

Abstract
The microRNA (miRNA)-guided RNA silencing pathway is a central and well-defined cellular process involved in messenger RNA (mRNA) translational control. This complex regulatory process is achieved by a well orchestrated machinery composed of a relatively few protein components, among which the ribonuclease III (RNase III) Dicer and Argonaute 2 (Ago2) play a central role. These two proteins are essential and it is of particular interest to measure and detect their catalytic activity under various situations and/or conditions. In this chapter, we describe different protocols that aim to study and determine the catalytic activity of Dicer and Ago2 in cell extracts, immune complexes and size-fractionated cell extracts. Another protocol aimed at assessing miRNA binding to Ago2 is also described. These experimental approaches are likely to be useful to researchers investigating the main steps of miRNA biogenesis and function in human health and diseases.
Keywords: Dicer, Argonaute 2, enzyme activity, microRNA, microRNA precursor, messenger RNA target, gene regulation, method
1. Introduction
The microRNA (miRNA)-guided RNA silencing pathway is a recently discovered gene regulatory process present in almost all eukaryotic cells and based on miRNAs. These small RNA species of approximatively 21 to 23 nucleotides (nt) are encoded by the genome and are responsible for the recognition and translational control of specific messenger RNA (mRNAs). Involving relatively few protein components, this complex and well integrated regulatory pathway plays a key role in recognizing a multitude of mRNA targets (1). Recent estimates suggest that up to 90% of the genes may be regulated by miRNAs in humans (2). Understanding the biological role and importance, as well as the possible defects of the miRNA-guided RNA silencing pathway is of great interest, and some protein components have been already implicated in some human diseases (3).
MiRNA genes are transcribed by RNA polymerase II (RNA polII) into primary miRNAs (pri-miRNA) transcripts that adopt hairpin folds. The pri-miRNAs are recognized by the nuclear microprocessor complex, composed of the ribonuclease III (RNase III) Drosha and the DiGeorge syndrome Critical Region gene 8 (DGCR8) protein (4–7), and processed into a miRNA precursor (pre-miRNA). After being exported to the cytoplasm via Exportin-5 (8), the pre-miRNA is recognized by the pre-miRNA processing complex, composed of the RNAse III Dicer (9, 10), the TAR RNA binding protein (TRBP) (11, 12) and the PKR-activating protein (PACT) (13), to generate a miRNA:miRNA* duplex. The complex is then joined by the Argonaute 2 (Ago2) protein, and the miRNA guide strand is selected based on the relative stability of the duplex extremities, to form a miRNA-containing ribonucleoprotein (miRNP) complex (12). The associated miRNA confers to the miRNP complex the ability to recognize specific binding sites generally located in the 3′ untranslated region (UTR) of different mRNAs. The mRNA will be cleaved if the complementarity between the miRNA and its binding site is perfect, or its translation regulated if the complementarity is imperfect (14). In this latter case, the repressed mRNA is translocated to the P-bodies, after which the mRNA can either be degraded or returned to the translational machinery for expression upon a specific cellular signal (15, 16).
Two of the major components of the miRNA-guided RNA silencing pathway are the RNase III Dicer and Ago2. These proteins are essential, and deregulation of their expression can have a major impact on normal cell functions (for a recent review, see Perron and Provost, (3)). Dicer recognizes its pre-miRNA substrates via its PAZ domain through the characteristic extremity harboring of pre-miRNA, formed by a 5′ phosphate and a 3′ hydroxylated end with 2-nt overhang, which represent the cleavage signature of members of the RNases III family of enzymes (17). The miRNA:miRNA* duplex is then excised by Dicer through the concerted intramolecular homodimerization of its two RNase III domains (18, 19).
As for Ago2, it is a member of the PAZ and PIWI domain (PPD) protein family expressed in metazoans and fungi, with the notable exception of the budding yeast Saccharomyces cerevisiae (20, 21). Ago2 harbors a binding pocket for miRNAs, in its PAZ domain, that mediate recognition of the characteristic 2-nt 3′ overhangs of miRNA duplexes (22–25). Acting in concert with the PAZ domain, the PIWI domain cleaves the mRNA strand between the nucleotides paired with the miRNA nt 10 and 11 if the complementarity is perfect. An active miRNP complex can then be regenerated and initiate a new round of mRNA cleavage, along a process known to amplify RNA silencing (26, 27).
In this chapter, we describe different protocols aimed to study and assess the specific catalytic activity of Dicer and Ago2 under various situations and/or conditions. The protocols can be easily transposed to different experimental contexts, i.e. cell types, cell lysates or immune complexes, and use various RNA substrates. Therefore, it is possible to compare wild-type and mutated proteins, as well as cells and tissues related to human diseases. We first propose a protocol for measuring Dicer and Ago2 catalytic activities in cell extracts and immune complexes, followed by the analytical methods (denaturing PAGE and an efficient Northern blot protocol to detect miRNAs) required to visualize the results. We also present a variation of these protocols to facilitate the study of fractionated cell extracts. Finally, we describe an efficient method to validate the presence of our miRNA of interest in Ago2 complexes.
2. Materials
It is important to use diethyl pyrocarbonate (DEPC)-treated water (see Note 1) in all preparation of the different solutions. Use RNase/DNase-free material in all conditions and always wear gloves for protection.
2.1 Cell culture
Dulbecco’s modified Eagle’s medium (DMEM) or Roswell Park Memorial Institute 1640 medium (RPMI) supplemented with 10% (v/v) fetal bovine serum (FBS), 1 mM sodium pyruvate, 100 units/mL penicillin, 100 μg/mL streptomycin and 2 mM L-glutamine.
Phosphate-buffered saline (PBS).
1X Trypsin- ethylenediaminetetraacetic acid (EDTA) solution.
2.2 In vitro transcription and radiolabeling of RNA transcript
MEGAshort script T7 kit (Ambion).
α 32P UTP, (10 μCi/μL, ~ 3000 Ci/mmol) (Perkin Life Science, 250μCi).
γ 32P ATP (10 μCi/μL, ~ 3000 Ci/mmol) (Perkin Life Science, 250μCi).
RNase/DNase-free screw-cap tubes (VWR).
0.5 M EDTA pH 8.0.
Calf intestine alkaline phosphatase (CIAP) with its 10X reaction buffer (GE Healthcare)
Opti-kinase with its 10X reaction buffer (USB Affimetrix).
RNase/DNase-free microcentrifuge tubes (VWR).
Sephadex-G25 column (GE Healthcare).
Glycogen (20 mg/m).
RNA annealing buffer: 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 25 mM NaCl)
RNA gel extraction buffer: 0.5 M Ammonium Acetate, 1 mM EDTA, 0.2% SDS.
2.3 Dicer RNase assay
2.3.1 S10 cell extracts
Dicer lysis buffer: 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 0.25% NP-40 (stored at 4°C). Add 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease cocktail inhibitor without EDTA 1X (Roche), prior to use.
2X Dicer assay buffer: 20 mM Tris-HCl pH 7.5, 2 mM MgCl2, 75 mM NaCl, 10% glycerol. Add 1 mM PMSF and protease inhibitor cocktail mix without EDTA 1X, prior to use.
2.3.2 Immune complexes
Dicer immunoprecipitation (IP) lysis buffer: 50 mM Tris-HCl pH 8.0, 137 mM NaCl, 1% Triton X-100 (stored at 4°C). Add 1 mM PMSF and protease cocktail inhibitor without EDTA 1X, prior to use.
Dicer IP washing buffer: 20 mM Tris-HCl pH 7.5, 2 mM MgCl2 (stored at 4°C).
2X Dicer IP assay buffer: 20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM dithiothreitol (DTT), 2 mM adenosine triphosphate (28) (see Note 5), 10% Superase•In (Ambion). Prepare immediately prior to use.
Protein G agarose beads (Roche).
Control IgG and a suitable anti-Dicer antibody, such as our rabbit polyclonal anti-Dicer antibody (9).
2.4 Ago2 cleavage assay
2.4.1 S100 cell extracts
2X Ago2 lysis buffer: 100 mM KOAc, 40 mM HEPES, 5 mM MgCl2, 2 mM DTT, 0.35% Triton X-100, adjust pH to 7.6 (stored at 4°C). Add 1 mM PMSF and protease cocktail inhibitor without EDTA 1X, prior to use.
10 mM ATP/2 mM guanosine triphosphate (GTP) solution (see Note 5).
Superase·In (Ambion).
2.4.2 Immune complexes
Ago2 IP lysis buffer: 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 0.25% NP-40 (stored at 4°C). Add 1 mM PMSF and protease cocktail inhibitor without EDTA 1X, prior to use.
Ago2 IP washing buffer: 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 0.1% NP-40 (stored at 4°C).
2x Ago2 lysis buffer (see Subheading 3.4.1).
10 mM ATP/2 mM GTP solution (see Note 5).
Superase·In (Ambion, cat. no. 2694).
Control IgG and anti-Ago2/EIF2C2 antibody. Our laboratory uses a mouse monoclonal antibody against human Ago2 (Abnova).
2.5 miRNA detection in Ago2 immune complex
Ago2 IP lysis buffer (see Subheading 2.4.2).
Ago2 IP washing buffer (see Subheading 2.4.2).
Yeast tRNA (5 μg/mL) (Ambion).
Protein G agarose beads (Roche).
Control IgG and Ago2 antibody (Abnova).
2.6 Size-fractionation of cell extracts using a Fast Protein Liquid Chromatography (FPLC) system
FPLC lysis buffer: 50 mM Tris-HCl pH 8.0, 137 mM NaCl, 1% Triton X-100 (stored at 4°C). Add 1 mM PMSF and protease cocktail inhibitor without EDTA 1X, prior to use.
0.2 μm filter (Pall).
Tris elution buffer: 20 mM Tris-HCl pH 7.5, 150 mM NaCl (stored at 4°C).
2X Dicer FPLC assay buffer: 20 mM Tris-HCl pH 7.5, 4 mM MgCl2, 75 mM NaCl, 10% Glycerol. Add 2 mM PMSF and protease cocktail inhibitor without EDTA 2X, prior to use.
2X Ago2 FPLC assay buffer: 100 mM KOAc, 40 mM HEPES, 5 mM MgCl2, adjust pH to 7.6 (stored at 4°C) Add 4 mM DTT, 2 mM ATP and 0.2 mM GTP, prior to use.
2.7 RNA extraction
RNase/DNase-free screw-cap tubes (VWR).
Glycogen (20 mg/mL) (Roche).
Yeast tRNA (5 μg/mL) (Ambion).
0.5 M EDTA pH 8.0.
Proteinase K (20 mg/mL) (Ambion).
Proteinase K buffer: 200 mM NaCl, 20 mM Tris pH 8.0, 2 mM EDTA, 1% SDS.
Acid Phenol:CHCl3 5:1 solution pH 4.5 (Ambion).
3 M sodium acetate, pH 5.5.
5 M ammonium acetate.
70% and 100% ethanol (ETOH).
2.8 Denaturing PAGE and Northern Blot analysis of small RNAs with EDC cross-linking
Acrylamide/Bis-Acrylamide 19:1 40% (Bio-Rad) (this is a neurotoxin when unpolymerized and care should be taken to avoid exposure).
Urea.
10X Tris-borate-EDTA (TBE 10X): 89 mM Tris, 89 mM boric acid, 2 mM EDTA.
10 X MOPS/NaOH solution: 200 mM MOPS, 0.05 M sodium acetate, 0.1 M EDTA pH 8.0, adjust at pH 7.0 with NaOH. Keep at room temperature (RT) for two weeks. Protect from light.
N,N,N,N′-Tetramethyl-ethylenediamine (TEMED). TEMED is very corrosive and flammable, and should be handled with care. Wear gloves and work in a chemical hood.
Ammonium persulfate (APS) (prepare a 10% stock solution in water and stored at −20°C).
Hybond NX nylon membrane (GE Healthcare) and 3MM chromatography paper.
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) (Thermo Scientific).
1-methylimidazole (Sigma).
20X SSC: 3 M NaCl, 0.3 M sodium citrate, adjust at pH 7.0 with HCl.
Prehybridization solution: 2X SSC, 1% SDS, 100 μg/mL ssDNA.
Wash solution: 0.2X SSC, 2% SDS.
Stripping buffer: 10 mM Tris/HCl, pH 8.5, 5 mM EDTA, 0.1% SDS.
Decade marker (Ambion) (prepare as described in the company protocol with γ 32P ATP).
2.9 Equipment
Fast protein liquid chromatography (FPLC) Akta system.
Refrigerated ultracentrifuge and microcentrifuge.
Spectrophotometer.
Vertical gel electrophoresis system with a small or large tank (GE Healthcare).
Semidry electroblotter (Fisher Biotech).
Roller for radioactivity. Hybridization oven.
End-over-end mixer.
Phosphorimager and storage phosphor screen, or X-ray film with intensifying screen, and an X-ray film processor.
Beta scintillation counter and Geiger counter.
3. Methods
3.1 In vitro transcription and radiolabeling of RNA transcript
These protocols are useful for different RNA substrates as well for RNA probes for Northern blot analysis. DNA templates will be different and prepared according to your conditions and specific requirements. T7-mediated in vitro transcription requires a DNA template with a T7 RNA polymerase promoter site (see the MEGAshort script T7 kit protocol). The template can be prepared by annealing of two complementary oligonucleotides, or by PCR filling with a T7 RNA polymerase promoter site oligonucleotide and a complementary oligonucleotide containing this sequence and the sequence to be transcribed. For Dicer RNA substrate, select your pre-miRNA sequence of interest (see Note 2) in miRBase http://microrna.sanger.ac.uk, which is a searchable database of published miRNA sequences and annotation (29), from which a complementary probe can be prepared (see Note 3). Finally, to detect Ago2 catalytic activity, the most efficient RNA substrate is the open-open RNA probe described by Ameres et al. (30).
3.1.1 In vitro transcription for RNA radiolabeling with [α 32P] UTP
Since working with radioactive material requires particular attention, be sure to have all the relevant information at hand (see Note 6).
Mix, according to the MEGAshort script T7 kit protocol, water, DNA template, ATP, CTP, GTP, transcription buffer, T7 RNA polymerase and add, at the end, the 20 μCi of [α 32P] UTP. Incubate for 3 h at 37°C. Add DNase I, mix and incubate for 15 min at 37°C. Stop the reaction by adding 1 μL of EDTA 0.5 M, mix and incubate for 2 min at RT (see Note 7 RNA probe for Northern blot analysis). Add 20 μL of gel loading buffer (GLB) II (provided in the MEGAshort script T7 kit). Heat for 5 min at 95°C and quickly put on ice for 5 min.
Load all 42 μL on a denaturing PAGE (see the protocol below, Subheading 3.6).
Run at 275 V in TBE 1X as gel running buffer, until the bromophenol blue reaches 2 inches from bottom of the gel (duration: ~ 2h30).
When the migration is finished, keep the gel on one of the two glass plates and wrap in plastic wrap. Put the gel in a plexiglass box (see Note 8).
In the dark room, expose the gel to an X-ray film for 30 sec and 2 min. Back in the radioactivity room, cut out the band corresponding to your RNA probe.
Cut the gel slice in small pieces and put into a 1.5 mL screw-cap tube. Add 400 μL of gel extraction buffer and incubate overnight at 37°C.
Centrifuge at 600 g for 1 min at 4°C. Transfer the supernatant into a new 1.5 mL screw-cap tube. Add 1 μL of glycogen and 1 mL of ice-cold ethanol 100%. Incubate for 30 min at −80°C, then centrifuge at 16,000 g for 30 min at 4°C.
Remove the supernatant with a pipet and wash with 900 μL of ice-cold ethanol 70%, then centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet and resuspend the Dicer substrate probe in 20 μL of annealing buffer. Anneal the RNA probe by heating for 5 min at 85°C and remove the block from the heating block and allow temperature to cool down to RT.
Remove 1 μL, add to 5 mL of scintillation liquid and count in the beta scintillation counter.
Aliquot in different tubes (make a tube ready to use at 40,000 cpm/μL) and store at −20°C.
3.1.2 In vitro transcription for RNA radiolabeling with [γ-32P] ATP
Mix, according to the MEGAshort script T7 kit protocol, water, DNA template, ATP, CTP, GTP, UTP, transcription buffer and T7 RNA polymerase to obtain a 20 μL final volume. Incubate for 3 h at 37°C. Add DNase I, mix and incubate for 15 min at 37°C. Stop the reaction by adding 1 μL of EDTA 0.5 M, mix and incubate for 2 min at RT. Keep the reaction on ice.
Prepare a Sephadex-G25 column according to the manufacturer’s protocol.
Put the RNA on the column and centrifuge at 600 g for 2 min at 4°C.
Add 180 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Centrifuge at 16,000 g for 4 min at 4°C. Transfer the supernatant into a new 1.5 mL screw-cap tube.
Add 1 μL of glycogen and 1 mL of ice-cold ethanol 100%. Incubate for 30 min at −80°C, then centrifuge at 16,000 g for 30 min at 4°C.
Remove the supernatant with a pipet and wash with 900 μL of ice-cold ethanol 70%, then centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet and resuspend in 40 μL of water. Add 5 μL of 10X CIAP buffer, 4 μL of water and 1 μL of CIAP. Incubate for 1 h at 37°C.
Add 150 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Centrifuge at 16,000 g for 4 min at 4°C. Transfer the supernatant into a new 1.5 mL screw-cap tube.
Add 1 μL of glycogen, 50 μL of 5 M ammonium acetate and 700 μL of ice-cold ethanol 100%. Incubate for 30 min at −80°C, then centrifuge at 16,000 g for 30 min at 4°C.
Remove the supernatant with a pipet and wash with 900 μL of ice-cold ethanol 70%, then centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet and resuspend it in 20 μL of water. Evaluate the RNA concentration (see Note 9).
To 5 pmol of dephosphorylated RNA, add 2 μL of 10X kinase buffer, 20 μCi of [γ-32P] ATP, 10 U of Opti-kinase and complete with water to have a 20-μL final volume. Incubate for 1 h at 37°C. Add 20 μL of GLB II. Heat for 5 min at 95°C and quickly put on ice for 5 min.
Purify the RNA on a denaturing PAGE as described in Subheading 3.1.1, steps 3 to 8.
If the RNA is an Ago2 RNA target, resuspend it in 20 μL of RNase free water. If the RNA is a Dicer substrate, resuspend it in 20 μL of annealing buffer and anneal the probe by heating for 5 min at 85°C and remove the block from the heating block and allow temperature to cool down to RT.
Remove 1 μL, add to 5 mL of scintillation liquid and count in the beta scintillation counter.
Aliquot in different tubes (make a tube ready to use at 40,000 cpm/μL for Dicer RNase assay or 10,000 cpm/μL for Ago2 cleavage assay) and store at −20°C.
3.2 Detection of human Dicer activity
3.2.1 Detection of human Dicer activity in S10 cell extracts
This protocol is adapted from Haase et al. (11).
Lyse mammalian cells (e.g. HEK293) from one 100-mm petri dish with 250 μL of Dicer lysis buffer, incubate for 15 min on ice, and centrifuge at 10,000 g for 10 min at 4°C. Determine the protein concentration by the method of Bradford (31) using the Bio-Rad dye reagent, with bovine serum albumin as standard. Adjust protein concentration to 5 mg/mL with Dicer lysis buffer.
Prepare the 50-μL reaction on ice in 1.5 mL screw-cap tubes as follows: 25 μL of S10 cell extract at 5 mg/mL (125 μg protein in total), 24 μL of 2X Dicer assay buffer and 1 μL of a radiolabeled dsRNA substrate probe (40,000 cpm/μL). Vortex.
Incubate for 10, 30, 60 and 120 min at 37°C.
To stop the reaction, quickly add 150 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C.
Precipitate RNA by adding 50 μL of 5 M ammonium acetate, 700 μL of ice-cold ethanol 100%, 1 μL of yeast tRNA and 1 μL of glycogen. Mix well and incubate for at least 1 h at −80°C.
Centrifuge at 16,000 g for 30 min at 4°C. Wash with 400 μL of ice-cold ethanol 70%. Centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet, add 10 μL of GLB II, and heat for 5 min at 95°C.
Load RNA on a denaturing PAGE (see the detailed protocol in Subheading 3.6). A typical result is shown in Figure 1A.
Figure 1. Detection of human Dicer activity in HEK293 cells.

(A) S10 protein extracts were incubated in the presence of a 32P-UTP labeled human let-7a-3 pre-miRNA substrate for the indicated period of time. (B) Endogenous Dicer immune complexes were incubated with a 32P-UTP or 32P-ATP labeled human let-7a-3 pre-miRNA substrate for 60 min. Lanes 1 and 5 represent the untreated probe (−). (C) Anti-Flag immune complexes derived from cells overexpressing Flag-Dicer, or transfected with empty plasmid, and endogenous Dicer immune complexes derived from untransfected HEK293 cells (−) were incubated in the presence of 32P-UTP-labeled TAR RNA substrate for 60 min. (A–C) The reactions were analyzed by denaturing PAGE and autoradiography. A 10-nt RNA ladder was used as a size marker. (D) The immune complexes from (C) were analyzed for the presence of Dicer protein by 7% SDS-PAGE and immunoblotting using anti-Dicer antibody (9).
3.2.2 Detection of human Dicer activity in immune complexes
This protocol is adapted from Provost et al. (9).
For IP of endogenous proteins, plate the cells and incubate in a CO2 incubator overnight at 37°C. For IP of overexpressed proteins, plate the cells so they reach appropriate confluency for transfection by calcium phosphate, or another transfection procedure, on the following day and incubate the cells an additional 24 to 48 h.
Lyse cells from one 100-mm petri dish with 1 mL of Dicer IP lysis buffer, incubate for 15 min on ice, and centrifuge at 10,000 g for 10 min at 4°C. Determine the protein concentration by the method of Bradford (31).
Prepare the IP by incubating 1 mg of protein extract with the appropriate antibody (see Note 10) for 1 h at 4°C under continuous rotation. During this time, pre-wash Protein-G agarose beads extensively (100 volumes, 2 times) with Dicer IP lysis buffer and resuspend the beads in 1 volume of buffer. Then, add 20 μL of beads (50% slurry) to the reaction and continue the incubation for an additionnal 3 h.
Wash the immune complexes 3 times with 1 mL of Dicer IP lysis buffer. Then, wash once with the Dicer IP washing buffer and transfer the beads into a clean 1.5 mL screw-cap tube. Be sure to leave ~10 μL of beads in the tube, by gently removing the supernatant with a pipette.
Prepare the 20-μL reaction on ice in a 1.5 mL screw-cap tube as follows: To the beads, add 9 μL of 2X Dicer IP assay buffer and 1 μL of a radiolabeled dsRNA substrate probe (40,000 cpm/μL). Vortex.
Incubate for 60 min at 37°C.
Stop the reaction by quickly adding 180 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C.
Perform RNA precipitation and gel electrophoresis as described in Subheading 3.2.1, steps 5 to 8. Typical results are shown in Figure 1 B–C when using different RNA labeled probe.
3.3 Detection of Ago2 cleavage activity
3.3.1 Detection of Ago2 cleavage activity in S100 cell extracts
This protocol is adapted from Ameres et al. (30).
Lyse mammalian cells from one 100-mm petri dish with 150 μL 2X Ago2 lysis buffer, incubate for 15 min on ice and perform an ultracentrifugation at 100,000 g for 1 h at 4°C. Determine the protein concentration by the method of Bradford (31). Adjust the protein concentration of the extract at 5 mg/mL with Ago2 cleavage assay lysis buffer.
Prepare the 20-μL reaction on ice in a 1.5 mL screw-cap tube as follows: 10 μL of S100 cell extract at 5 mg/mL (50 μg protein in total), 2 μL of 10 mM ATP/2 mM GTP solution, 0.5 μL of Superase·In, 6.5 μL of water and, finally, 1 μL of a radiolabeled RNA target probe (10,000 cpm/μL). Vortex.
Incubate for 90 min at 30°C.
Add 20 μL of proteinase K buffer and 1 μL of proteinase K. Incubate for 20 min at 50°C.
Stop the reaction by quickly adding 160 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C.
Precipitate RNA by adding 20 μL of 3 M sodium acetate pH 5.2, 500 μL of ice-cold ethanol 100%, 1 μL yeast tRNA and 1 μL of glycogen. Mix well and incubate for at least 1 h at −80°C.
Centrifuge at 16,000 g for 30 min at 4°C. Wash with 500 μL of ice-cold ethanol 70%. Centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet and add 10 μL of GLB II.
Load RNA on a denaturing PAGE (see the detailed protocol in Subheading 3.6). A typical result is shown in Figure 2 A.
Figure 2. Detection of human Ago2 activity in megakaryocytes.
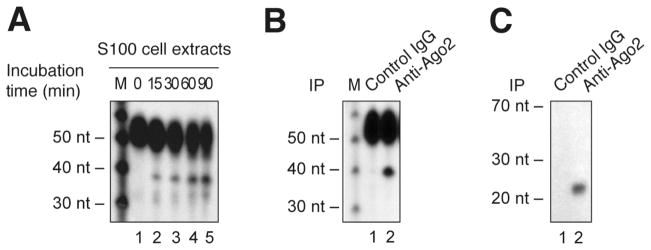
RISC activity assays were performed using S100 protein extracts (A) or Ago2 immune complex (B) from MEG-01 cell line and incubated in the presence of a 32P-labeled sensor RNA bearing a binding site complementary to human miR-223. (C) Ago2 immune complexes were analyzed by Northern blot for the presence of miR-223. The reactions were analyzed by denaturing PAGE and autoradiography. A 10-nt RNA ladder was used as a size marker (M). Adapted from Landry et al. (35), with permission from The Nature Publishing Group.
3.3.2 Detection of Ago2 cleavage activity in immune complexes
This protocol is adapted from Ameres et al. (30), Ender et al. (32), Rudel et al. (33).
For IP of endogenous proteins, plate the cells and incubate in a CO2 incubator overnight at 37°C. For IP of overexpressed proteins, plate the cells so they reach the appropriate confluency for transfection by calcium phosphate, or another transfection procedure, on the following day and incubate the cells for an additional 24 to 48 h.
Lyse cells from one 100-mm petri dish with 1 mL of 2X Ago2 IP lysis buffer, incubate for 15 min on ice, and centrifuge at 10,000 g for 10 min at 4°C. Determine the protein concentration by the method of Bradford (31).
Prepare the IP by incubating 1 mg of protein extract with the appropriate antibody (see Note 11) for 1 h at 4°C under continuous rotation. During this time, pre-wash Protein-G agarose beads extensively (100 volumes, 2 times) with 2X Ago2 lysis buffer and resuspend the beads in 1 volume of buffer. Then, add 20 μL of beads (50% slurry) to the reaction and continue the incubation for an additional 3 h.
Wash the immune complexes 3 times with 1 mL of Ago2 IP washing buffer. Then, wash a last time with the 2X Ago2 lysis buffer and transfer the beads into a new 1.5 mL screw-cap tube. Be sure to leave ~ 10 μL of beads in the tube, by gently removing the supernatant with a pipette.
Prepare the reaction on ice in a 1.5 mL screw-cap tube as follows: To the beads, add 10 μL of 1X Ago2 lysis buffer (a dilution of the 2X Ago2 lysis buffer), 2 μL of 10 mM ATP/2 mM GTP solution, 0.5 μL of Superase·In and finally 1 μL of a radiolabeled RNA target probe (10,000 cpm/μL). Vortex.
Incubate for 90 min at 30°C.
Add 20 μL of proteinase K buffer and 1 μL of proteinase K. Incubate for 20 min at 50°C.
Stop the reaction by quickly adding 180 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C.
Perform RNA precipitation as described in Subheading 3.3.1, steps 6 to 9. A typical result is shown in Figure 2 B.
3.4 Detection of miRNAs bound to Ago2 immune complexes
This protocol is adapted from Ameres et al. (30), Ender et al. (32), Rudel et al. (33).
Prepare cell extracts as described in Subheading 3.3.2, steps 1 and 2.
To 1 mg of proteins, add 6 μL of yeast tRNA, 1.5 μL of Superase·In and adjust the final reaction volume to 300 μL with 2X Ago2 IP lysis buffer (used in Subheading 3.3.2). Pre-clear the reaction mixture with 10 μL of Protein-G agarose beads pre-washed with 2X Ago2 IP lysis buffer. Incubate for 45 min at 4°C under continuous rotation.
Centrifuge at 600 g for 1 min at 4°C, and transfer the supernatant to a new 1.5 mL screw-cap tube. Add 10 μL of fresh pre-washed Protein-G agarose beads and the appropriate antibody (see Note 11) and incubate for 3 h at 4°C under continuous rotation.
Wash 6 times with 1 mL of Ago2 IP washing buffer.
Add 20 μL of proteinase K buffer and 1 μL of proteinase K. Incubate for 20 min at 50°C.
Stop the reaction by quickly adding 150 μL of water and 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C.
Precipitate RNA by adding 20 μL of 3 M sodium acetate pH 5.2, 500 μL of ice-cold ethanol 100% and 1 μL of glycogen. Mix well and incubate for at least 1 h at −80°C.
Centrifuge at 16,000 g for 30 min at 4°C. Wash with 500 μL of ice-cold ethanol 70%. Centrifuge at 16,000 g for 5 min at 4°C.
Dry the RNA pellet, add 10 μL of GLB II and heat for 5 min at 95°C.
Load RNA on a MOPS denaturing PAGE to perform Northern blot analysis. (see the detailed protocol in Subheading 3.6). A typical result is shown in Figure 2 C.
3.5 Detection of human Dicer and Ago2 activity in size-fractionated cell extracts
Lyse cells from 3 to 4 100-mm petri dishes with FPLC lysis buffer, incubate for 15 min on ice and perform an ultracentrifugation at 100,000 g for 45 min at 4°C. Filter the supernatant through a 0.2 μm filter and determine the protein concentration by the method of Bradford (31). Load 1 mg (100 μL of 10 mg/mL) of proteins derived from this S100 cell extract on a Superose 6 column (10/300 GL) using an ÄKTA FPLC system to give fractions of 400 μL in the Tris elution buffer (0.3 mL/min) (see Note 12).
Divide each fraction as follows; transfer 100 μL for Dicer RNase activity assay and 100 μL for Ago2 cleavage assay in separate 1.5 mL screw-cap tubes. For the input, take 5 μL of the S100 cell extract (10 mg/mL) and add 95 μL of elution buffer. Elution buffer (100 μL) will serve as a negative loading control (see Note 13).
For Dicer RNase activity assay: To prepare the 200-μL reaction mixture, add 99 μL of 2X Dicer FPLC assay buffer and 1 μL of a radiolabeled dsRNA substrate, mix and incubate for 60 min at 37°C. To stop the reaction quickly add 200 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C. Perform RNA precipitation, as described in Subheading 3.2.1, steps 5 to 8. A typical result is shown in Figure 3 B.
For Ago2 cleavage assay: To prepare the 200-μL reaction mixture, add 1 μL of Superase·In to each tube and incubate for 10 min at RT. Then, add 99 μL of 2X Ago2 FPLC assay buffer and 1 μL of a radiolabeled RNA target, mix and incubate for 90 min at 30°C. To stop the reaction, add 200 μL of proteinase K buffer and 1 μL of proteinase K, and incubate for 15 min at 45°C. Then, add 400 μL of Acid Phenol:CHCl3 (5:1 solution, pH 4.5). Vortex for 20 sec. Separate the aqueous and organic phases by centrifugation at 16,000 g for 4 min at 4°C. Perform RNA precipitation, as described in Subheading 3.3.1, steps 6 to 9. A typical result is shown in Figure 3 C.
Figure 3. Characterization of an Ago2-containing effector complex competent in RNA silencing in HeLa cells.

(A) Extracts from HeLa cells were separated by gel filtration on a Superose 6 column and the fractions were analyzed by immunoblot analysis for the presence of Dicer and Ago2. (B) Selected (odd) fractions were tested for their intrinsic Dicer activity upon addition of a 32P-labeled human let-7a-3 pre-miRNA substrate. # indicates the expected miRNA product. (C) RISC activity assays were performed by using a 32P-labeled let-7c sensor RNA transcript. * Indicates the expected 38-nt cleavage products. The reactions were analyzed by denaturing PAGE and autoradiography. A 10-nt RNA ladder was used as a size marker. Adapted from Landry et al. (35), with permission from The Nature Publishing Group.
3.6 Denaturing PAGE
Prepare a 0.75-mm thick 10% polyacrylamide gel (19:1) containing 7 M urea. For a 30 mL preparation, mix 7.5 mL of polyacrylamide (19:1) (stock 40%), 12.6 g of urea, 3 mL of 10X TBE solution and complete with DEPC water. Incubate at 37°C and mix until urea is completely dissolved. Add 150 μL of 10% APS, 30 μL of TEMED, mix gently, and pour the gel immediately. The gel should polymerize in about 30 min.
Pre-run the gel at 250 V for 30 min in 1X TBE at RT. Use of an electrophoresis apparatus with a big tank will contribute to a constant migration temperature.
Rince 2 times the wells with the 1X TBE buffer before loading the RNA samples on the denaturating 10% polyacrylamide gel. Load a radiolabeled 10-nt Decade size marker in order to estimate the size of RNA species under study. Electrophorese at 275 V for 2 to 3 h.
Stop the electrophoresis when the bromophenol blue reaches 2 inches from bottom of the gel (duration: ~ 2h30).
Wrap the gel in a platic wrap and expose it to an X-ray film with an intensifying screen at −80°C or analyze with the phosphorimager.
3.7 Northern Blot analysis of small RNAs with EDC cross-linking
Protocol adapted from Pall et al. (34).
Prepare a 0.75-mm thick 10% polyacrylamide gel (19:1) containing 7 M urea. For a 30 mL preparation, mix 7.5 mL of polyacrylamide (19:1) (stock 40%), 12.6 g of urea, 3 mL of 10X MOPS solution and complete with DEPC water. Incubate at 37°C and mix until urea is completely dissolved. Add 150 μL of APS 10%, 30 μL of TEMED, mix gently, and pour the gel immediately. The gel should polymerize in about 30 min.
Pre-run the gel at 250 V for 30 min in 1X MOPS solution at RT. Use of an electrophoresis apparatus with a big tank will contribute to a constant migration temperature.
Rince 2 times the wells with the 1X MOPS buffer before loading the RNA samples on the denaturating 10% polyacrylamide gel. Load a radiolabeled 10-nt Decade size marker in order to estimate the size of RNA species under study. Electrophorese at 275 V for 2 to 3 h.
Stop the electrophoresis when the bromophenol blue reaches 2 inches from bottom of the gel (duration: ~ 2h30). Wash the gel with DEPC water for 1 min.
Prepare 6 filter papers and a membrane Hybond-NX that fit the size of the gel. Hydrate the filter papers and the membrane in the water for at least 5 min.
Place 3 filter papers on the bottom half of the semidry electroblotter. Eliminate air bubbles by rolling over with a glass pipette. Place the gel on the filter papers, cover with the membrane and remove air bubbles by rolling over the membrane with a glass pipette. Complete the sandwich with other 3 filter papers. Remove air bubbles. Place the top of the electroblotter and screw the notches not too tightly.
Transfer at 500 mA for 1 h at 4°C.
After the transfer, prepare the cross-linking reaction. Cut a sheet of filter paper slightly larger than the membrane.
Immediatly prior to use, prepare a fresh solution of EDC in 0.13 M 1-methylimidazole at pH 8.0. For a 20 × 16 cm Hybond NX nylon membrane, add 122.5 μL of 1-methylimidazole to 10 mL of water and adjust to pH 8.0 with 1 M HCl. Then add 0.373 g of EDC and complete to 12 mL.
Saturate the filter paper with the EDC solution. On a plastic wrap, place the nylon membrane face down and the EDC saturated paper over, and wrap it. Incubate for 2 h at 60°C.
Wash residual EDC with distilled water prior to pre-hybridization or dry and store the membrane at −20°C.
Prepare the probe, as described in Subheading 3.1.1.
Prehybridize the membrane in 10 mL of prehybridization solution with gentle agitation for at least 1 h at 50°C.
Hybridize the membrane in 10 mL of hybridization solution (same as pre-hybridization) containing at least 1 × 107 cpm of labeled antisens RNA probe with gentle agitation for 8 to 24 h at 50°C.
After the hybridization, wash the membrane 5 times with 25 mL of wash solution with gentle agitation for 10 min at 50°C.
With the Geiger counter, ensure that the membrane has been washed sufficiently. If not, wash once for 5 min at 60°C (or ~10°C lower than the estimated melting temperature of the probe) under agitation.
Wrap the blot in plastic wrap and expose to an X-ray film in the presence of an intensifying screen at −80°C or analyze with a phosphorimager.
If stripping is needed, place the membrane in stripping buffer and incubate for 1 min at 100°C.
Acknowledgments
M.P.P. was supported by a doctoral studentship from Natural Sciences and Engineering Research Council of Canada (NSERC). P.P. is a Senior Scholar from the Fonds de la Recherche en Santé du Québec (FRSQ). This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) (262938-2008), an Operating Grant from Health Canada/Canadian Institutes of Health Research (CIHR) (HOP-83069) and a CIHR/Rx&D Collaborative Research Grant (IRO-86239) to P.P.
Footnotes
Although DEPC water is commercially available, it can be prepared by adding 1 mL of DEPC to 1 L of milliQ water, mixing overnight and autoclaving. DEPC is carcinogenic and should be handled with care. Wear gloves and work under a chemical hood.
Human pre-let-7a-3 DNA template for in vitro transcription was prepared by PCR amplification of the following oligonucleotides: The human pre-let-7a-3 sequence oligonucleotide 5′-GGAAAGACAGTAGATTGTATAGTTATCCCATAGCAGGGCAGAGCCCCA AACTATACAACCTACTACCTCATATAGTGAGTCGTATTA-3′ and the T7 promoter oligonucleotide 5′-TAATACGACTCACTATA-3′. The PCR product was isolated on a 1% agarose gel and utilized as a DNA template. (see Figure 1 AB).
The DNA template used for in vitro transcription of the human miR-223 Northern blot probe was prepared by PCR amplification of the following oligonucleotides: The miR-223 detection probe oligonucleotide 5′-TGTCAGTTTGTCAAATACCCCACCCTATAGTGAGTCGTATTA-3′ and the T7 promoter oligonucleotide: 5′-TAATACGACTCACTATA-3′. The PCR product was isolated on a 1% agarose gel and utilized as a DNA template (see Figure 2 C).
The DNA template used for in vitro transcription of the sensor bearing a binding site complementary to human miR-223 was prepared by PCR amplification of the following oligonucleotides: The miR-223 binding site oligonucleotide 5′-TGTTCTAGTTGTCTATGTTAATCTGATTGTCAGTTTGTCAAATACCCCAG TGTTGTTGTGTCTATGTTAATGTGCCTATAGTGAGTCGTATTAAATT-3′ and the T7 promotor oligonucleotide: 5′-TAATACGACTCACTATA-3′. The PCR product was isolated on a 1% agarose gel and utilized as a DNA template (see Figure 2 A–B).
Preparation of the 1 M stock of ATP (200 μL final): 110 mg of ATP (ATP-5′-triphosphate, disodium salt, Sigma, cat. no. A-2383) are dissolved in 150 μL of 1 M Tris, pH 8.0. The pH should be verified on a pH paper strip and adjusted to 7.0 by adding 10 N NaOH, 10 μL at a time. Complete by adding water to 200 μL. Preparation of the 200 mM stock of GTP (400 μL final): 45 mg of GTP (GTP-5′-triphosphate, disodium salt) are dissolved in 320 μL of water. The pH should be verified on a pH paper strip and adjusted to 7.0 by adding 10 N NaOH, 10 μL at a time. Complete by adding water to 400 μL. Freeze at −20°C in small aliquots.
All radioactive substances should be used in accordance with local regulation and safety recommandations. Work in a safe environment behind plexiglass protection. The level of radioactivity used in these protocols is important enough to monitor any possible contamination at every step with a Geiger counter.
For Northern blot RNA probes, prepare a Sephadex-G25 column according to the manufacturer’s instructions. Place the labeled RNA on the column and centrifuge at 600 g for 2 min at 4°C. Remove 1 μL and add 5 mL of scintillation liquid for counting in a beta scintillation counter. The probe is then ready to be used in Northern blot analyses (see Subheading 3.7).
Be sure that the plastic wrap is not contaminated by carefully applying it on the gel. If necessary, change your glove protection before each manipulation. Verify that a piece of paper rubbed softly on the plastic wrap covering the gel is not contaminated with a Geiger counter to avoid contaminating the X-ray film processor in the dark room.
To measure RNA concentration, dilute your RNA sample 1/100 (3 μL RNA + 297 μL water) and read the absorbance at 260 nm and 280 nm on a spectrophotometer. For short nucleic acids (<200 nt), the concentration is determined by the following formula: A260 x dilution factor x 33 μg/mL. The purity of the RNA can be estimated from the A260/A280 ratio. A ratio of >1.8 and up to 2.1 is expected from highly pure RNA.
Determine the appropriate amount of antibody to be used for IP. The IP can be divided in half for concomitant Dicer RNase activity assay and Western blot analysis of the IP content in Dicer protein (see Figure 1 C–D).
Determine the appropriate amount of antibody to be used for IP. When using the anti-Ago2 antibody from Abnova, 2 μL was optimal.
It is important to utilize a Tris elution buffer in order not to interfere with RNA precipitation.
Reserve an additional 75 μL for the detection of protein(s) of interest in each fraction by Western blot. Another alternative is to perform IP of Dicer or Ago2 protein on each fraction and document the activity concealed in these immune complexes in vitro (see Subheadings 3.2.2 and 3.3.2).
References
- 1.Perron MP, Provost P. Protein interactions and complexes in human microRNA biogenesis and function. Front Biosci. 2008;13:2537–2547. doi: 10.2741/2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 3.Perron MP, Provost P. Protein components of the microRNA pathway and human diseases. Methods Mol Biol. 2009;487:369–385. doi: 10.1007/978-1-60327-547-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. Embo J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 7.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 8.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. Embo J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. Embo J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005;6:961–967. doi: 10.1038/sj.embor.7400509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO J. 2006;25:522–532. doi: 10.1038/sj.emboj.7600942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Rivas FV, Wohlschlegel J, Yates JR, 3rd, Parker R, Hannon GJ. A role for the P-body component GW182 in microRNA function. Nat Cell Biol. 2005;7:1261–1266. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macrae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Kataoka Y, Takeichi M, Uemura T. Developmental roles and molecular characterization of a Drosophila homologue of Arabidopsis Argonaute1, the founder of a novel gene superfamily. Genes Cells. 2001;6:313–325. doi: 10.1046/j.1365-2443.2001.00427.x. [DOI] [PubMed] [Google Scholar]
- 21.Cerutti L, Mian N, Bateman A. Domains in gene silencing and cell differentiation proteins: the novel PAZ domain and redefinition of the Piwi domain. Trends Biochem Sci. 2000;25:481–482. doi: 10.1016/s0968-0004(00)01641-8. [DOI] [PubMed] [Google Scholar]
- 22.Lingel A, Simon B, Izaurralde E, Sattler M. Nucleic acid 3′-end recognition by the Argonaute2 PAZ domain. Nat Struct Mol Biol. 2004;11:576–577. doi: 10.1038/nsmb777. [DOI] [PubMed] [Google Scholar]
- 23.Yan KS, Yan S, Farooq A, Han A, Zeng L, Zhou MM. Structure and conserved RNA binding of the PAZ domain. Nature. 2003;426:468–474. doi: 10.1038/nature02129. [DOI] [PubMed] [Google Scholar]
- 24.Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–1437. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- 25.Lingel A, Simon B, Izaurralde E, Sattler M. Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature. 2003;426:465–469. doi: 10.1038/nature02123. [DOI] [PubMed] [Google Scholar]
- 26.Yuan YR, Pei Y, Ma JB, Kuryavyi V, Zhadina M, Meister G, Chen HY, Dauter Z, Tuschl T, Patel DJ. Crystal structure of A. aeolicus argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Mol Cell. 2005;19:405–419. doi: 10.1016/j.molcel.2005.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 28.Leu YW, Rahmatpanah F, Shi H, Wei SH, Liu JC, Yan PS, Huang TH. Double RNA interference of DNMT3b and DNMT1 enhances DNA demethylation and gene reactivation. Cancer Res. 2003;63:6110–6115. [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130:101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.Ender C, Krek A, Friedlander MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. A human snoRNA with microRNA-like functions. Mol Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Rudel S, Flatley A, Weinmann L, Kremmer E, Meister G. A multifunctional human Argonaute2-specific monoclonal antibody. Rna. 2008;14:1244–1253. doi: 10.1261/rna.973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pall GS, Codony-Servat C, Byrne J, Ritchie L, Hamilton A. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Res. 2007;35:e60. doi: 10.1093/nar/gkm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landry P, Plante I, Ouellet DL, Perron MP, Rousseau G, Provost P. Existence of a microRNA pathway in anucleate platelets. Nat Struct Mol Biol. 2009;16:961–966. doi: 10.1038/nsmb.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]