibody and intracellular staining of a known YKL‐40‐negative cell type (CD3‐positive lymphocytes). The anti‐biotin antibody showed minor binding to macrophages, but the YKL‐40‐expressing cells were sufficiently distinct to allow detection of YKL‐40 binding.
Flow cytometry
Sample acquisition was performed using a FACSCalibur (BD, Franklin Lakes, NJ, USA). A four‐color compensation matrix was created by six single‐stained macrophage samples. Macrophages were gated based on forward scatter and side scatter log characteristics. The YKL‐40‐positive and YKL‐40‐negative populations were then gated separately to determine expression of phenotypic markers. For each sample, 30 000 events were collected. Proper gating was established using appropriate isotype and fluorescence‐minus‐one controls. Data analysis and graphic representations were done using FlowJo v.8.8.4 (TreeStar, Ashland, OR, USA). Comparison of the fluorescence intensities of each phenotypic marker in the YKL‐40‐positive and YKL‐40‐negative populations was also performed with FlowJo using a probability binning algorithm for the multivariate parameter to calculate the T(x) value (47).
Primary human microglia cultures
Microglial cultures were isolated from adult human post‐mortem brains without history of a neurological disorder (age 52.6 ± 15.2 years; two female, five male; post‐mortem interval 21.2 ± 2.2 h). The specimens were received either from the Brain Tissue Donation Program of the Department of Psychiatry at the University of Pittsburgh or from the University of Pittsburgh Medical Center autopsy service. After removal of meninges, tissue from parietal cortex was sliced into small pieces and then homogenized using gentleMACS dissociator (Miltenyi Biotec, Auburn, CA, USA). Homogenates were incubated with TryplE (Invitrogen) and DNase (Worthington Biochemical, Lakewood, NJ, USA) for 45 minutes at 37°C and then filtered through a 100 µm mesh cell strainer (BD Biosciences, Franklin Lakes, NJ, USA). After centrifugation, the cell pellet was resuspended in 25% Percoll solution and layered over 75% Percoll solution. Following density centrifugation at 800 g for 30 minutes, the cells at the 25/75 interface were collected and plated at 1 × 106/mL in DMEM with high glucose and glutamine, 10% FBS and penicillin/streptomycin. Forty‐eight hours after plating, nonadherent cells were removed by washing. Using this protocol, the purity of microglial cultures is generally >99% as assessed by Iba‐1 and GFAP immunocytochemistry or lack of GFAP amplification by quantitative RT‐PCR.
Ten days after plating, cells were switched to FBS‐free medium for 48 h before induction of classical or alternative activation. Another 48 h later, supernatant and RNA were collected. Appropriate M1 and M2 induction was verified by assessment of chemokine secretion into the supernatant (25). M1‐stimulated cultures showed a 74.0 ± 20.5‐fold (mean ± standard deviation [SD]) increase in CXCL10 release (R&D Systems) compared with non‐stimulated control cells, whereas M2 cultures showed 17.5 ± 31.5‐fold induction of CCL17 release (R&D Systems) (25).
Primary human astrocyte cultures
Primary human astrocyte cultures were obtained from human fetal tissue collected as per University of Pittsburgh ethics and biosafety guidelines and established protocols as described previously (3). The cells were cultured in flasks with Dulbecco's modified Eagle's medium/F12 supplemented with 10% FBS (Mediatech). The cells were grown to 80% confluence, trypsinized, aliquoted and frozen in liquid nitrogen for further analysis. Each culture was characterized for the percent of intensely GFAP‐positive cells using immunohistochemistry and GFAP mRNA using RT‐PCR normalized to GAPDH. Only preparations with at least 80% of cells strongly stained for GFAP were used in these studies. CD68 and Iba‐1 staining confirmed that the passaged astrocyte cultures did not contain microglia contamination.
Scratch‐wound assay
Astrocytes were grown in 12‐well plates until confluent. The monolayer was scratched with a sterile plastic pipette tip. Duplicate wells were washed and growth medium was added in the absence or presence of YKL‐40 (6 µg/mL) or macrophage culture medium (MCM) collected at day 6 in vitro. After 24 h, the cells were fixed and stained with crystal violet. Ten random fields were captured from each duplicate well, and mean gap width along the scratch was assessed in each well. A one‐way analysis of variance followed by Bonferroni test was used to evaluate differences between groups.
RESULTS
Neuroinflammatory diseases showed abundant in vivo expression of YKL‐40 by reactive astrocytes but not macrophages/microglia
Paraffin sections of human brain tissue from patients that died of acute bacterial meningitis, PML, PRES and CJD were triple immunostained for YLK‐40, GFAP and CD68 (Figure 1). In bacterial meningitis, subpial gray matter showed considerable YKL‐40 staining associated with GFAP‐positive astrocytes but not with CD68‐positive microglia. PML demyelinated areas showed abundance of activated YKL‐40‐negative macrophages between YKL‐40‐positive reactive astrocytes. PRES showed reactive astrocytosis and microgliosis with YKL‐40 staining limited to astrocytic elements. The noninflammatory degenerative disease, CJD, showed abundant reactive astrocytosis with minimal microgliosis and the absence of any YKL‐40 staining.
Figure 1.

Localization of YKL‐40 in neuroinflammatory and neurodegenerative conditions. Paraffin sections of human brain tissue were triple immunostained for YLK‐40 (first column, green), GFAP (second column, red) and CD68 (third column, blue). The final column displays merged images. In bacterial meningitis (first row), subpial (and perivascular) gray matter astrocytes show reactive changes staining for GFAP (red) with a modest parenchymal activation of microglia expression of CD68 (blue). All YKL‐40 (green) staining is associated with the soma of reactiveastrocytes. In progressive multifocal leukoencephalopathy (PML), demyelinated areas consist of a sea of activated macrophages with interspersed reactive astrocytes. YKL‐40 staining is limited to the reactive astrocytes. In the third row, posterior reversible encephalopathy syndrome (PRES) shows reactive astrocytosis and microgliosis with YKL‐40 staining again limited to astrocytic elements. In the noninflammatory degenerative disease Creutzfeldt–Jakob disease (CJD), severe reactive astrocytosis with minimal microgliosis is seen in the absence of any YKL‐40 staining.
In bacterial meningitis, abundant immune cells were observed in the meninges with hematoxylin and eosin (H&E) staining (Figure 2A). ISH showed that those meningeal immune cells were strongly positive for IL‐1β mRNA (black grains; Figure 2B) and weakly positive for YKL‐40 mRNA (Figure 2C). In the parenchyma, only astrocytes were strongly positive for YKL‐40 mRNA (Figure 2C).
Figure 2.

YKL‐40 transcription in meningeal immune cells and parenchymal astrocytes in acute bacterial meningitis. Paraffin sections from acute bacterial meningitis were stained with H&E or hybridized with 35S‐labeled RNA probes for IL‐1β and YKL‐40 as described in Materials and Methods. Abundant immune cells in the meninges (A) are positive for IL‐1β mRNA (B). YKL‐40 ISH localized weakly with immune cells in the meninges and strongly with parenchymal astrocytes (C).
Macrophages in vitro express abundant YKL‐40
Primary human macrophage cultures were prepared as described in the Materials and Methods section. Immunohistochemical analysis showed that differentiating macrophages expressed YKL‐40 that was most pronounced in fully differentiated macrophages at day 10 in culture (Figure 3). MCM and lysates were collected at days 4–18 and subjected to YKL‐40 ELISA analysis or sodium dodecyl sulfate—polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blot analysis, respectively. The results showed that differentiating macrophages secreted YKL‐40 into the culture medium reaching a peak at day 10–13 in culture (Figure 4A). Western blot analysis showed constant cellular YKL‐40 levels over the first 9 days in culture that declined when the cells senesced in culture (Figure 4B). Luminex analysis showed that the concentration of macrophage secreted inflammatory factors diminished over time in culture. In particular, the concentration of the inflammatory cytokines IL‐1β (6.2 ± 0.9 pg/mL) and TNFα (114.8 ± 31.4 pg/mL) diminished at day 14 in culture to 1.8 ± 0.9 pg/mL and 7.8 ± 2.5 pg/mL, respectively (Figure 4C,D).
Figure 3.

YKL‐40 expression in differentiating macrophages. Primary human monocyte‐derived macrophages were grown in culture as described in Materials and Methods. The cells were fixed at day 4, 6, 8 and 10 in culture and stained with the macrophage marker CD68 and YKL‐40. The right panels show the merged images of CD68 and YKL‐40 staining.
Figure 4.

Time course of YKL‐40 and cytokine secretion in culture. Macrophages were grown as described in Materials and Methods. Time course of YKL‐40 levels released into the culture medium was determined by YKL‐40 ELISA kit (A). Cellular YKL‐40 levels were determined by Western blot analysis (B). Time course of IL‐1β (C) and TNFα (D) levels in the culture medium were determined by Luminex analysis. Data shown are mean ± SEM from three donors using triplicate samples.
YKL‐40 transcription in macrophages was induced by classical activation (M1) and inhibited by alternative activation (M2) but was not affected by anti‐inflammatory drugs
Treatment of macrophage cultures with known inducers of macrophage classical activation like IFNγ (20 ng/mL) and lipopolysaccharide (LPS) (100 ng/mL) induced YKL‐40 transcription by 9.3 ± 0.5‐fold at day 5 post‐stimulation (Figure 5A). Conversely, IL‐4 (20 ng/mL), an alternative macrophage activation inducer, inhibited YKL‐40 transcription by 90%. IL‐1β transcription was used as a measure for treatment effectiveness showing 558 ± 131 and 247.5 ± 40‐fold induction 24 h and 5 days post‐stimulation with IFNγ and LPS, respectively, and 90% inhibition with IL‐4 at day 5 post‐stimulation (Figure 5B). Treatment with the anti‐inflammatory drug dexamethasone (100 ng/mL) for 24 h inhibited YKL‐40, as well as IL‐1β transcription, while the anti‐inflammatory drugs simvastatin (100 ng/mL) and minocycline (100 µg/mL) did not affect YKL‐40 transcription in macrophages despite inhibiting IL‐1β transcription by 70% (Figure 5C,D).
Figure 5.

YKL‐40 transcription in macrophages is induced by classical activation (M1) and inhibited by alternative activation (M2). Macrophages were grown as described in Materials and Methods and were treated with IFNγ (20 ng/mL) and LPS (100 ng/mL) or IL‐4 (20 ng/mL) for 1–5 days, then RNA was extracted and assessed for YKL‐40 (A) and IL‐1β (B) transcription by real‐time PCR. Macrophages were treated with dexamethasone (100 ng/mL), simvastatin (100 ng/mL) or minocycline (100 µg/mL) for 24 h, then RNA was extracted and assessed for YKL‐40 (C) and IL‐1β (D) transcription by real‐time PCR. Data shown are from a single experiment using duplicate samples and are representative of three independent experiments.
YKL‐40 transcription in microglia in vitro was minimally changed by classical or alternative activation
Primary human microglial cultures were prepared as described in the Materials and Methods section. Classical activation with IFNγ (20 ng/mL) and LPS (100 ng/mL) induced minimal induction of YKL‐40 transcription after 48 h (1.3 ± 0.3‐fold increase compared with non‐stimulated cells), while alternate activation with IL‐4 (20 ng/mL) inhibited YKL‐40 transcription by 46% (Figure 6A). These changes in gene expression were associated with similar changes in YKL‐40 release into the supernatant with a 2.2 ± 1.7‐fold induction following classical activation and a −1.5 ± 1.9 decrease following alternative activation (data not shown). Conversely, IL‐1β transcription showed 408 ± 125‐fold induction 48 h poststimulation with IFNγ and LPS with no significant inhibition with IL‐4 (Figure 6B).
Figure 6.

Minimal induction of YKL‐40 transcription in microglial cultures by classical activation. Microglia were grown as described in Materials and Methods and were treated with IFNγ (20 ng/mL) and LPS (100 ng/mL) or IL‐4 (20 ng/mL) for 48 h, then RNA was extracted and assessed for YKL‐40 (A) and IL‐1β (B) transcription by real‐time PCR.
Correlation between YKL‐40 expression and activation state in macrophages
As YKL‐40 production in macrophages increases as a function of time in culture, macrophage phenotypic markers were examined in macrophages from days 4, 7 and 11 in culture to determine if YKL‐40‐positive macrophages showed a distinct signature compared with YKL‐40‐negative macrophages. The percentage of macrophages expressing YKL‐40 increased from 34% on day 4 in culture, peaked at 89% on day 7 and decreased to 67% by day 11, while the intensity of YKL‐40 expression increased slightly from day 4 to 7 (MFI = 125 vs. 147) in culture and decreased by day 11 (MFI = 50.2) (Table 1). Of the markers examined, only CD206 (M2 marker) expression robustly distinguished the YKL‐40‐positive and YKL‐40‐negative populations at all time points, with no CD206 expression on YKL‐40‐negative macrophages (Figure 7). Despite comparable TNFα (M1 marker) expression for YKL‐40‐positive and YKL‐40‐negative populations on day 4, a greater percentage of YKL‐40‐positive macrophages retained higher TNFα expression through days 7 and 11. Conversely, the intensity of CD14 and HLA‐DR expression decreased only on YKL‐40‐positive macrophages on days 7 and 11. IL‐1β expression was not detected in either population, while IL‐12 p40/p70 (M1 marker) expression was observed only on day 4 in both YKL‐40‐positive and YKL‐40‐negative populations. Similarly, CD16 and CD68 did not clearly distinguish the different populations.
Table 1.
Median fluorescence intensity (MFI) for YKL40‐positive and YKL40‐negative macrophages. Macrophages were stained with anti‐YKL40 and antibodies against CD14, IL‐1β, TNFα, IL‐12 p40/p70, CD206, CD16, CD68, HLA‐DR and isotype controls. Macrophages were gated based on forward scatter and side scatter log parameters. Additional gates for YKL40‐positive and YKL40‐negative macrophage populations were established using isotype and fluorescence‐minus‐one controls. The T(x) values were determined to compare the fluorescence intensities of each marker between the YKL40‐positive and YKL40‐negative populations. Each comparison had a T(x) value >4, with the exception of IL‐1β. A value T(x) >4 implies the two distributions are different with a P < 0.01.
| D4 | D7 | D11 | Control | ||||
|---|---|---|---|---|---|---|---|
| YKL‐40+ | YKL‐40− | YKL‐40+ | YKL‐40− | YKL‐40+ | YKL‐40− | — | |
| YKL‐40 | 125 | 9 | 147 | 7 | 50 | 4 | 1 |
| IL1β | 16 | 16 | 17 | 18 | 28 | 24 | 23 |
| TNFα | 305 | 252 | 42 | 4 | 55 | 6 | 5 |
| CD14 | 252 | 370 | 70 | 293 | 103 | 389 | — |
| IL12 p40/p70 | 43 | 35 | 3 | 4 | 4 | 5 | 3 |
| CD206 | 71 | 17 | 35 | 10 | 57 | 10 | — |
| CD68 | 38 | 31 | 87 | 79 | 55 | 40 | — |
| CD16 | 6 | 14 | 114 | 72 | 17 | 41 | — |
| CD11b | 825 | 700 | 858 | 341 | 136 | 41 | — |
| CD11c | 258 | 222 | 323 | 302 | 386 | 364 | — |
| HLA‐DR | 1233 | 1019 | 429 | 922 | 260 | 943 | — |
Figure 7.
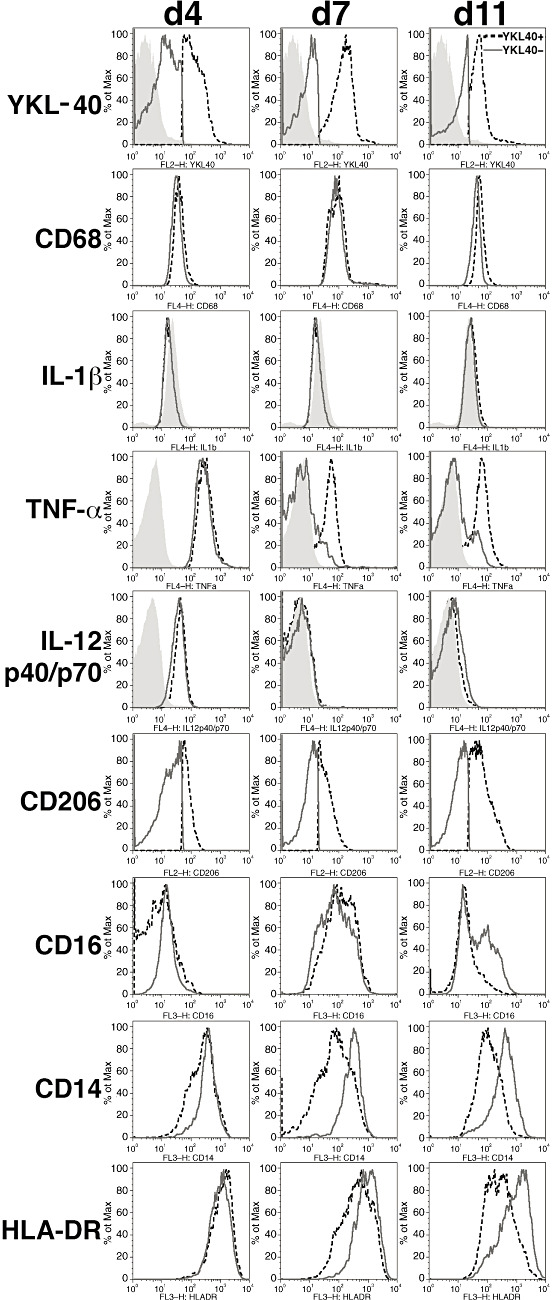
CD206, TNFα, HLA‐DR and CD14 expression distinguish YKL‐40‐positive from YKL‐40‐negative macrophages. Macrophages were stained with anti‐YKL‐40 and antibodies against CD14, IL‐1β, TNFα, IL‐12 p40/p70, CD206, CD16, CD68, HLA‐DR and isotype controls. Macrophages were gated based on forward scatter and side scatter log parameters. Additional gates for YKL‐40‐positive and YKL‐40‐negative macrophages populations were established using isotype and fluorescence‐minus‐one controls. Staining for each phenotypic marker listed on the left was evaluated for YKL‐40‐positive (black‐dashed line) and YKL‐40‐negative (dark gray solid line) macrophages on days 4, 7 and 11 in culture. Control staining is represented by the filled histogram.
YKL‐40 transcription was induced in astrocytes by proinflammatory mediators released from macrophages
Astrocytes were exposed to MCM collected at different days in culture for 24 h, and then RNA was extracted and used to measure YKL‐40 transcription by RT‐PCR. MCM collected after 4–9 days in culture induced a 7 to 18‐fold induction of YKL‐40 transcription compared with control non‐treated astrocytes (Figure 8A). With more prolonged macrophage culture (MCM collected at days 10–17), this induction was substantially reduced. Exposure of astrocytes to MCM collected from M1‐ or M2‐differentiated macrophages showed 278% induction and 66.5% inhibition of YKL‐40 transcription, respectively (Figure 8B). In order to identify mediators, we screened a panel of cytokines for their ability to stimulate YKL‐40 transcription in astrocytes compared with MCM. Primary astrocytes were treated with a variety of proinflammatory interleukins (10 ng/mL of IL‐1β, IL‐2, IL‐6, IL‐12, IL‐13, IL‐17 and IL‐18) and other cytokines and mediators like TNFα and IFNγ for 24 h, then RNA was extracted and YKL‐40 transcription was evaluated by RT‐PCR. This analysis showed that IL‐1β induced YKL‐40 transcription (19.4‐fold) although not as robust as MCM (51‐fold). TNFα induced a more modest YKL‐40 transcription (4.8‐fold) (Figure 8C). Other cytokines and mediators did not induce substantial YKL‐40 transcription compared with control non‐treated astrocytes. Dose response analysis showed that IL‐1β could induce YKL‐40 transcription after 24 h at low concentrations of 0.1 pg/mL, while TNFα began inducing YKL‐40 transcription at 10 000 pg/mL (Figure 8D). The kinetics of IL‐1β‐induced YKL‐40 transcription showed that acute treatment (24 h) resulted in a transient YKL‐40 induction peaking at day 4 post‐stimulation and declining to control levels by day 7 post‐stimulation. Prolonged IL‐1β stimulation (7 days) showed an initial 12‐fold induction of YKL‐40 transcription followed by an 85‐fold induction at day 7 (Figure 8E). IL‐1β‐induced YKL‐40 transcription was completely inhibited by IL‐1 receptor antagonist (2 µg/mL), and TNFα‐induced transcription was completely inhibited by the TNFα antagonist Enbrel® (Amgen, Thousand Oaks, CA, USA) (1 mg/mL) (Figure 8F). In order to study if IL‐1β and TNFα were responsible for all of the stimulatory activity found in the MCM, we tested whether IL‐1 receptor antagonist and TNFα antagonist Enbrel® were able to inhibit MCM‐induced YKL‐40 transcription in astrocytes. As those cytokines are known to stimulate NF‐κB cell signaling, we also tested two NF‐κB cell signaling inhibitors [Bay 11‐7082 (660 ng/mL) and IKK inhibitor (2 mM)]. IL‐1 receptor antagonist, Enbrel® and the combined treatment of both inhibited YKL‐40 transcription by 21%, 31.5% and 35%, respectively. Bay 11‐7082 and IKK inhibitor inhibited YKL‐40 transcription by 72% and 91%, respectively (Figure 8G). Thus, other MCM mediators in addition to IL‐1β and TNFα could induce YKL‐40 transcription in astrocytes through NF‐κB stimulation.
Figure 8.

YKL‐40 transcription is induced in astrocytes by mediators released from differentiating macrophages. Astrocytes were treated for 24 h with macrophage culture medium (MCM) collected at different time points in culture, then RNA was extracted from the astrocytes and assessed for YKL‐40 transcription by real‐time PCR (A). Non‐treated or non‐stimulated or M1‐ and M2‐stimulated MCM was used to treat astrocytes for 24 h, then RNA was extracted and analyzed for YKL‐40 transcription by real‐time PCR (B). Astrocytes were treated with MCM, IL‐1β, TNFα, IL‐2, IL‐6 in the presence of sIL‐6R, IL‐12, IL‐13, IL‐17, IL‐18, IFNγ, LPS or bFGF (10 ng/mL, 24 h) and assessed for YKL‐40 transcription by real‐time PCR (C). Astrocytes were treated with increasing concentrations of IL‐1β and TNFα (24 h) and assessed for YKL‐40 transcription by real‐time PCR (D). Astrocytes were treated acutely with IL‐1β (10 ng/mL, 24 h), then the medium was changed to growth medium without IL‐1β, and RNA was extracted 1, 4, 7 and 10 days post‐stimulation. YKL‐40 transcription increased up to 4 days in culture and declined to baseline by 7 days. For chronic stimulation, astrocytes were treated with IL‐1β (10 ng/mL) for 7 days, and RNA was extracted 1, 2, 3 and 7 days poststimulation (E). Astrocytes were treated with IL‐1β (10 ng/mL, 24 h) in the presence or absence of IL‐1 receptor antagonist (2 µg/mL). Alternatively, astrocytes were treated with TNFα (10 ng/mL, 24 h) in the presence or absence of the TNFα antagonist Enbrel® (1 mg/mL). Both IL‐1 receptor antagonist and Enbrel® blocked YKL‐40 stimulation in response to IL‐1β or TNFα, respectively (F). Astrocytes were treated with MCM (24 h) in the presence or absence of IL‐1 receptor antagonist (2 µg/mL) and/or Enbrel® (1 mg/mL). Alternatively, astrocytes were treated with MCM in the presence of the NF‐κB inhibitors Bay 11‐7082 (660 ng/mL) or IKK inhibitor (2 mM). Both IL‐1 receptor antagonist and Enbrel® blocked YKL‐40 transcription by 35% and the NF‐κB inhibitors Bay 11‐7082 and IKK inhibitor YKL‐40 transcription by 72% and 91%, respectively (G). Data shown are from a single experiment using duplicate samples and are representative of three independent experiments.
Combined YKL‐40 ISH and GFAP immunohistochemistry of astrocyte cultures treated with MCM showed that only a fraction of astrocytes were positive for YKL‐40 transcription at 4 days post‐stimulation (Figure 9D–F), while at day 7 post‐stimulation, more than 50% of the astrocytes were positive for YKL‐40 mRNA (Figure 9G–I). Stimulated astrocytes demonstrated induction of YKL‐40 mRNA and were morphologically distinct, adopting a more spindle morphology (Figure 9G–I) compared with control non‐treated astrocytes (Figure 9A–C).
Figure 9.

Soluble mediators released from macrophages induce YKL‐40 transcription and morphology changes in astrocytes. Astrocytes were treated with MCM (collected after 5 days in vitro) and fixed at day 0 (A–C), 4 (D–F) and 7 days (G–I) post‐stimulation. The cells were hybridized with 35S‐labeled RNA probe for YKL‐40 (B,E,H) followed by immunofluorescent staining for GFAP (A,D,G) as described in Materials and Methods. The right panels (C,F,I) show the merged images of the ISH and immunohistochemistry, scale bar = 100 µm. The number of YKL‐40‐expressing astrocytes increased over time and was associated with morphology change from quiescent to reactive phenotype.
YKL‐40‐treated astrocytes show induced migratory capacity
As YKL‐40 is a secreted protein and induced in reactive astrocytes, we wanted to test whether it can induce migration in naïve astrocytes. Confluent primary astrocytes cultures were stimulated to migrate using the scratch‐wound assay in the absence or presence of YKL‐40 (6 µg/mL) or MCM as described in the Materials and Methods. Wound closure was significantly induced when astrocyte cultures were treated with YKL‐40 or exposed to MCM from separate macrophage cultures (P < 0.0001) (Figure 10). YKL‐40‐treated and MCM‐treated astrocytes showed 52% and 67% reduction in gap width compared with non‐treated astrocytes, respectively.
Figure 10.
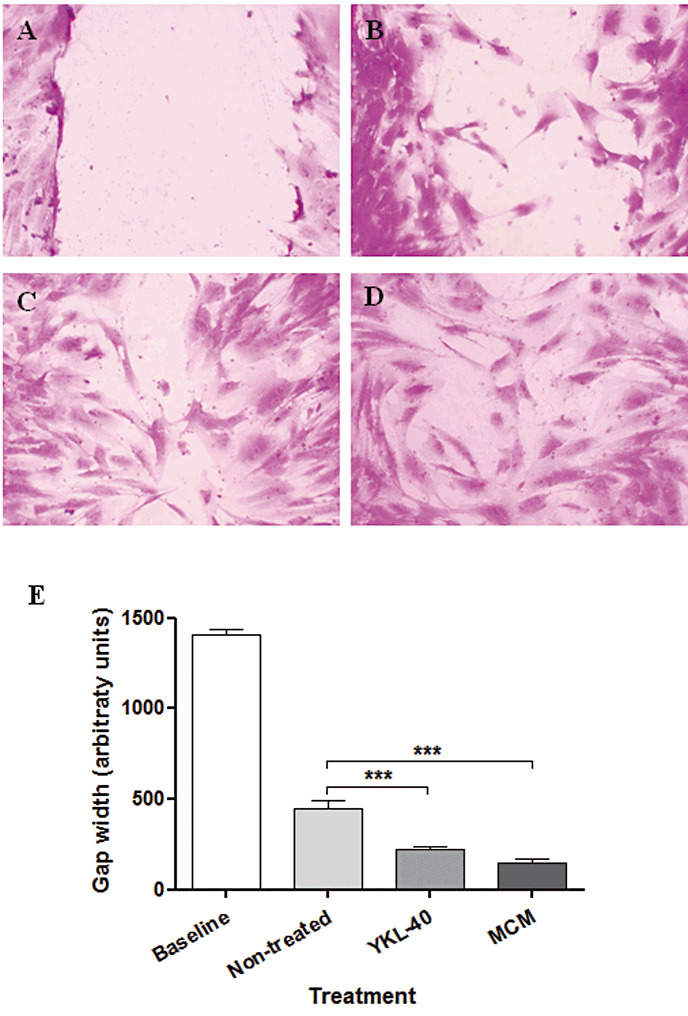
YKL‐40‐treated astrocytes show induced migratory capacity. Astrocyte cultures were grown to confluence, and then a scratch wound was made in the presence or absence of YKL‐40 (6 µg/mL) or macrophage culture medium. Twenty‐four hours later, the cells were fixed and stained with crystal violet. Baseline gap width (A), gap width after 24 h without any treatment (B), gap width after 24 h in the presence of YKL‐40 (6 µg/mL) (C) and gap width after 24 h in the presence of MCM (D). Data shown are from a single experiment using duplicate samples and are representative of three independent experiments. Quantitation of scratch width was performed, and data were subjected to statistical analysis as described in Materials and Methods, ***P < 0.0001 (E).
Coculture of macrophages and astrocytes neither blocked macrophage nor induced astrocyte YKL‐40 expression
Surprisingly, coculturing of macrophages with astrocytes did not induce YKL‐40 expression although they looked healthy and were phenotypically functional as measured by TNFα secretion (Figure 11, Supporting Information Figure S2). Cocultured astrocytes did not suppress YKL‐40 expression in macrophages.
Figure 11.
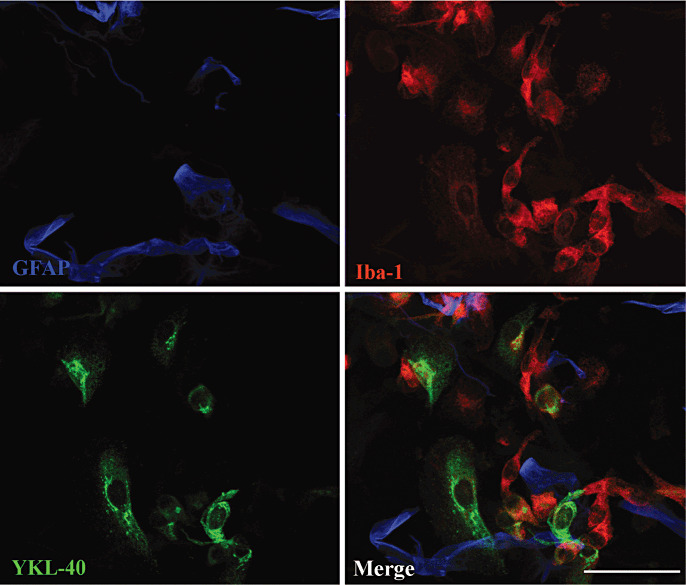
Coculture of macrophages and astrocytes neither blocked macrophage nor induced astrocyte YKL‐40 expression. Astrocytes were plated in chamber slides and were grown for 2 days before macrophages were added to the culture. Cocultured cells were grown for 7 days, then fixed and stained for YKL‐40 (green), GFAP (blue) and Iba‐1 (red).
DISCUSSION
We have shown a discrepancy between in vivo and in vitro expression of YKL‐40 in macrophages and microglia. In vitro macrophages expressed high levels of YKL‐40 that was attenuated by alternate activation; however, in inflammatory conditions, in vivo macrophages/microglia did not express abundant YKL‐40. Conversely, in vitro astrocytes expressed high levels of YKL‐40 when induced by MCM factors but not when placed in contact with macrophages as would be expected to mimic in vivo environments. Expression of YKL‐40 by astrocytes was associated with morphological changes characteristic of a reactive phenotype and increased migratory capacity. These results suggest that in vivo YKL‐40 expression is intricately regulated and could play a role in the neuroinflammatory gliotic response of neurological diseases.
In vivo YKL‐40 expression is induced in reactive astrocytes by the presence of macrophages, while conversely, YKL‐40 expression in macrophages is diminished
Previous studies have shown that YKL‐40 is expressed by macrophages in vitro 6, 27, 45, 46 and in vivo in inflammatory conditions like rheumatoid arthritis and atherosclerosis 6, 24. Screening additional examples of neuroinflammatory conditions (eg, acute bacterial meningitis, PML and PRES) confirmed that YKL‐40 expression is mostly restricted to astrocytes. This paradoxical reversal of YKL‐40 expression in vivo in neuroinflammation was dramatically illustrated in PML where despite abundant macrophage infiltration, YKL‐40 expression was not detected in the macrophages but was restricted to the lesional reactive astrocytes. Reactive astrocytes in the absence of neuroinflammation (eg, CJD) did not express YKL‐40. In acute bacterial meningitis, meningeal immune cells showed modest YKL‐40 transcription (although not as robust as parenchymal astrocytes) implying that the meninges are a more permissive environment for macrophage YKL‐40 transcription than the CNS parenchyma.
Macrophages in vitro express abundant YKL‐40 that can be induced by classical activation and inhibited by alternative activation
Macrophages expressed and released YKL‐40 into the culture medium as measured by ELISA and as detected by immunohistochemistry and Western blotting. Time course analysis showed that YKL‐40 release was induced over the differentiation period in culture in agreement with previous studies 6, 46. With prolonged culture, macrophages showed diminished secretion of YKL‐40 [as well as cytokines (eg, IL‐1β and TNFα)] as shown by ELISA and Luminex analyses. These results were confirmed by flow cytometry analysis showing increased numbers of YKL‐40‐expressing macrophages during differentiation in vitro. The phenotype of YKL‐40‐producing macrophages was distinguished from macrophages that did not produce YKL‐40 by increased TNFα production and decreased HLA‐DR and CD14 expression. This suggests that YKL‐40 production is associated with macrophages exhibiting immunostimulatory functions with decreased ability to present antigen or recognize pathogen‐associated molecular patterns. YKL‐40‐producing macrophages were also characterized by dim expression of CD206, the mannose receptor 1 that was not present in the YKL‐40 null population. This is interesting as CD206 is considered a marker for alternative activation (49). Tumor‐infiltrating macrophages also demonstrate pleiotropic expression of classical and alternative markers including TNFα and CD206 (52). It will be interesting to examine how various cells and mediators, especially ones derived from the neuroinflammatory environment, alter the phenotype and function of macrophages/microglia in context of YKL‐40 production.
Classical activation of macrophages with LPS and IFN‐γ induced 10‐fold transcription of YKL‐40, while alternative activation with IL‐4 inhibited YKL‐40 transcription by 90% suggesting that YKL‐40 may be involved in promotion rather than resolution of inflammation. Interestingly, a recent study showed that monocytes incubated with IL‐13 increased the secretion of YKL‐40 (10). It is well accepted that IL‐4 and IL‐13 have overlapping activities and both induce alternative activation of macrophages; however, IL‐4 and IL‐13 pathways have convergent and divergent features, which can account for the differences in the phenotype induced in monocytes or macrophages 36, 37.
Activated microglia showed a similar pattern of YKL‐40 changes compared with macrophages although the magnitude was less pronounced. The discrepancy between our in vivo findings in neuroinflammatory conditions and the abundance of YKL‐40 transcription in macrophages in vitro could be the result of tissue‐specific inhibitory pathways that diminish YKL‐40 transcription in infiltrating macrophages in the CNS. We have shown that IL‐4 can inhibit YKL‐40 transcription in macrophages, thus it is reasonable to speculate that other inhibitory cytokines might be responsible for inhibiting YKL‐40 transcription in infiltrating macrophages in the CNS. Recent studies showed that neuroimmunoregulatory proteins expressed by CNS cells can control neuroinflammation, which is vital for brain homeostasis 11, 13. These neuronal cell surface molecules (eg, CD200, CD47, CD45, CD22 and semaphorin 3A) can modulate the activation of inflammatory cells contributing to the reduction of tissue inflammation 35, 39. Because in vitro macrophage/microglia cultures and combination astrocyte/macrophage cultures did not recapitulate the tissue regulation that attenuated YKL‐40 expression in vivo, we would hypothesize that neuronal contact with these cells may be critical in modulating their neuroinflammatory response.
Anti‐inflammatory drugs had a mixed effect on YKL‐40 transcription in macrophages. Dexamethasone is a known potent anti‐inflammatory drug that decreases production of inflammatory mediators such as IL‐1β(18). Indeed, our results showed that dexamethasone can reduce IL‐1β transcription as well as YKL‐40. Other anti‐inflammatory drugs (eg, simvastatin, which inhibits macrophage proliferation and suppresses macrophage infiltration 48, 57, and minocycline, which reduces neuroinflammation 23, 29, 33, 40, 50), despite modulating macrophage activation, did not affect YKL‐40 transcription. The differential effects of various drugs on the neuroinflammatory responses of macrophages offer potential avenues to modulate the glial scar.
YKL‐40 transcription was induced in astrocytes by proinflammatory mediators released from macrophages
The current study showed that exposing astrocytes to MCM induced YKL‐40 transcription. These findings are consistent with our previous studies showing abundant YKL‐40 expression in vivo in reactive astrocytes in acute and chronic neuroinflammatory diseases, specifically in areas adjacent to inflammatory cells 3, 4, 5. Of the many proinflammatory cytokines released by macrophages, IL‐1β proved to be a potent inducer of astrocyte YKL‐40 transcription. Other proinflammatory interleukins like IL‐2, IL‐6 (with or without the soluble IL‐6 receptor), IL‐12, IL‐13, IL‐17 and IL‐18 did not induce substantial YKL‐40 transcription in astrocytes. Interestingly, previous studies addressed the possibility that IL‐13 regulates BRP‐39 and demonstrated that the levels of BRP‐39 mRNA and protein were significantly higher in lungs from IL‐13 overexpressing mice compared with wild‐type controls (30). BRP‐39 was most prominent in airway epithelial cells and alveolar macrophages. This implies that IL‐13 may be involved with YKL‐40 transcription in certain cells but does not play a role in inducing YKL‐40 transcription in astrocytes. Although macrophages can be a source of IFNγ(2), we did not observe IFNγ induction of YKL‐40 transcription in astrocytes. LPS and bFGF, which are reported activators of astrocytes 22, 34, 42, also did not induce YKL‐40 transcription in astrocytes. TNFα was capable of inducing some YKL‐40 transcription, however, only at concentrations that were 10 000‐fold higher than IL‐1β. These findings are in agreement with previous studies showing that IL‐1β and TNFα induced YKL‐40 mRNA and protein secretion in rat chondrocytes (44). A 1‐day IL‐1β exposure of astrocytes was sufficient to induce YKL‐40 transcription for several subsequent days that then diminished to control levels after 7 days. Similar findings were observed in rat chondrocytes that were exposed to TNFα for 4 h, elevated YKL‐40 mRNA and protein were evident up to 72 h after removal of the cytokine (44). This transient pattern of YKL‐40 expression was also seen in vivo in our previous study in a model of TBI (5). Our previous time course analysis of TBI showed that YKL‐40 transcription began 1 day after injury, remained elevated for several days and then declined by day 12. YKL‐40 transcription in astrocytes was coincident with IL‐1β expression. However, the current in vitro studies show that additional factors other than IL‐1β and TNFα are involved in YKL‐40‐induced transcription in astrocytes through NF‐κB cell signaling activation. Thus, YKL‐40 is induced in astrocytes during neuroinflammation by IL‐1β and TNFα, as well as other factors released from infiltrating macrophages. Not surprisingly, a longer exposure of astrocytes to IL‐1β induced a more robust YKL‐40 transcription, which may explain the robust YKL‐40 transcription seen in active neuroinflammatory conditions like lentiviral encephalitis and MS.
In vitro non‐treated astrocytes have a typical flat morphology without abundant YKL‐40 mRNA (3). Treatment with MCM (or IL‐1β, data not shown) induced a change in astrocyte morphology with individual cells adopting an elongated spindle shape at the same time they start producing YKL‐40 mRNA. This morphological change and induction of YKL‐40 transcription progressively involved more astrocytes until by 7 days post‐stimulation, more than 50% of the astrocytes expressed abundant YKL‐40 mRNA. This morphological change has been described as a reactive phenotype associated with inflammatory stimulation (51). Previous studies show that IL‐1β induced reorganization of F‐actin and dephosphorylation of focal adhesion kinase and myosin light chain 2 in primary human astrocytes, which implies cytokine regulation of the Rho‐ROCK pathway in the generation of a reactive astrogliosis (20). Treating astrocytes with exogenous YKL‐40 induced migration in the scratch‐wound assay. The phenomenon of YKL‐40 induction was not unique to astrocytes and has been observed in other cell types such as chondrocytes, where neither YKL‐40 protein nor mRNA were detectable in normal human articular cartilage, while both were abundant in synovial specimens from patients with rheumatoid arthritis (44). Thus, chondrocyte and astrocyte expression of YKL‐40 may be a general response to inflammation.
Coculturing of macrophages and astrocytes does not replicate YKL‐40 expression in monocultures or the in vivo findings in neurological conditions
Several studies have shown that astrocytes play a key role in deactivating microglial cells by releasing immunomodulatory factors. Cytokines like IL‐4 and IL‐10 reduced inflammation by inhibiting microglial production of proinflammatory cytokines, reactive oxygen intermediates and nitric oxide 16, 32, 53, 56. However, coculturing macrophages with astrocytes did not show inhibition of YKL‐40 expression in macrophages. This could be caused by the different cytokines and mediators that are produced in vivo vs. those produced in monocultures in vitro or may result from differences in the cell–cell contacts or extracellular matrix in vitro and in vivo. Interestingly, although we expected that the presence of macrophages would induce YKL‐40 expression in astrocytes, we did not observe abundant YKL‐40 expression in cocultures, implying that the presence of astrocytes can attenuate the repertoire of mediators released from macrophages toward an anti‐inflammatory phenotype and inhibit YKL‐40 expression. The fact that the in vitro studies failed to recapitulate the in vivo findings interjects a note of caution regarding the applicability of combining astrocyte and microglial cultures to mimic all of the cell signaling present in the CNS in vivo.
In summary, YKL‐40 transcription is induced in astrocytes by proinflammatory factors released from macrophages. YKL‐40 transcription in astrocytes is associated with cell migration and morphological changes characteristic of reactive gliosis. Further studies are required to elucidate the role of astrocyte YKL‐40 expression in neuroinflammation and the inhibition of its production in CNS macrophages. Modulation of YKL‐40 expression in these two cell types would be expected to affect glial scar formation and potentially affect neurodegeneration and regeneration.
Supporting information
Figure S1. Validation of YKL‐40 staining procedure. Macrophages were gated based on forward scatter (FS) and side scatter (SS) log parameters as shown in the left graph. The arrow points to analysis of macrophages that were stained using different protocols. The histogram shows YKL‐40 staining (black‐dashed line) of macrophages, which is easily distinguishable from the following control stains: YKL‐40 is not detected without intracellular staining (ICS) permeablization of the cell (dark gray solid line). Unstained, permeabilized macrophages are similar to the other controls (filled dark gray peak). Staining of permeabilized macrophages with an irrelevant biotin‐conjugated antibody (filled black peak) (A). Histogram shows the distinction of macrophages that were: not treated with GolgiStop and stained with YKL‐40 (filled light gray peak), stained with anti‐biotin antibody only (filled dark gray peak), stained with 0.5 µg anti‐YKL‐40 biotin‐conjugated antibody (dashed line) and stained with 5 µg anti‐YKL‐40 biotin‐conjugated antibody (black line) (B). Expression of YKL‐40 was not observed in CD3+ lymphocytes (C).
Figure S2. Validation of TNFα secretion in cocultured macrophages. Astrocytes were plated in chamber slides and were grown for 2 days before macrophages were added to the culture. Cocultured cells were grown for 14 days and the culture medium was collected every 2 days and analyzed for TNFα. Time course of TNFα levels released into the culture medium was determined by TNFα ELISA kit. Data shown are from a single experiment using duplicate samples and are representative of three independent experiments.
Supporting info item
Supporting info item
ACKNOWLEDGEMENTS
We thank Peter D. Wu and Mark Stauffer for valuable technical assistance. This work was supported by a grant from the Emmerling Foundation to DB‐B and grants K24 MH01717 and RO1 MH071151 to (CAW).
REFERENCES
- 1. Bissel SJ, Wang G, Trichel AM, Murphey‐Corb M, Wiley CA (2006) Longitudinal analysis of activation markers on monocyte subsets during the development of simian immunodeficiency virus encephalitis. J Neuroimmunol 177:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bogdan C, Schleicher U (2006) Production of interferon‐gamma by myeloid cells—fact or fancy? Trends Immunol 27:282–290. [DOI] [PubMed] [Google Scholar]
- 3. Bonneh‐Barkay D, Bissel SJ, Wang G, Fish KN, Nicholl GC, Darko SW et al (2008) YKL‐40, a marker of simian immunodeficiency virus encephalitis, modulates the biological activity of basic fibroblast growth factor. Am J Pathol 173:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonneh‐Barkay D, Wang G, Starkey A, Hamilton RL, Wiley CA (2010) In vivo CHI3L1 (YKL‐40) expression in astrocytes in acute and chronic neurological diseases. J Neuroinflammation 7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonneh‐Barkay D, Zagadailov P, Zou H, Niyonkuru C, Figley M, Starkey A et al (2010) YKL‐40 expression in traumatic brain injury—an initial analysis. J Neurotrauma 27:1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boot RG, van Achterberg TA, van Aken BE, Renkema GH, Jacobs MJ, Aerts JM, de Vries CJ (1999) Strong induction of members of the chitinase family of proteins in atherosclerosis: chitotriosidase and human cartilage gp‐39 expressed in lesion macrophages. Arterioscler Thromb Vasc Biol 19:687–694. [DOI] [PubMed] [Google Scholar]
- 7. Brasso K, Christensen IJ, Johansen JS, Teisner B, Garnero P, Price PA, Iversen P (2006) Prognostic value of PINP, bone alkaline phosphatase, CTX‐I, and YKL‐40 in patients with metastatic prostate carcinoma. Prostate 66:503–513. [DOI] [PubMed] [Google Scholar]
- 8. Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S et al (2007) A chitinase‐like protein in the lung and circulation of patients with severe asthma. N Engl J Med 357:2016–2027. [DOI] [PubMed] [Google Scholar]
- 9. Cintin C, Johansen JS, Christensen IJ, Price PA, Sorensen S, Nielsen HJ (2002) High serum YKL‐40 level after surgery for colorectal carcinoma is related to short survival. Cancer 95:267–274. [DOI] [PubMed] [Google Scholar]
- 10. Correale J, Fiol M (2011) Chitinase effects on immune cell response in neuromyelitis optica and multiple sclerosis. Mult Scler 17:521–531. [DOI] [PubMed] [Google Scholar]
- 11. Griffiths MR, Gasque P, Neal JW (2010) The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: a brief review. Int J Inflamm 2010:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gruber MF, Weih KA, Boone EJ, Smith PD, Clouse KA (1995) Endogenous macrophage CSF production is associated with viral replication in HIV‐1‐infected human monocyte‐derived macrophages. J Immunol 154:5528–5535. [PubMed] [Google Scholar]
- 13. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM et al (2000) Down‐regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290:1768–1771. [DOI] [PubMed] [Google Scholar]
- 14. Hoji A, Connolly NC, Buchanan WG, Rinaldo CR Jr (2007) CD27 and CD57 expression reveals atypical differentiation of human immunodeficiency virus type 1‐specific memory CD8+ T cells. Clin Vaccine Immunol 14:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hormigo A, Gu B, Karimi S, Riedel E, Panageas KS, Edgar MA et al (2006) YKL‐40 and matrix metalloproteinase‐9 as potential serum biomarkers for patients with high‐grade gliomas. Clin Cancer Res 12:5698–5704. [DOI] [PubMed] [Google Scholar]
- 16. Hu S, Chao CC, Ehrlich LC, Sheng WS, Sutton RL, Rockswold GL, Peterson PK (1999) Inhibition of microglial cell RANTES production by IL‐10 and TGF‐beta. J Leukoc Biol 65:815–821. [DOI] [PubMed] [Google Scholar]
- 17. Jensen BV, Johansen JS, Price PA (2003) High levels of serum HER‐2/neu and YKL‐40 independently reflect aggressiveness of metastatic breast cancer. Clin Cancer Res 9:4423–4434. [PubMed] [Google Scholar]
- 18. Jeon YJ, Han SH, Lee YW, Lee M, Yang KH, Kim HM (2000) Dexamethasone inhibits IL‐1 beta gene expression in LPS‐stimulated RAW 264.7 cells by blocking NF‐kappa B/Rel and AP‐1 activation. Immunopharmacology 48:173–183. [DOI] [PubMed] [Google Scholar]
- 19. Johansen JS (2006) Studies on serum YKL‐40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan Med Bull 53:172–209. [PubMed] [Google Scholar]
- 20. John GR, Chen L, Rivieccio MA, Melendez‐Vasquez CV, Hartley A, Brosnan CF (2004) Interleukin‐1beta induces a reactive astroglial phenotype via deactivation of the Rho GTPase‐Rock axis. J Neurosci 24:2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Junker N, Johansen JS, Andersen CB, Kristjansen PE (2005) Expression of YKL‐40 by peritumoral macrophages in human small cell lung cancer. Lung Cancer 48:223–231. [DOI] [PubMed] [Google Scholar]
- 22. Kalmar B, Kittel A, Lemmens R, Kornyei Z, Madarasz E (2001) Cultured astrocytes react to LPS with increased cyclooxygenase activity and phagocytosis. Neurochem Int 38:453–461. [DOI] [PubMed] [Google Scholar]
- 23. Kielian T, Esen N, Liu S, Phulwani NK, Syed MM, Phillips N et al (2007) Minocycline modulates neuroinflammation independently of its antimicrobial activity in staphylococcus aureus‐induced brain abscess. Am J Pathol 171:1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirkpatrick RB, Emery JG, Connor JR, Dodds R, Lysko PG, Rosenberg M (1997) Induction and expression of human cartilage glycoprotein 39 in rheumatoid inflammatory and peripheral blood monocyte‐derived macrophages. Exp Cell Res 237:46–54. [DOI] [PubMed] [Google Scholar]
- 25. Kofler JBS, Stauffer M, Starkey A, Wiley CA (2010) Classical and alternative activation states of human adult microglia. J Neuropathol Exp Neurol 69:525–526. [Google Scholar]
- 26. Koyanagi Y, O'Brien WA, Zhao JQ, Golde DW, Gasson JC, Chen IS (1988) Cytokines alter production of HIV‐1 from primary mononuclear phagocytes. Science 241:1673–1675. [DOI] [PubMed] [Google Scholar]
- 27. Krause SW, Rehli M, Kreutz M, Schwarzfischer L, Paulauskis JD, Andreesen R (1996) Differential screening identifies genetic markers of monocyte to macrophage maturation. J Leukoc Biol 60:540–545. [DOI] [PubMed] [Google Scholar]
- 28. Kuepper M, Bratke K, Virchow JC (2008) Chitinase‐like protein and asthma. N Engl J Med 358:1073–1075, author reply 5. [DOI] [PubMed] [Google Scholar]
- 29. Kure I, Nishiumi S, Nishitani Y, Tanoue T, Ishida T, Mizuno M et al (2010) Lipoxin A(4) reduces lipopolysaccharide‐induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor‐kappaB activation. J Pharmacol Exp Ther 332:541–548. [DOI] [PubMed] [Google Scholar]
- 30. Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA et al (2009) Role of breast regression protein 39 (BRP‐39)/chitinase 3‐like‐1 in Th2 and IL‐13‐induced tissue responses and apoptosis. J Exp Med 206:1149–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Letuve S, Kozhich A, Arouche N, Grandsaigne M, Reed J, Dombret MC et al (2008) YKL‐40 is elevated in patients with chronic obstructive pulmonary disease and activates alveolar macrophages. J Immunol 181:5167–5173. [DOI] [PubMed] [Google Scholar]
- 32. Lodge PA, Sriram S (1996) Regulation of microglial activation by TGF‐beta, IL‐10, and CSF‐1. J Leukoc Biol 60:502–508. [DOI] [PubMed] [Google Scholar]
- 33. Maderna P, Cottell DC, Berlasconi G, Petasis NA, Brady HR, Godson C (2002) Lipoxins induce actin reorganization in monocytes and macrophages but not in neutrophils: differential involvement of rho GTPases. Am J Pathol 160:2275–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahler M, Ferhat L, Gillian A, Ben‐Ari Y, Represa A (1996) Tenascin‐C mRNA and tenascin‐C protein immunoreactivity increase in astrocytes after activation by bFGF. Cell Adhes Commun 4:175–186. [DOI] [PubMed] [Google Scholar]
- 35. Majed HH, Chandran S, Niclou SP, Nicholas RS, Wilkins A, Wing MG et al (2006) A novel role for Sema3A in neuroprotection from injury mediated by activated microglia. J Neurosci 26:1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. [DOI] [PubMed] [Google Scholar]
- 37. Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483. [DOI] [PubMed] [Google Scholar]
- 38. Michelsen AE, Rathcke CN, Skjelland M, Holm S, Ranheim T, Krohg‐Sorensen K et al (2010) Increased YKL‐40 expression in patients with carotid atherosclerosis. Atherosclerosis 21:589–595. [DOI] [PubMed] [Google Scholar]
- 39. Mott RT, Ait‐Ghezala G, Town T, Mori T, Vendrame M, Zeng J et al (2004) Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 46:369–379. [DOI] [PubMed] [Google Scholar]
- 40. Nikodemova M, Watters JJ, Jackson SJ, Yang SK, Duncan ID (2007) Minocycline down‐regulates MHC II expression in microglia and macrophages through inhibition of IRF‐1 and protein kinase C (PKC)alpha/betaII. J Biol Chem 282:15208–15216. [DOI] [PubMed] [Google Scholar]
- 41. Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R et al (2008) Effect of variation in CHI3L1 on serum YKL‐40 level, risk of asthma, and lung function. N Engl J Med 358:1682–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pang Y, Cai Z, Rhodes PG (2001) Analysis of genes differentially expressed in astrocytes stimulated with lipopolysaccharide using cDNA arrays. Brain Res 914:15–22. [DOI] [PubMed] [Google Scholar]
- 43. Rand V, Prebble E, Ridley L, Howard M, Wei W, Brundler MA et al (2008) Investigation of chromosome 1q reveals differential expression of members of the S100 family in clinical subgroups of intracranial paediatric ependymoma. Br J Cancer 99:1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Recklies AD, Ling H, White C, Bernier SM (2005) Inflammatory cytokines induce production of CHI3L1 by articular chondrocytes. J Biol Chem 280:41213–41221. [DOI] [PubMed] [Google Scholar]
- 45. Rehli M, Niller HH, Ammon C, Langmann S, Schwarzfischer L, Andreesen R, Krause SW (2003) Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J Biol Chem 278:44058–44067. [DOI] [PubMed] [Google Scholar]
- 46. Renkema GH, Boot RG, Au FL, Donker‐Koopman WE, Strijland A, Muijsers AO et al (1998) Chitotriosidase, a chitinase, and the 39‐kDa human cartilage glycoprotein, a chitin‐binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur J Biochem 251:504–509. [DOI] [PubMed] [Google Scholar]
- 47. Roederer M, Treister A, Moore W, Herzenberg LA (2001) Probability binning comparison: a metric for quantitating univariate distribution differences. Cytometry 45:37–46. [DOI] [PubMed] [Google Scholar]
- 48. Senokuchi T, Matsumura T, Sakai M, Yano M, Taguchi T, Matsuo T et al (2005) Statins suppress oxidized low density lipoprotein‐induced macrophage proliferation by inactivation of the small G protein‐p38 MAPK pathway. J Biol Chem 280:6627–6633. [DOI] [PubMed] [Google Scholar]
- 49. Stein M, Keshav S, Harris N, Gordon S (1992) Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD et al (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Summers L, Kangwantas K, Nguyen L, Kielty C, Pinteaux E (2010) Adhesion to the extracellular matrix is required for interleukin‐1 beta actions leading to reactive phenotype in rat astrocytes. Mol Cell Neurosci 44:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K et al (2008) Tumor‐infiltrating myeloid‐derived suppressor cells are pleiotropic‐inflamed monocytes/macrophages that bear M1‐ and M2‐type characteristics. J Leukoc Biol 83:1136–1144. [DOI] [PubMed] [Google Scholar]
- 53. Vincent VA, Tilders FJ, Van Dam AM (1997) Inhibition of endotoxin‐induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor beta. Glia 19:190–198. [DOI] [PubMed] [Google Scholar]
- 54. Wahl SM, Allen JB, McCartney‐Francis N, Morganti‐Kossmann MC, Kossmann T, Ellingsworth L et al (1991) Macrophage‐ and astrocyte‐derived transforming growth factor beta as a mediator of central nervous system dysfunction in acquired immune deficiency syndrome. J Exp Med 173:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang G, Achim CL, Hamilton RL, Wiley CA, Soontornniyomkij V (1999) Tyramide signal amplification method in multiple‐label immunofluorescence confocal microscopy. Methods 18:459–464. [DOI] [PubMed] [Google Scholar]
- 56. Wirjatijasa F, Dehghani F, Blaheta RA, Korf HW, Hailer NP (2002) Interleukin‐4, interleukin‐10, and interleukin‐1‐receptor antagonist but not transforming growth factor‐beta induce ramification and reduce adhesion molecule expression of rat microglial cells. J Neurosci Res 68:579–587. [DOI] [PubMed] [Google Scholar]
- 57. Yoshimura A, Inui K, Nemoto T, Uda S, Sugenoya Y, Watanabe S et al (1998) Simvastatin suppresses glomerular cell proliferation and macrophage infiltration in rats with mesangial proliferative nephritis. J Am Soc Nephrol 9:2027–2039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Validation of YKL‐40 staining procedure. Macrophages were gated based on forward scatter (FS) and side scatter (SS) log parameters as shown in the left graph. The arrow points to analysis of macrophages that were stained using different protocols. The histogram shows YKL‐40 staining (black‐dashed line) of macrophages, which is easily distinguishable from the following control stains: YKL‐40 is not detected without intracellular staining (ICS) permeablization of the cell (dark gray solid line). Unstained, permeabilized macrophages are similar to the other controls (filled dark gray peak). Staining of permeabilized macrophages with an irrelevant biotin‐conjugated antibody (filled black peak) (A). Histogram shows the distinction of macrophages that were: not treated with GolgiStop and stained with YKL‐40 (filled light gray peak), stained with anti‐biotin antibody only (filled dark gray peak), stained with 0.5 µg anti‐YKL‐40 biotin‐conjugated antibody (dashed line) and stained with 5 µg anti‐YKL‐40 biotin‐conjugated antibody (black line) (B). Expression of YKL‐40 was not observed in CD3+ lymphocytes (C).
Figure S2. Validation of TNFα secretion in cocultured macrophages. Astrocytes were plated in chamber slides and were grown for 2 days before macrophages were added to the culture. Cocultured cells were grown for 14 days and the culture medium was collected every 2 days and analyzed for TNFα. Time course of TNFα levels released into the culture medium was determined by TNFα ELISA kit. Data shown are from a single experiment using duplicate samples and are representative of three independent experiments.
Supporting info item
Supporting info item