

Abstract
The risk of radionuclide release in terrorist acts or exposure of healthy tissue during radiotherapy demand potent radioprotectants/radiomitigators. Ionizing radiation induces cell death by initiating the selective peroxidation of cardiolipin in mitochondria by the peroxidase activity of its complex with cytochrome c leading to release of hemoprotein into the cytosol and commitment to the apoptotic program. Here we design and synthesize mitochondria-targeted triphenylphosphonium-conjugated imidazole-substituted oleic and stearic acids which blocked peroxidase activity of cytochrome c/cardiolipin complex by specifically binding to its heme-iron. We show that both compounds inhibit pro-apoptotic oxidative events, suppress cyt c release, prevent cell death, and protect mice against lethal doses of irradiation. Significant radioprotective/radiomitigative effects of imidazole-substituted oleic acid are observed after pretreatment of mice from 1 hr before through 24 hrs after the irradiation.
Introduction
Despite having evolved from organisms adapted to massive irradiation during the early development of Earth’s biosphere, the human body – with its abundance of water - is vulnerable to radiolysis by high-energy (ionizing) irradiation. Medical applications of irradiation critically consider this sensitivity of normal tissues, particularly in the use of total body exposure for bone marrow transplantation patients. However, in uncontrolled situations of exposure to radiation such as during a terrorist attack, or the unavoidable radiation exposure of flight crews during extended space missions, the development of protective measures is lagging behind and there is an immediate need for the stockpiling of safe and effective radioprotectors/radiomitigators.
Acute radiation syndrome is associated with damage to the hematopoietic system and gastrointestinal tract due to massive cell loss in radiosensitive tissues occurring largely via apoptosis1–3. Along with radicals generated by radiolysis of water, the execution of mitochondria-mediated apoptosis is universally associated with the production of reactive oxygen species (ROS)4,5. Therefore, development of radiomitigators/radioprotectors for biodefense applications and radiotherapy has mostly focused on non-specific thiol-based antioxidants that have shown clinically insignificant results6–8. Recently, ROS production has been identified as a required step in selective peroxidation of a mitochondria-specific phospholipid, cardiolipin (CL), whose oxidation products are essential for the outer membrane permeabilization and release of pro-apoptotic factors4,9. The catalyst of the peroxidation reaction is cytochrome c (cyt c) that forms a high affinity complex with CL exhibiting potent peroxidase activity towards polyunsaturated CLs9. In normal mitochondria, CL and cyt c are spatially separated: the former is confined almost exclusively to the inner mitochondrial membrane, while the latter is located in the intermembrane space10. Early in apoptosis, CL migrates from the inner to the outer mitochondrial membrane – a process likely facilitated by one of the four candidate mitochondrial proteins: scramblase-3, nucleoside diphosphate kinase (NDPK-D), mitochondrial isoforms of creatine kinase (m–CPK) and t-Bid11–13. Trans-membrane re-distribution of CL makes physical interaction of CL and cyt c possible resulting in the formation of cyt c/CL complexes9. Several previous studies have proposed that there are two types of interaction of cyt c with anionic phospholipids: an electrostatic interaction and a specific hydrophobic interaction. While the electrostatic interaction is mainly driven by the charges between the protein and anionic lipids, the hydrophobic interaction involves the insertion of the lipid acyl chain in a hydrophobic channel present in the structure of cyt c. It has been shown that both interactions are essential for initiating the peroxidase activity of cyt c9 leading to peroxidation of bound polyunsaturated molecular species of CL. Notably, accumulation of peroxidized CL is essential for the execution of the apoptotic program. Conversely, prevention of CL peroxidation leads to inhibition of apoptosis14.
We reasoned that the new “pro-oxidant” enzymatic activity of cyt c/CL complexes represents a target for anti-apoptotic radioprotective drugs. Specifically, the peroxidase activity is due to CL-induced partial unfolding of the protein in the complex resulting in a “loosened” liganding capacity of heme-iron by a distal Met8015. We hypothesized that “locking” of the heme-iron coordination bond with a strong ligand delivered through the hydrophobic channel to the immediate proximity of the heme catalytic site would block the peroxidase activity, inhibit CL peroxidation and prevent the progressions of apoptosis. Indeed, we demonstrated that mitochondria-targeted 3-hydroxypropyl-triphenylphosphonium (TPP)-conjugated imidazole-substituted oleic acid (TPP-IOA) and stearic acid (TPP-ISA) exerted strong specific liganding of heme-iron in cyt c/CL complex, effectively suppressed its peroxidase activity and CL peroxidation thus preventing cyt c release and cell death, and protecting mice against lethal doses of irradiation.
Results
Molecular modeling and docking studies
We have designed (Fig. 1) and synthesized (Supplementary Fig. S1) imidazole-substituted fatty acids and their esters - imidazole substituted oleic acid (IOA), stearic acid (ISA) (Fig. 1a, b), imidazole substituted methyl ester of oleic acid (IEOA, Fig. 1c) and imidazole substituted dodecanoic acid (IDA) (Fig. 1d) - assuming that these compounds will interact with cyt c by providing the imidazole group as a sixth ligand to the heme. A similar strategy was used to inhibit the terminal oxidation of fatty acids by CYP4A116. Molecular modeling and docking studies of partially unfolded cyt c showed that 18-carbon long IOA and ISA (Fig. 2a, b, c) as well as IEOA (Fig. 2d) with the imidazole moiety 7-carbon atoms away from the terminal methyl group indeed positioned the heterocycle close to heme such that the nitrogen atom of imidazole was within 2.4 Å (IOA), 2.5 Å (IEOA) and 2.7 Å (ISA) from the heme-iron. A hydrophobic channel formed by the displacement of the Met80 containing loop in the partially unfolded cyt c structure stabilized the interaction (Fig. 2b, colored in yellow). Notably, IDA, with the imidazole-moiety attached to the terminal CH2-group, this specific positioning was not achieved and the imidazole nitrogen was 6.9 Å away from the heme-iron (Fig. 2e).
Figure 1. Structural formulas of synthesized compounds.

(a) 12-(1H-imidazol-1-yl)-(Z)-octadec-9-enoic acid (IOA); (b) 12-(1H-imidazol-1-yl)octadecanoic acid (ISA); (c) methyl 12-(imidazol-1-yl)-(Z)-octadec-9-enoate (IEOA); (d) 12-(imidazol-1yl)-dodecanoic acid (IDA). Synthetic and experimental details are provided in supplementary methods.
Figure 2. Modeling of the binding of imidazole substituted fatty acids.

(a, b) IOA, (c) ISA, (d) IEOA, and (e) IDA. Cyt c is colored in green and represented as cartoon in (a, c, d, e) and as surface in (b). IOA, IEOA, ISA, TPP-IOA, TPP-ISA, IDA and heme are represented as sticks. IOA, IEOA, are colored in cyan, ISA is colored in yellow, and IDA is colored in wheat. The hydrophobic surface corresponding to the predicted IOA/ISA binding site is colored in yellow (b).
Suppression of peroxidase activity of cyt c complexes
We next evaluated the effect of ISA, IOA and IEOA on the peroxidase activity of cyt c complexes with tetra-oleoyl-cardiolipin (TOCL) towards H2O2-driven oxidation of two prototypical phenolic substrates, Amplex Red (Fig. 3a) and etoposide (Fig. 3b). We found that ISA, IOA and IEOA acted as potent inhibitors of the peroxidase activity of cyt c/TOCL complexes with both substrates (Fig. 3). Computer modeling showed that IOA (Fig. 2a), IEOA (Fig. 2c) and ISA (Fig. 2d) bind to cyt c in a similar fashion. The truncated derivative IDA, did not exert any inhibitory effect (Fig. 3a), in line with our computer modeling data. Because catalytic reactive intermediates of cyt c/CL peroxidase complexes – protein-immobilized (tyrosyl) radicals (Tyr•) – can be detected by EPR spectroscopy17, we studied the effect of IEOA and IOA on H2O2-dependent formation of radicals (Fig. 3c). Both IOA and IEOA (but not IDA) effectively quenched generation of Tyr• radicals. Finally, we assessed the ability of ISA and IEOA to inhibit peroxidase activity using isolated mouse liver mitochondria. To avoid decomposition of H2O2 by catalase, we used tert-butyl hydroperoxide (tBOOH) as a source of oxidizing equivalents. Both ISA and IEOA suppressed peroxidase activity in a concentration-dependent manner (Supplementary Fig. S2).
Figure 3. Inhibition of peroxidase activity of cyt c/TOCL complexes.

(a) Assessments of peroxidase activity of cyt c/TOCL by H2O2-induced oxidation of Amplex Red to resorufin. Data are means ± S.D., n = 4, *p<0.01 (Student’s t-test) vs control (TOCL/cyt c/H2O2 with no ISA or IOA or IEOA or TPP-IOA or TPP-ISA added). (b) A typical EPR spectrum of etoposide phenoxyl radicals (left panel). Assessments of peroxidase activity of cyt c/TOCL by H2O2-induced oxidation of etoposide using EPR spectroscopy (right panel). (c) A typical low temperature EPR spectrum of protein-immobilized (tyrosine) radicals (left panel). Assessments of protein-immobilized (tyrosine) radicals by low temperature (77 K) EPR spectroscopy (right panel). (d) ISA and IEOA limit accessibility of heme to small molecules. A typical low temperature (77 K) EPR spectrum of cyt c/TOCL complexes in the presence of Angeli’s salt (left panel). Effects of ISA and IEOA on heme-nitrosylation of cyt c/TOCL induced by nitroxyl (HNO) generated from Angeli’s salt (right panel). Data are means ± S.D., n=3, *p<0.05 vs control (no ISA and IEOA added). (e) Liquid-He EPR evidence for ligation change in cyt c heme-iron. X-band liquid-He (20 K) EPR spectra of cyt c.
Liganding of heme-iron in cyt c/TOCL complexes
To address whether ISA and IEOA act as strong ligands of heme-iron in cyt c/TOCL complexes, we used low temperature EPR spectroscopy18. In the reduced form, cyt c/TOCL binds NO• and produces heme-nitrosylated complexes with characteristic EPR spectra9. Indeed, at 77 K, typical spectra of penta-, and hexa-coordinated cyt c were detectable upon incubation of cyt c/TOCL complexes in the presence of a source of nitroxyl (HNO), Angeli’s salt (Fig. 3d). ISA and IEOA (Fig. 3d) caused a concentration-dependent decrease of the EPR signal. To provide evidence for coordination changes of the heme-iron in cyt c we performed liquid-He EPR measurements. The X-band EPR spectrum of native ferri-cyt c at pH 7.4, recorded at 20 K, exhibited anisotropic, low spin signals, with gz=3.09, gy=2.24 and gx~1 (usually unobserved) (Fig. 3e) indicative of His/Met axial coordination at the heme19,20. Upon the addition of IOA, there was no change in the EPR spectrum indicating retention of the native His/Met axial coordination (Fig. 3e). However, when IOA was added to the cyt c/TOCL complex, the EPR spectrum revealed the presence of another low-spin species (gz =2.97, gy=2.27, gx~1.5, Fig. 3f) with g-values entirely consistent with His/imidazole coordination20,21. This signal was broadened on the low-field side, suggesting a combination of the native ferri-cyt c and a His/imidazole form. Spectral simulations (not shown) confirm an approximately 50:50 mixture of the native structure and the form in which Met80 has been replaced by the imidazole moiety of IOA. These were the only signals observed; in particular there were none at g ~ 6, indicating the absence of any penta- or hexacoordinate high-spin species. Similarly, ISA was able to change heme-iron coordination in cyt c whereby Met80 was substituted by the imidazole moiety (Fig. 3e). These results confirm experimentally that the imidazole moiety of imidazole fatty acids can indeed serve as a coordinating ligand for the heme substituting for Met80 ligation.
Inhibition of apoptosis by TPP-ISA and TPP-IOA
We then explored the ability of imidazole substituted fatty acids to inhibit apoptosis in cells. To target imidazole substituted fatty acids into mitochondria, we esterified them with TPP salt (Fig. 4 a, b), an organic cationic alcohol with delocalized electron density – known to be effectively “electrophoresed” due to the negative potential on the inside of the organelle’s membrane22. By using computer modeling we confirmed that conjugation of ISA and IOA with TPP did not significantly affect their positioning within the immediate proximity of the cyt c’s heme-iron, i.e. the nitrogen atom of the imidazole of both TPP-IOA and TPP-ISA was within 2.9 Å from the heme-iron (Fig. 4 c, d). Moreover, TPP-ISA and TPP-IOA were as effective as non-conjugated ISA and IOA in inhibiting peroxidase activity of cyt c/TOCL complexes (Fig. 3).
Figure 4. Structures and molecular modeling of TPP-IOA and TPP-ISA.
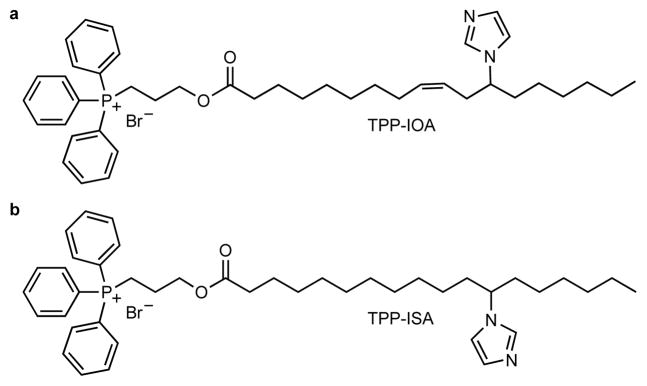

(a, c) 9-(Z)-(3-(12-imidazol-1-yl)octadeca-9-enoyloxy)propyl)triphenylphosphonium bromide (TPP-IOA); (b, d) (3-(12-imidazol-1-yl)-octadecanoyl)propyl)triphenylphosphonium bromide (TPP-ISA); TPP-IOA is colored in cyan, TPP-ISA is colored in yellow
We further estimated the amounts of accumulated TPP-IOA in mitochondria of mouse embryonic cells using high performance liquid chromatography (HPLC) and electrospray ionization mass spectrometry (ESI-MS) (Supplementary Fig. S3). We found that most of TPP-IOA was present in mitochondria (Supplementary Fig. S3). Assuming that the volume of mitochondria constitutes ~15–25% of the total volume of a cell, the mitochondrial enrichment factor becomes even greater such that the concentration of TPP-IOA in mitochondria may be as high as ~5 mM. It is likely that endogenous esterases can hydrolyze the ester-bond and release IOA and TPP. To test this, we performed assessments of esterase activity of mitochondria and the cytosolic fractions isolated from mouse embryonic cells based on HPLC measurements of TPP-IOA (Supplementary Fig. S3). We found that hydrolysis of TPP-IOA takes place in both mitochondria and the cytosol. The hydrolysis rate in mitochondria was comparable to that in the cytosolic fraction (Supplementary Fig. S3). Thus, both TPP-IOA and its de-esterifed form, IOA, could be present in mitochondria of mouse embryonic cells. Because TPP-conjugated imidazole substituted fatty acids effectively partition into mitochondria, we examined whether they affected bioenergetic functions, particularly ATP production. Neither TPP-IOA nor TPP-ISA had any effect on ATP levels in mouse embryonic cells. Normally, mitochondria are the major source of superoxide radicals in mouse embryonic cells23,24. Assessments of intracellular superoxide production using dihydroethidium (DHE) showed that neither TPP-IOA nor TPP-ISA had any effect on superoxide production in cells with or without radiation treatment (Supplementary Fig. S4).
For the anti-apoptotic action, it is essential that proposed heme-iron ligation is effective in inhibiting peroxidation of polyunsaturated species of CL. Therefore, we used an oxidizable tetra-linoleoyl-cardiolipin (TLCL), and conducted experiments on its cyt c/H2O2-induced oxidation (Fig. 5). In the presence of H2O2, we detected accumulation of characteristic TLCL peroxidation products with multiple oxygenated linoleic acid residues detectable by ESI-MS (Fig. 5a). We found that both TPP-IOA and TPP-ISA inhibited cyt c/H2O2 induced oxidation of TLCL in a concentration dependent manner. No difference in inhibition of TLCL oxidation between TPP-IOA and TPP-ISA was detected. The oxidation of TLCL was completely blocked at a ratio of cyt c to TPP-IOA or TPP-ISA of 1:20 (Fig. 5b).
Figure 5. Inhibition of H2O2-induced TLCL peroxidation.

(a) Typical ESI mass spectra of doubly-charged molecular ions of non-oxidized TLCL and TLCL oxidized by CL/cyt c and H2O2. Mass-to-charge (m/z; negative MS mode) values of 723.5 and 731.5, 739.5, 747.5, 755.5, 763.5, 771.5 and 779.5 were assigned to molecular clusters of non-oxidized TLCL and TLCL enriched with 1 – 7 oxygen atoms, respectively. (b) Quantitative assessment of TLCL and its oxidation products by ESI-MS. Data are means ± S.D., n=3, *p<0.05 (ANOVA) vs TLCL/cyt c/H2O2.
We then employed a model of intrinsic apoptosis induced in mouse embryonic cells by γ-irradiation and assessed several biomarkers of apoptosis. We found that TPP-IOA and TPP-ISA had similar radiation mitigating effects on mouse embryonic cells as evidenced by PS externalization (Fig. 6a), caspase 3/7 activation (Fig. 6b) and cyt c release (Fig. 6c). In contrast, IEOA or ISA - devoid of a mitochondria-targeting TPP-moiety - exerted no protection against apoptosis in mouse embryonic cells (Fig. 6d). To verify that the protective effects were realized during mitochondrial stages of apoptosis, TPP-ISA were added to the in vitro “caspase activation system” containing S100 fraction from mouse embryonic cells25. Caspase activation caused by exogenously added cyt c was completely insensitive to TPP-ISA and TPP-IOA (Fig. 6e) thus confirming that anti-apoptotic effects of TPP-IOA and TPP-ISA were realized in mitochondria. Furthermore, the anti-apoptotic activity was neither cell- nor stimulus- specific as TPP-IOA effectively inhibited apoptosis induced by a mitochondrial complex I inhibitor, rotenone, in cultured mouse lung endothelial cells (Fig. 6f).
Figure 6. Mitigative effects of TPP-ISA and TPP-IOA against apoptosis.

Effect of TPP-ISA and TPP-IOA on γ-irradiation induced PS externalization (dark color – Annexin V positive, PI positive cells; light color – Annexin V positive, PI negative cells) (a), caspase 3/7 activation (b), and cyt c release from mitochondria into the cytosol (anti-cyt c antibody, 0.2 μg/mL, 1:3000, PharMingen) (c) in mouse embryonic cells. Cells were γ-irradiated to a dose of 10 Gy, and then incubated in the presence of different concentrations of TPP-ISA or TPP-IOA for 48 h. Data are means ± S.D., n = 3. *p<0.01 (Student’s t-test) vs irradiated only cells. TPP-ISA or TPP-IOA was added to cells 30 min after γ-radiation. (d) Effect of IEOA and ISA on actinomycin D induced cell death in mouse embryonic cells. IEOA or ISA were incubated in the presence of fatty acid free bovine serum albumin (BSA) (Sigma) at a molar ratio of 5:1 at 37 °C for 30 min. Mouse embryonic cells were incubated with IEOA/BSA or ISA/BSA (100 μM) complexes for 30 min before the addition of 100 ng/ml actinomycin D (ActD). After 18 h incubation with ActD, cell viability was analyzed by flow cytometry using an Annexin V/propidium iodide (PI) kit. ActD induced cell death in ~34.2% of cells. Treatment of cells with ISA or IEOA in concentrations ranging from 1 to 100 μM exerted no detectable protection against ActD induced cell death. Representative data with 100 μM IEOA/BSA and ISA/BSA complexes are shown. Data are means ± S.D., n=3. (e) Effect of TPP-ISA and TPP-IOA on caspase-3 activation in S-100 from mouse embryonic cells. Data are means ± S.D., n = 3. (f) Effect of TPP-IOA on mouse lung endothelial cells treated with rotenone (apoptosis was assessed by PS externalization). Data are means ± S.D., n = 3. *p<0.05 (Student’s t-test) vs rotenone challenged cells. (g) Effect of TPP-ISA and TPP-IOA on clonogenic survival of mouse embryonic cells after γ-irradiation. The data were fitted to a single-hit multi-target model. Data are means ± S.D., n = 3. TPP-IOA or TPP-ISA was added to cells 30 min after γ-radiation.
Mitochondria are believed to be involved in orchestration of different cell death pathways26,27. We found that necrotic cells (PI-positive, Annexin V-positive) represented 12.9% while apoptotic cells (PI-negative, Annexin V-positive) were accountable for 28.9% of total PS positive cells detectable after irradiation. Treatment with TPP-IOA caused a 2-fold decrease in the number of apoptotic cells (to 14.8%) and 1.6-fold reduction in the number of necrotic cells (to 8.2%). Similarly, TPP-ISA protected via both anti-apoptotic and anti-necrotic mechanisms (to 13.9% of apoptotic and 7.8% necrotic cells, respectively). Overall, these results suggest that TPP-IOA and TPP-ISA afforded the radiomitigation in mouse embryonic cells acting through both anti-apoptotic and anti-necrotic pathways. In addition to apoptosis, and necrosis, mitotic cell death can be also triggered in irradiated cells. Using clonogenic assay, that includes the mitotic cell death component, we demonstrated that post-irradiation treatment of mouse embryonic cells with TPP-ISA or TPP-IOA resulted in a significant protection. Using a single-hit multi-target model, we estimated that TPP-ISA or TPP-IOA increased D0 - the dose needed to reduce cell surviving fraction to 37% (1/e) - to 1.67 ± 0.06 and 1.71 ± 0.05, respectively, compared to 1.33 ± 0.08 in untreated cells (Fig. 6g).
Radiomitigative effects of of TPP-IOA and TPP-ISA
Given the ability of TPP-IOA and TPP-ISA to act as radiomitigators in vitro, we assessed their potential to act as radioprotectors/radiomitigators in vivo. C57BL/6NTac female mice were exposed to 9.25 Gy total body irradiation at the dose rate of 80 cGy/min using the cesium irradiator. Three independent experiments (the total number of mice in each group was 31 to 35) yielded similar results: irradiation resulted in death of animals within 13–15 days (with survival of only 20% of animals by day 30). TPP-IOA or TPP-ISA (i.p. injection, 5 mg/kg body weight, 10 min after irradiation) showed a strong radiomitigative effect for both compounds (Fig. 7a). There was no statistically significant difference in radiomitigative potency of TPP-IOA and TPP-ISA (p=0.6389, a two-sided log-rank test). We also used a clinical linear accelerator to deliver the radiation dose. The mice (three groups with 22–23 animals in each) were irradiated to 9.25 Gy at the dose rate of 100 cGy/min using a Varian TrueBeam linear accelerator (Varian Medical Systems, Palo Alto, CA) and injected i.p. with 5 mg/kg body weight of either TPP-ISA or TPP-IOA 10 min after irradiation. Similarly to the results with cesium irradiator, the survival curves over 52 days were statistically different for TPP-IOA (5 mg/kg body weight) and TPP-ISA (5 mg/kg body weight) vs irradiated controls (Table 1); however, there was no statistically significant difference between TPP-IOA and TPP-ISA (p = 0.4567, a two-sided log-rank test). Thus, after multiple experiments, using two different irradiators, we demonstrated a high potency of both TPP-IOA and TPP-ISA in mitigating radiation damage without significant difference in radiomitigative activity between them. Therefore, all subsequent in vivo experiments were conducted with TPP-IOA and linear accelerator as the radiation source. We chose a dose of 5 mg/kg body weight of TPP-IOA because a lower dose (2.5 mg/kg body weight) of drug was not effective in mitigating the mice against irradiation. When mice (10 per group) were irradiated and administered 2.5 mg/kg body weight (10 min after irradiation), there was a trend towards a greater survival that, however, did not reach the level of significance (p=0.0525, a two-sided log-rank test) (Table 1). We further tested whether TPP-IOA was protective if given at later time points (than 10 min) after irradiation (10 mice per group). Administration of TPP-IOA at 5 hrs or 24 hrs after irradiation resulted in a significant increase in survival (Fig. 7b, Table 1). TPP-IOA was also protective if given 10 min or 1 hr before irradiation (Fig. 7b; Table 1).
Figure 7. Radiation protection and mitigation by TPP-IOA and TPP-ISA.

C57BL/6NTac female mice were exposed to total body irradiation to a dose of 9.25 Gy using a cesium source (n = 31 to 35 mice per group) (a) or a linear accelerator (n = 10 to 23 mice per group) (b). (a) - The mice were irradiated and injected i.p. with TPP-IOA or TPP-ISA (5 mg/kg body weight in 100 μl of water containing 25% ethanol) 10 min after irradiation. Mice exposed to: total body irradiation at the dose of 9.25 Gy only (black circles); to total body irradiation at the dose of 9.25 Gy and injected with TPP-ISA (5 mg/kg body weight) 10 min (blue triangles) or TPP-IOA (5 mg/kg body weight) 10 min (red squares) thereafter. p < 0.0001 (a two-sided log-rank test) - TPP-IOA or TPP-ISA injected and exposed to total body irradiation mice vs mice exposed to total body irradiation only. (b) - The mice were injected i.p. with TPP-IOA (5 mg/kg body weight in 100 μl of water containing 25% ethanol) at 1 hr or 10 min before irradiation or 10 min, 5 hrs or 24 hrs after irradiation. Mice exposed to: total body irradiation at the dose of 9.25 Gy only (black circles); to total body irradiation at the dose of 9.25 Gy and injected with TPP-IOA (5 mg/kg body weight) 10 min (blue circles), 5 hrs (blue triangles) and 24 hrs (blue squares) thereafter. Mice injected with TPP-IOA (5 mg/kg body weight) 10 min (red squares) and 1hr (red triangles) before total body irradiation (9.25 Gy). For assessments of significance see Table 1.
Table 1.
TPP-IOA and TPP-ISA increase survival of mice following irradiation. The two-sided log-rank test was used to examine the differences between irradiated mice with or without treatment with TPP-ISA or TPP-IOA.
| Treatment Group | Overall Survival (52 days) | Survival Over First 20 Days | N | ||
|---|---|---|---|---|---|
| Median Survival (95% Confidence Interval) | p* | Median Survival (95% Confidence Interval) | p* | ||
| 9.25Gy | 13 (11,14) | 13 (11,14) | 22 | ||
| TPP-IOA 5 mg/kg body weight, 1hr before 9.25Gy | - (12, -) | 0.0230 | - (12, -) | 0.0230 | 10 |
| TPP-IOA 5 mg/kg body weight, 10min before 9.25Gy | - (18, -) | 0.0009 | - (18, -) | 0.0009 | 10 |
| TPP-IOA 5 mg/kg body weight, 10min after 9.25Gy | - (-, -) | <0.00010 .4567# |
- (-, -) | <0.0001 | 23 |
| TPP-IOA 5 mg/kg body weight, 5hr after 9.25Gy | - (17, -) | 0.0030 | - (17, -) | 0.0030 | 10 |
| TPP-IOA 5 mg/kg body weight, 24hr after 9.25Gy | 18 (13, -) | 0.0416 | 18 (13, -) | 0.0416 | 10 |
| TPP-IOA 2.5 mg/kg body weight, 10 min after 9.25 Gy | 17 (14, -) | 0.0525 | 17 (14, -) | 0.0525 | 10 |
| TPP-ISA 5.0 mg/kg body weight 10 min after 9.25 Gy | - (13, -) | 0.0114 | - (13, -) | 0.0114 | 22 |
Mice were irradiated to 9.25 Gy using a Varian TrueBeam linear accelerator (Varian Medical Systems, Palo Alto, CA) and injected i.p. with either TPP-ISA or TPP-IOA (2.5 or 5 mg/kg body weight in 100 μl of water containing 25% ethanol) at 1 hr or 10 min before irradiation or 10 min, 5 hrs or 24 hrs after irradiation.
TPP-IOA and TPP-ISA vs irradiated mice.
TPP-IOA (5 mg/kg body weight 10 min after irradiation) vs TPP-ISA (5 mg/kg body weight 10 min after irradiation).
Given the high radioprotective/radiomitigative activity of TPP-IOA, we determined the extent to which it would be absorbed to reach radiosensitive tissues. This required the development of new LC-MS/MS protocols to quantitate TPP-IOA in tissue samples. These involved the establishment of a selective reaction monitoring (SRM) protocol for TPP-IOA that provides high selectivity and sensitivity. Our direct assessments clearly demonstrated the presence of TPP-IOA in plasma (Supplementary Fig. S5) as well as in the two most important radiosensitive tissues, bone marrow and small intestine. TPP-IOA levels were highest in plasma (54.0 ng/ml of plasma), followed by small intestine (1.5 ng/g of tissue) and bone marrow (0.2 ng/g of tissue), at the 10 min time point after i.p. injection. The levels measured in plasma are consistent with those found for decyl-TPP, a closely related compound, as seen in the study by Porteous, et al.28. The authors showed that after i.v. injection, the TPP-compounds are distributed rapidly to various tissues and less than 1% remains in plasma after 15 min. In addition, we assessed the hydrolysis of TPP-IOA in tissues (plasma, bone marrow and small intestine) yielding TPP and non-esterified IOA utilizing two different LC-MS/MS approaches: a selected reaction monitoring (SRM) method to determine the levels of IOA, a likely hydrolysis products of TPP-IOA, in tissue samples, and multiple reaction monitoring (MRM) offering extremely high selectivity and sensitivity (Supplementary Fig. S6). Using these approaches, we determined that bone marrow had the least amount of hydrolysis, exhibiting a level of 0.6 ng/g tissue/min of the TPP-propyl hydrolysis product. This was followed by a level of 4.1 ng/g tissue/min and 19.0 ng/g tissue/min of the TPP hydrolysis product in plasma and small intestine, respectively. Simple calculations show that the total amounts of non-hydrolyzed TPP-IOA plus hydrolyzed (IOA + TPP) will be significantly (approximately two orders of magnitude) higher than those of the non-hydrolyzed compound measured in the small intestine and bone marrow. The importance of this is underscored by our data that both TPP-IOA and IOA are effective inhibitors of peroxidase activity of cyt c/CL complexes (Fig. 3) and CL peroxidation (Fig. 5).
Discussion
In this work, we designed, synthesized and explored a new class of mechanism-based mitochondria-targeted inhibitors that act as strong ligands of iron in complexes of cyt c with CL. The compounds “lock” the catalytic site of the enzymatic complex, inhibiting its ability to facilitate the development of pro-apoptotic oxidative events, and suppress release of cyt c from mitochondria into the cytosol thus inhibiting apoptotic cell death. We demonstrated that imidazole fatty acids specifically interact with partially unfolded cyt c and not with intact cyt c. Indeed, low-temperature (He) EPR experiments indicate that liganding of heme-iron in cyt c by IOA was dependent on the presence of CL. While it is possible that imidazole fatty acids may interact with other hemoproteins – e.g., cytochromes P45016 their effects on the oxygenase/peroxidase activity of the hemoproteins might depend on several parameters such as redox potential, heme-coordination state, spin states. Notably, many hemoproteins with peroxidase function – cytochromes P450, myeloperoxidase, cyclooxygenase - do not require anionic (phospho)lipids for their activation.
Importantly, TPP-ISA and TPP-IOA exerted strong radoprotective/radiomitigative effects in vivo against lethal doses of irradiation of mice (from 1 hr before through 24 hrs after the irradiation). One can envision two possible applications of our findings in clinical practice. One of them may be associated with the employment of the newly discovered radiomitigators for the treatment of victims of terroristic attacks with nuclear devices as well as individuals inadvertently exposed to irradiation resulting from catastrophic nuclear accidents. Intraperitoneal injections of TPP-IOA and TPP-ISA employed in this study is not the preferred route of drug administration in clinical practice although it has been used for selected drugs in several disease states29–31. It is likely that pharmacologists would have to derive new formulations that could be given orally or as a skin patch. For radiation counterterrorism, a topical or trans-dermal delivery system would be preferred.
The second kind of applications may be relevant to organ specific delivery of TPP-IOA and TPP-ISA to protect normal tissues from ionizing irradiation effects in clinical radiotherapy. Several highly effective chemotherapy drugs are known to interact with ionizing irradiation to promote tumor cell killing, but unfortunately also exacerbate normal tissue toxicity. Local, tissue specific delivery of TPP-IOA and TPP-ISA may be also important in amelioration of the toxicity of this combined modality cancer therapy. The design of a pharmacological formulation by which to facilitate delivery of TPP-IOA and TPP-ISA to the oral cavity or oropharynx during radiotherapy of head and neck cancer; to the esophagus during radiotherapy of non-small cell lung cancer; to the bladder during brachytherapy or fractionated pelvic radiotherapy of endometrial or cervix cancer; and to the rectum during fractionated radiotherapy of prostate cancer, could provide both normal tissue protection and decreased morbidity for conventional treatment protocols and potentially allow radiation dose escalation to improve local control.
γ-Irradiation is a potent carcinogen by virtue of its ability to cause single- and double-strand DNA breaks32 - the effect that is also associated with a massive p53-dependent cell death in radiosensitive targets (primary lymphoid organs and intestinal epithelium) leading to acute injury33,34. While it is known that p53 acts as a potent and important tumor suppressor35, it is believed that this tumor suppressor activity is not directly related to p53’s acute pathological response to irradiation induced systemic genotoxicity34. Indeed there is substantial pharmacologic36 and genetic34 evidence supporting the notion that temporary and reversible suppression of p53, resulting in massive rescue of cells in radiosensitive tissues, is not associated with an increase in carcinogenicity. Rather, the tumor suppressor activity of p53 is related to activities in cells several days after the irradiation induced systemic genotoxicity. Thus, p53 inhibitors are expected to represent tissue protective drugs to be used under pathological conditions associated with massive apoptotic cell death37. Although p53 inhibitors hold therapeutic promise, their potential applications are limited by the tissue specificity of p53-dependent radiosensitivity. For example, despite the pronounced p53-dependent apoptosis that occurs among epithelial cells of the small intestine after irradiation, clinical gastrointestinal acute radiation syndrome develops independently of p53. Thus radioprotection of the gastrointestinal tract should be based on p53-independent strategies38, possibly other types of apoptosis inhibitors. The current work capitalizes on this principle by utilizing mitochondria-targeted inhibitors of cyt c/CL peroxidase activity – TPP-IOA and TPP-ISA – that demonstrated effectiveness in protecting against radiation induced apoptosis as well as radioprotective and radiomitigative effects in vivo. The mechanism of radiation mitigation proposed herein will prevent acute radiation sickness while having no impact on p53-mediated tumor suppression. When treatment with the mitochondrial-targeted inhibitors of cyt c is withdrawn, those few cells that have acquired a radiation-induced oncogenic chromosome aberration will be eliminated by p53-mediated apoptosis. This acquired oncogenic stress will be present in these cells for many weeks prior to tumor onset allowing ample time for p53 to execute its function as a tumor suppressor.
This study was aimed at the discovery, rather than the development, of novel mitochondria-targeted radiomitigators and radioprotectors. However, further improvements of structural features and pharmacological characteristics of the proposed inhibitors may lead to an optimized series of radioprotectors and radiomitigators with a broad spectrum of biomedical applications in the biodefense area as well as in radiotherapy of cancer by achieving increased resistance of normal tissues surrounding the tumor.
Methods
Animals
C57BL/6NTac female mice were anesthetized with Nembutal (1 mg/20 gm mouse), irradiated to a total body dose of 9.25 Gy using either a Shepherd Mark 1 Model 68 cesium irradiator at a dose rate of 80 cGy/min (31–35 mice per group) or a Varian TrueBeam Linear Accelerator (Varian Medical Systems, Palo Alto, CA) at 100 monitor units or approximately 100 cGy/min using 6 MV photons with a 40 cm × 40 cm field at 100 SSD (10–23 mice per group). Mice were injected intraperitoneally (i.p.) with 5 mg/kg body weight of TPP-IOA in a 100 μl volume of water containing 25% ethanol at 1 hr before irradiation, 10 min before irradiation, 10 min after irradiation, 5 hrs after irradiation and 24 hrs after irradiation. Other groups were injected i.p. with 2.5 mg/kg body weight of TPP-IOA or 5 mg/kg body weight of TPP-ISA 10 min after irradiation. The mice were followed for the development of hematopoietic syndrome (at which time they were sacrificed). The health of the non-irradiated mice was unexceptional and no adverse side effects were noticed over the period of study (52 days) after i.p. injection of either TPP-IOA or TPP-ISA (5 mg/kg body weight). The log-rank test was used for three analyses: the comparison of overall survival which is defined as the time from the date of radiation to the date of death for all mice under study; the comparison of short term survival over the first 20 days, i.e., the overall survival count at 20 days; and the comparison of conditional survival in mice surviving 20 or more days, i.e., the time from the date of radiation to the date of death for all mice who survived 20 days or longer after radiation. All these comparisons were made between each of the treated groups and the radiation-only control group. All procedures were pre-approved and performed according to the protocols established by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Cells
Mouse embryonic cells (courtesy of Dr. X. Wang, University of Texas, Dallas) were cultured in DMEM supplemented with 15% FBS, 25 mM Hepes, 50 mg/liter uridine, 110 mg/liter pyruvate, 2 mM glutamine, 1 × nonessential amino acids, 50 μM β-mercaptoethanol, 0.5 × 106 U/liter mouse leukemia inhibitory factor, 100 U/liter penicillin, and 100 mg/liter streptomycin in a humidified atmosphere of 5% CO2/95% air at 37°C. Mouse lung endothelial cells were obtained as previously described39. Briefly, lungs were flushed with HBSS containing 10 U/mL heparin, then homogenized and digested in type I collagenase using a gentle MACsTM dissociator (Miltenyi, CA). Pulmonary endothelial cells were isolated by magnetic beads coated with antibody (rat anti-mouse) to PECAM-1 (BD Pharmingen, San Diego, CA), and seeded for subculture (passage 1). At approximately passage 2, cells were incubated with fluorescently-labeled diacetylated LDL (diI-LDL) followed by FACS sorting for further purification. The enriched PECAM and diI-LDL population was sub-cultured on a collagen/gelatin matrix in 2% O2, 5% CO2, 93% nitrogen in a Coy Hypoxic Glove Box/Chamber in Opti-MEM (Gibco), 10% FBS, 2 mM glutamine, 0.2% retinal derived growth factor (Vec Technologies, Rensselaer, NY), 10U/mL heparin, 0.1 mM non-essential amino acid supplement (Gibco) and 55 μM β-mercaptoethanol (Counted as passage 3). Cells at passage 4 to 6 were applied to do the experiments.
Phopshatidylserine externalization
Phosphatidylserine externalization was determined by Annexin V–FITC apoptosis detection kit (Biovision, Mountain View, CA) according to the manufacturer’s instruction.
Caspase 3/7 activity
Caspase–3/7 activity was measured using a luminescence Caspase–Glo™ 3/7 assay kit (Promega, Madison, WI) according to the manufacturer’s instruction.
Measurement of cyt c release by Western blot analysis
Cells were harvested after 48 hrs post-irradiation incubation and resuspended in lysis buffer containing 0.05% digitonin for 4 min on ice. Supernatants were collected after centrifugation for 10 min at 10,000×g. Equal amounts of protein were subject to 15% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with antibodies against cyt c (clone 7H8.2C12, BD Pharmingen, San Diego, CA) or actin (Novus, Littleton, CO) (loading control) followed by horseradish peroxidase-coupled detection. The protein band profile was analyzed by densitometry using Labworks Image Acquisition and Analysis Software (UVP, Upland, CA).
Liquid-He EPR measurements of ISA and IEOA liganding
Conditions: Native cyt c (300 μM), IEOA/cyt c (2:1), TOCL/IEOA/cyt c (20:2:1), 20 mM HEPES buffer, pH 7.4. Samples (200 μL) were placed in 3 mm o.d. suprasil quartz EPR tubes and frozen in liquid nitrogen for subsequent spectroscopic measurements. X-band (9 GHz) EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments ESR 910 flow cryostat for ultra-low-temperature measurements. Spectra were recorded at 9.8 G modulation amplitude and 200 μW microwave power. The microwave frequency was calibrated by a frequency counter and the magnetic field was calibrated with a gaussmeter. The temperature was calibrated with carbon-glass resistors (CGR-1-1000) from LakeShore. This instrument and the software (SpinCount) to analyze the EPR spectra were graciously provided by Professor Mike Hendrich, Carnegie Mellon University.
Molecular docking
The structures of the ligands ISA, IOA, IEOA, IDA, TPP-ISA and TPP-IOA were docked to the partially unfolded structure of horse heart cyt c using the Lamarckian genetic algorithm provided by the Autodock 4.0 software40,41. The partially unfolded structure of cyt c, where the Met80 containing loop is displaced was obtained using the molecular dynamics simulation approach as described below. The X-ray crystal structure (Protein Database accession code: 1HRC) was used as the base structure for performing the MD simulations. The docking procedure used was similar to the studies performed earlier42, except for the following changes. A cubic box positioned at x, y, and z 0.032, −0.399, and −0.36, respectively as the center was built around the protein with 70 × 52 × 54 points and a spacing of 0.375 Å between the grid points was used. A total of 25 genetic algorithm runs were considered in each case with an initial population of 300 and a maximum number of 5,000,000 energy evaluations. The top 25 resulting orientations that have less than or equal to 0.5 Å root mean square deviation were clustered together. The best ligand bound to the partially unfolded cyt c structure in each case was chosen based on lowest energy as well as the conformation where the imidazole group was closest to the heme iron.
Molecular dynamics simulation
Molecular Dynamics simulations were performed using the MD software package NAMD43 using the VEGA ZZ 2.3.244 package as the user interface. The model structure of horse heart cyt c (PDB ID: 1HRC) was downloaded from the Protein Data Bank, and explicit hydrogens were added using the package VEGA ZZ 2.3.244. A spherical solvent cluster with a radius of 48.5 Å positioned at geometric center of the protein was built and Cl− ions were added to neutralize the system. This system was energy minimized for 2000 steps using conjugate gradient method. The energy-minimized structure was slowly heated from 0 to 310 K over a period of 100ps. Following this 2ns molecular dynamics simulation was performed at 310 K to obtain the partially unfolded cyt c structure.
Statistics
For the in vitro experiments, the results are presented as means ± S.D. values from at least three experiments, and statistical analyses were performed by either paired/unpaired Student’s t-test or one-way ANOVA. As an exploratory analysis, p-values were not adjusted for multiple comparisons For the survival data, median survival and its 95% confidence interval were calculated for each group, and the two-sided log-rank test was used to examine the differences between irradiated mice and irradiated mice treated with TPP-ISA or TPP-IOA. In all these tests, a p-value of less than 0.05 was regarded as significant.
Supplementary Material
Acknowledgments
Supported by NIH: U19AIO68021, HL70755, HL094488, CA119927; by NIOSH OH008282 and by La Junta de Extremadura y el Fondo Social Europeo (2010063090).
Footnotes
Author contributions
J.A. designed and synthesized the compounds, J.F. and Y.W contributed to synthesis of chemical compounds. A.A.K., A.M. and I.I.V. performed experiments on peroxidase activity of cytc/CL complexes, L.P. and J.P. performed liquid-He EPR experiments, N.Y. and J.K.S. performed computer modeling, Y.Y.T. and V.A.T performed MS analysis of cardiolipin oxidation, A.A.A. and A.K.S. performed MS analysis of compounds in tissues. N.A.B. participated in HPLC analysis of compounds in cells, D.A.S. performed HPLC analysis of compounds in cells, participated in the design of in vivo experiments on radioprotection, Z.H. J.J, W.F., and. K.W., perfomed cell experiments, H.B., B.R.P., participated in design of experiments, discussion of results and writing the maniuscript, M.W.E. and J.S.G. contributed to the design and performance of the in vivo experiment, H.W. performed statistical analysis of in vivo data, V.E.K. suggested the idea, designed the study, wrote the manuscript.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Paris F, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–297. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 2.Merritt AJ, et al. The role of p53 in spontaneous and radiation-induced apoptosis in the gastrointestinal tract of normal and p53-deficient mice. Cancer Res. 1994;54:614–617. [PubMed] [Google Scholar]
- 3.Komarova EA, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23:3265–3271. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- 4.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 5.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic byproduct or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 6.Weiss JF, Landauer MR. Protection against ionizing radiation by antioxidant nutrients and phytochemicals. Toxicology. 2003;189:1–20. doi: 10.1016/s0300-483x(03)00149-5. [DOI] [PubMed] [Google Scholar]
- 7.Dziegielewski J, et al. WR-1065, the active metabolite of amifostine, mitigates radiation-induced delayed genomic instability. Free Radic Biol Med. 2008;45:1674–1681. doi: 10.1016/j.freeradbiomed.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorr RT. Radioprotectants: pharmacology and clinical applications of amifostine. Semin Radiat Oncol. 1998;8:10–13. [PubMed] [Google Scholar]
- 9.Belikova NA, et al. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry. 2006;45:4998–5009. doi: 10.1021/bi0525573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs JJ, Hauser H, Carafoli E. Asymmetric distribution of phospholipids in the inner membrane of beef heart mitochondria. J Biol Chem. 1979;254:5308–5316. [PubMed] [Google Scholar]
- 11.Liu J, et al. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res. 2003;1:892–902. [PubMed] [Google Scholar]
- 12.Schlattner U, et al. Mitochondrial kinases and their molecular interaction with cardiolipin. Biochim Biophys Acta. 2009;1788:2032–2047. doi: 10.1016/j.bbamem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Sorice M, et al. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11:1133–1145. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 14.Tyurina YY, et al. Oxidative lipidomics of hyperoxic acute lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L73–85. doi: 10.1152/ajplung.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kagan VE, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 16.Lu P, Alterman MA, Chaurasia CS, Bambal RB, Hanzlik RP. Heme-coordinating analogs of lauric acid as inhibitors of fatty acid omega-hydroxylation. Arch Biochem Biophys. 1997;337:1–7. doi: 10.1006/abbi.1996.9768. [DOI] [PubMed] [Google Scholar]
- 17.Qian SY, et al. Identification of protein-derived tyrosyl radical in the reaction of cytochrome c and hydrogen peroxide: characterization by ESR spin-trapping, HPLC and MS. Biochem J. 2002;363:281–288. doi: 10.1042/0264-6021:3630281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kon H. Paramagnetic resonance study of Nitric Oxide hemoglobin. J Biol Chem. 1968;243:4350–4357. [PubMed] [Google Scholar]
- 19.Gadsby PM, Peterson J, Foote N, Greenwood C, Thomson AJ. Identification of the ligand-exchange process in the alkaline transition of horse heart cytochrome c. Biochem J. 1987;246:43–54. doi: 10.1042/bj2460043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brautigan DL, et al. Multiple low spin forms of the cytochrome c ferrihemochrome. EPR spectra of various eukaryotic and prokaryotic cytochromes c. J Biol Chem. 1977;252:574–582. [PubMed] [Google Scholar]
- 21.Carraway AD, Miller GT, Pearce LL, Peterson J. The Alkaline Transition of Bis(N-acetylated) Heme Undecapeptide. Inorg Chem. 1998;37:4654–4661. doi: 10.1021/ic971391u. [DOI] [PubMed] [Google Scholar]
- 22.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 23.Du C, et al. Mitochondrial ROS and radiation induced transformation in mouse embryonic fibroblasts. Cancer Biol Ther. 2009;8:1962–1971. doi: 10.4161/cbt.8.20.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, et al. Loss of manganese superoxide dismutase leads to abnormal growth and signal transduction in mouse embryonic fibroblasts. Free Radic Biol Med. 2010;49:1255–1262. doi: 10.1016/j.freeradbiomed.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 26.Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009;11:1050–1062. doi: 10.1016/j.micinf.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 27.Nagley P, Higgins GC, Atkin JD, Beart PM. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta. 2010;1802:167–185. doi: 10.1016/j.bbadis.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Porteous CM, et al. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim Biophys Acta. 2010;1800:1009–1017. doi: 10.1016/j.bbagen.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Logtenberg SJ, et al. Health-related quality of life, treatment satisfaction, and costs associated with intraperitoneal versus subcutaneous insulin administration in type 1 diabetes: a randomized controlled trial. Diabetes Care. 2010;33:1169–1172. doi: 10.2337/dc09-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishigami H, et al. Phase II study of weekly intravenous and intraperitoneal paclitaxel combined with S-1 for advanced gastric cancer with peritoneal metastasis. Ann Oncol. 2010;21:67–70. doi: 10.1093/annonc/mdp260. [DOI] [PubMed] [Google Scholar]
- 31.Kahokehr A, Sammour T, Srinivasa S, Hill AG. Systematic review and meta-analysis of intraperitoneal local anaesthetic for pain reduction after laparoscopic gastric procedures. Br J Surg. 2011;98:29–36. doi: 10.1002/bjs.7293. [DOI] [PubMed] [Google Scholar]
- 32.Ulsh BA. Checking the foundation: recent radiobiology and the linear no-threshold theory. Health Phys. 2010;99:747–758. doi: 10.1097/HP.0b013e3181e32477. [DOI] [PubMed] [Google Scholar]
- 33.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–942. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–217. doi: 10.1038/nature05077. [DOI] [PubMed] [Google Scholar]
- 35.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leonova KI, et al. A small molecule inhibitor of p53 stimulates amplification of hematopoietic stem cells but does not promote tumor development in mice. Cell Cycle. 2010;9:1434–1443. doi: 10.4161/cc.9.7.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gudkov AV, Komarova EA. Prospective therapeutic applications of p53 inhibitors. Biochem Biophys Res Commun. 2005;331:726–736. doi: 10.1016/j.bbrc.2005.03.153. [DOI] [PubMed] [Google Scholar]
- 38.Gudkov AV, Komarova EA. Radioprotection: smart games with death. J Clin Invest. 2010;120:2270–2273. doi: 10.1172/JCI43794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang ZL, et al. Roles for metallothionein and zinc in mediating the protective effects of nitric oxide on lipopolysaccharide-induced apoptosis. Mol Cell Biochem. 2002;234/235:211–217. [Google Scholar]
- 40.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008;Chapter 8(Unit 8):14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 41.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 42.Yanamala N, Tirupula KC, Klein-Seetharaman J. Preferential binding of allosteric modulators to active and inactive conformational states of metabotropic glutamate receptors. BMC Bioinformatics. 2008;9(Suppl 1):S16. doi: 10.1186/1471-2105-9-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedretti A, Villa L, Vistoli G. VEGA--an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des. 2004;18:167–173. doi: 10.1023/b:jcam.0000035186.90683.f2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.