

Abstract
Myelin proteolipid protein gene (Plp1) expression is temporally regulated in brain, which peaks during the active myelination period of CNS development. Previous studies with Plp1-lacZ transgenic mice demonstrated that (mouse) Plp1 intron 1 DNA is required for high levels of expression in oligodendrocytes. Deletion-transfection analysis revealed the intron contains a single positive regulatory element operative in the N20.1 oligodendroglial cell line, which was named ASE (antisilencer/enhancer) based on its functional properties in these cells. To investigate the role of the ASE in vivo, the element was deleted from the native gene in mouse using a Cre/lox strategy. While removal of the ASE from Plp1-lacZ constructs profoundly decreased expression in transfected oligodendroglial cell lines (N20.1 and Oli-neu), the element was dispensable to achieve normal levels of Plp1 gene expression in mouse during development (except perhaps at postnatal day 15) and throughout the remyelination period following cuprizone-induced (acute) demyelination. Thus, it is possible that the ASE is nonfunctional in vivo, or that loss of the ASE from the native gene in mouse can be compensated for by the presence of other regulatory elements within the Plp1 gene.
Keywords: cuprizone demyelination, enhancer, gene regulation, gene targeting, myelin proteolipid protein, remyelination
INTRODUCTION
The myelin proteolipid protein gene (Plp1) encodes the most abundant protein found in mature myelin from the CNS (Eng et al. 1968; Norton et al. 1973). Two major gene products are generated via alternative splicing. The DM20 isoform is identical to PLP except it lacks an internal 35 amino acid stretch encoded by the distal portion of exon 3 (Macklin et al. 1987; Nave et al. 1987b; Simons et al. 1987). PLP has been proposed to function as an adhesive strut between apposing layers of the myelin sheath, aiding in the compaction of CNS myelin and conferring structural stability (Gow et al. 1997). However, more recently, additional functions have been ascribed for PLP/DM20 including roles in oligodendrocyte-neuron interactions (Yool et al. 2001), signaling (Gudz et al. 2003), and programmed cell death (Skoff et al. 2004; Hüttemann et al. 2009).
The gene is present in a single copy on the X chromosome, and as such, several X-linked recessive disorders – Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia type 2 (SPG2) – arise from mutations in the human PLP1 gene (for reviews see Inoue 2005; Garbern 2007). These dysmyelinating disorders can be caused by a lack of the protein due to deletion of the chromosomal region containing PLP1, or conversely from increased PLP1 gene dosage, resulting in PLP1 overexpression and oligodendrocyte dysfunction. Thus, PLP1 gene activity in oligodendrocytes must be tightly regulated, befitting a ‘Goldilocks scenario’ where too little or too much of the protein have negative consequences. Expression of the gene is temporally regulated in oligodendrocytes (reviewed in Wight and Dobretsova 2004), with peak levels being attained during the active myelinating period of CNS development.
Previous studies with mice that harbor the PLP(+)Z transgene [contains mouse Plp1 genomic DNA from the proximal 2.4 kb of 5′-flanking DNA to the first 37 bp of exon 2 linked in-frame with a lacZ reporter gene] demonstrated that the Plp1-lacZ fusion gene was expressed in brain, in a temporal manner, consistent to that of the endogenous Plp1 gene (Wight et al. 1993; Li et al. 2002b). Yet expression of a comparable transgene, PLP(−)Z, which altogether lacks Plp1 intron 1, was very much blunted, resulting in low levels of Plp1-lacZ gene activity in brain throughout development (Li et al. 2002b). Taken together, these results suggest that Plp1 intron DNA contain regulatory element(s) that are important in mediating the dramatic increase in Plp1 gene activity observed during the active myelination period. Subsequently, deletion-transfection analysis was performed with Plp1-lacZ constructs that lack a portion of Plp1 intron 1 DNA to map the location of these regulatory element(s). Several regulatory elements were found to be functionally active in N20.1 cells [immortalized oligodendroglial cell line derived from mouse which expresses the Plp1 gene (Verity et al. 1993)], consisting of a couple of negative regulatory elements (Dobretsova and Wight 1999; Li et al. 2002a) and a single positive element (Dobretsova and Wight 1999). The positive element, termed ASE for antisilencer/enhancer, is located within intron 1 positions 1083-1177 (Dobretsova et al. 2000), based on numbering the entire intron from positions 1 to 8140 (Wight and Dobretsova 1997). To test whether the ASE is the element responsible for mediating the surge in Plp1 gene activity during the active myelination period of CNS development, it was targeted for deletion from the native gene in mouse through homologous recombination, using a Cre/lox system. Results presented here suggest that the ASE is largely dispensable for the developmental regulation of Plp1 gene expression in vivo. Likewise, lack of the ASE did not seem to impair Plp1 gene expression in adult mice actively undergoing CNS remyelination.
METHODS
Cell culture
Two mouse oligodendroglial cell lines were used in the study. N20.1 cells were grown at 34°C in a 1:1 mixture of Ham’s F-12/Dulbecco’s modified Eagle’s low-glucose medium (Invitrogen, Carlsbad, CA, USA) supplemented with 15mM HEPES, 2.438 g/L sodium bicarbonate, 4 g/L glucose, 100 μg/mL G-418, 10% fetal bovine serum (HyClone, Logan, UT, USA), and maintained in an atmosphere of 5% CO2. Oli-neu cells (Jung et al. 1995) were grown at 37°C in SATO medium devoid of mitogens (ODM) according to the modifications by He et al. (2007), and supplemented with 1% horse serum (Invitrogen). Oli-neu cells were maintained in an atmosphere of 10% CO2.
Plp1-lacZ constructs
Details regarding the generation of Plp1-lacZ constructs, PLP(+)Z (Wight et al. 1993), PLP(−)Z (Wight and Dobretsova 1997), PLPΔ809-5807 (Dobretsova and Wight 1999), and PLPΔ809-5807 F(−AP4) (Dobretsova et al. 2004), referred to here as ‘PLPΔ809-5807 + ASE’, has been reported, previously. These constructs possess mouse Plp1 genomic DNA [proximal 2.4 kb of 5′-flanking DNA, all of exon 1 DNA, and the first 37 bases of exon 2] which is used to drive expression of a lacZ reporter gene cassette. The constructs differ only in the amount of Plp1 intron 1 DNA included; PLP(+)Z contains the entire intron (8140 bp), whereas PLP(−)Z totally lacks it. The PLPΔ809-5807 construct is missing intron 1 positions 809 to 5807, including the ASE sequence, based on numbering Plp1 intron DNA from positions 1 to 8140 (Wight and Dobretsova 1997. The ASE sequence (intron 1 positions 1093-1177) was reinstated at the deletion-junction site of PLPΔ809-5807 to generate the PLPΔ809-5807 + ASE construct.
The Plp1-lacZ construct, PLPΔASE, was produced with a PCR fragment (2214 bp) generated from genomic DNA of Plp1ΔASE mice (described below), and encompasses a region of Plp1 intron 1 in which the ASE sequence has been deleted. Plp1 sense (ΔASE-For; 5′-AGGAGTTCAACTTTGGGGCTTTG-3′) and antisense (ΔASE-Rev; 5′-AAAGCTCGAGGAACCAGGTGTC-3′) primers were utilized for PCR. The resulting amplicon was digested with SanDI and NheI, and exchanged for the corresponding region in PLP(+)Z. Hence, PLPΔASE is identical to PLP(+)Z except for missing 93 bp of Plp1 intron 1 DNA, and contains a loxP site in lieu of the ASE.
Transfection analysis
Methodology for transient transfection analysis in N20.1 cells using Lipofectamine Transfection Reagent (Invitrogen) has been described, previously (Dobretsova and Wight 1999). Briefly, N20.1 were seeded at a density of 2.8 × 105 cells in 2 mL of growth medium per 35-mm well (6-well plate; Costar, Cambridge, MA, USA). The following day, cells were treated with a mixture of 5 μL of Lipofectamine and 2 μg of total DNA per well. Wells contained equimolar amounts of a specific Plp1-lacZ construct plus 0.35 μg of RSVL to control for differences in transfection efficiency; the RSVL plasmid utilizes the RSV promoter to drive expression of a luciferase reporter gene (de Wet et al. 1987). Empty vector (pBluescript SK+; Stratagene, La Jolla, CA) was used either alone to determine the background with the reporter gene assays, or as filler to keep the total amount of DNA transfected constant. Cells were incubated for 6 h with the Lipofectamine/DNA mixture and lysates prepared approximately 72 h post-DNA addition in 190 μL of Reporter Lysis Buffer (Promega, Madison, WI, USA) as recommended by the supplier.
Oli-neu cells were seeded at 2.5 × 105 cells per 35-mm well in 2 mL of growth medium the day prior to transfection. The FuGENE 6 Transfection Reagent (Roche Diagnostics Corp., Indianapolis, IN, USA) was used to transfect the cells according to manufacturer’s specifications. The cells within a given well were treated with a mixture of FuGENE 6 (4.5 μL) and 3 μg of total DNA, which contained the same amount of Plp1-lacZ test construct and RSVL as used for N20.1 cells, with slightly more pBluescript SK+ to bring the total amount of DNA to 3 μg. The cells were left undisturbed for 48 h post-DNA addition, after which cell lysates were prepared as described for N20.1 cells.
Cell lysates (10-μl aliquots) were evaluated for reporter gene expression (in triplicate) using the Galacto-Light Plus Kit (Tropix, Bedford, MA, USA) and Luciferase Assay System (Promega) for determination of β-galactosidase (β-gal) and luciferase activity as described, previously (Dobretsova and Wight 1999). Luminescence was measured as relative light units (RLU) in an AutoLumat LB 953 luminometer (EG&G Berthold, Gaithersburg, MD, USA). Transfection results represent the mean ± SD of β-gal activity relative to that obtained for PLP(−)Z transfected cells, which was arbitrarily set at 100% (N20.1 cells) or 1-fold (Oli-neu cells) in every experiment. Results were compiled from three or more independent experiments.
Construction of the Plp1NeoΔASE targeting vector and associated plasmids
The plasmid, pOSDUPDEL [gift from Dr. Oliver Smithies, University of North Carolina, Chapel Hill], which contains a floxed (flanked by loxP sites) neomycin resistance (Neo) gene cassette from pMC1Neo (Thomas and Capecchi 1987) for positive selection and a herpes simplex virus (HSV)-thymidine kinase (TK) gene cassette driven by the mouse phosphoglycerate kinase (Pgk) promoter for negative selection, was used to generate the Plp1 targeting vector. The Plp1-specific sequences (regions of homology) in the targeting vector were obtained by PCR using mouse genomic DNA (129/SvJ) as the template. The 5′ homologous region (‘left arm’) includes Plp1 DNA from the proximal 2.4 kb of 5′-flanking DNA, downstream, to intron 1 position 1083. Approximately 200 ng of genomic DNA was amplified by PCR for 35 cycles [denaturation at 94°C for 30 s, annealing at 52.2°C for 30 s, and extension at 68°C for 230 s] using the AgeI sense (5′-CACCGGTCACGTTTTCTGGTTTACTGCTAGAACTG-3′) and XhoI antisense (5′-ACTCGAGCCAAACCTGGTTGTTTGGACCAGT-3′) primers which incorporate convenient restriction enzyme sites (underlined sequence) for cloning. The PCR-generated DNA was digested with AgeI and XhoI and the 3.7-kb fragment cloned immediately upstream of the Neo cassette in pOSDUPDEL in a like orientation. The resulting plasmid was designated ‘Clone A’. The 3′ homologous region (‘right arm’) contains Plp1 genomic DNA extending from intron 1 position 1177, downstream, to the first 37 bp of exon 2. As well, genomic DNA encompassing another region of Plp1 was amplified by PCR for 35 cycles [denaturation at 94°C for 30 s, annealing at 57.6°C for 30 s, and extension at 68°C for 7.5 min] using PacI sense (5′-GTTAATTAATTTTGTCTACTTTCAGGGTGGAAGATCAAGTAATAAGGATGTGGC-3′) and NotI antisense (5′-TGCGGCCGCCCCCTACCAGACATCTAGCACAACACT-3′) primers. The PCR product was digested with PacI and NotI restriction enzymes and the resulting 7.0-kb fragment inserted in Clone A immediately downstream of the Neo gene cassette to generate the Plp1NeoΔASE targeting vector (Clone B). The sequence for both arms of homology and the Neo gene cassette were verified by DNA sequencing. Hence, the successfully targeted Plp1 gene will contain the Neo gene cassette in lieu of the ASE sequence [i.e., missing Plp1 intron 1 DNA positions 1084-1176].
Clone P was generated to screen for homologous recombination between the targeting vector and the native Plp1 gene in embryonic stem (ES) cells (i.e., contains the Plp1NeoΔASE allele). To aid in the construction of Clone P, another plasmid was generated, called CK-7. CK-7 possesses a ClaI-KpnI fragment (~7 kb) of mouse Plp1 genomic DNA [5′-flanking DNA to the proximal third of intron 1] isolated from clone KK1 (Macklin et al. 1991) and inserted into the analogous sites in the pGEM-7Zf(−) vector (Promega). Clone A and CK-7 were digested with AatII and NgoM IV and the resulting 3.2-kb AatII-NgoM IV fragment from Clone A was ligated to the 5.1-kb fragment from CK-7 [includes pGEM-7Zf(−) vector backbone] to generate Clone P. Clone P contains Plp1 genomic DNA [proximal 3.6 kb of 5′-flanking DNA to intron 1 position 1083] fused to the 5′ portion of the Neo gene cassette.
Gene targeting and generation of knock in mouse lines
Pluripotent ES cells (W4) were electroporated with the targeting vector (NotI-linearized) at the Gene Targeting Facility (University of Iowa, Iowa City), and subsequently grown in the presence of G-418 and ganciclovir for positive/negative selection, respectively. The resulting ES colonies were isolated, expanded, and screened for homologous recombination. Initially, clones were screened for homologous recombination with the left arm of the targeting vector by PCR using a Plp1 sense (P1; 5′-ACTTCTCAATGCCCTGGTTCAGTGGAGC-3′) and Neo antisense (P1′; 5′-CGTCACCTTAATATGCGAAGTGGACCTGG-3′) primer pair which results in a 4.1-kb amplicon. PCR conditions were: denaturation at 94°C for 2 min, followed by 40 cycles of amplification [denaturation at 94°C for 30 s, annealing at 58.5°C for 30 s, extension at 68°C for 246 s], and final extension at 68°C for 5 min. The PCR products were fractionated by gel electrophoresis on a 0.8% agarose gel and visualized by staining with ethidium bromide. Subsequently, ES clones deemed positive in the initial (PCR) analysis were evaluated for homologous recombination at both ends through Southern blot analysis using standard techniques (Sambrook and Russell 2001). Briefly, genomic DNA (10 μg) from the ES clones was digested with either SphI or AseI for 3 h, fractionated on a 0.7% agarose gel, and transferred to a nitrocellulose membrane using 10× SSC. Homologous recombination with the left arm of the targeting vector was assessed using a 4-kb 32P-radiolabeled probe (BamHI fragment of mouse Plp1 genomic DNA located 1.4 kb upstream of exon 1). Homologous recombination with the right arm of the targeting vector was evaluated by hybridization to a 3-kb 32P-labeled probe prepared from an NdeI fragment of Plp1 intron 1 DNA (see Fig. 2a for further details). The blots were washed 3-4 times in a solution of 2× SSC and 0.1% SDS at 22°C for 1 h, and then twice in a solution of 0.1× SSC and 0.1% SDS at 42°C for 2 h. Following the washes, the blots were subjected to autoradiography. Hybridization of the BamHI probe with SphI digested genomic DNA results in 17.6-kb band from the native (wild-type; WT) Plp1 gene or a 7.7-kb band from the rearranged gene (Plp1NeoΔASE). Likewise, hybridization to AseI digested DNA results in a 14.6 kb-band for the WT allele and an 8.6 kb-band for Plp1NeoΔASE [7.4-kb band for Plp1ΔASE after excision the Neo cassette with Cre recombinase]. Hybridization of the NdeI probe to AseI cleaved DNA results in a 14.6-kb band for the WT gene and a 7.1-kb band with the rearranged allele (Plp1NeoΔASE).
Fig. 2.

Strategy and validation for removal of the ASE from the native mouse Plp1 gene. (a) Restriction map of the mouse Plp1 gene is diagrammed at the top with only the pertinent restriction enzymes sites indicated: ApaI (Ap), AseI (A), BamHI (B), NheI (N), and SphI (S). Numbered boxes depict the seven major exons. The terminal portion of exon 3 (gray box) encodes the PLP-specific region absent from DM20. The targeting vector contains sequences encoding selectable markers for negative (HSV-TK) and positive (Neo) selection to enrich for ES cells that underwent homologous recombination. The ‘left arm’ of the targeting vector contains 3.7 kb of Plp1 DNA from the proximal ApaI site in the 5′-flanking region to intron 1 position 1083, while the ‘right arm’ encompasses sequence from intron 1 position 1177 to the ApaI site in exon 2. Thus, recombination with the targeting vector will result in deletion of the ASE (gray star) from the native Plp1 gene, with a floxed Neo gene cassette in its place (Plp1NeoΔASE). Incorporation of the Neo cassette introduces a new SphI and AseI site to the locus (only these site are indicated in the Plp1NeoΔASE allele). The Neo cassette was subsequently removed by crossing Plp1NeoΔASE mice with Cre-deleter mice (EIIa-cre C57BL/6 mice) to generate mice with the Plp1ΔASE allele, which contains a single loxP site in lieu of the ASE. (b) Validation of ES clones that underwent homologous recombination with the targeting vector by Southern blot analysis. Genomic DNA was isolated from the indicated ES clones and a wild-type (WT) control, digested with SphI or AseI, and hybridized to the BamHI and NdeI probes, respectively. All of the ES clones shown contain the Plp1NeoΔASE allele (NeoΔASE). Clones ES920, ES993 and ES403 were also positive for the native Plp1 allele (WT) to varying degrees, indicating contamination with ES cells that had not undergone homologous recombination (perhaps due to incomplete selection with G418 and/or ganciclovir). Clones ES901 and ES 914 were injected into 129/SvJ blastocysts and subsequently implanted in pseudopregnant C57BL/6 females. Resulting male chimeras (F0 founders) were bred with C57BL/6 (WT) females and the F1 progeny screened for the rearranged gene (Plp1NeoΔASE). (c) PCR genotyping of progeny resulting from a parent harboring the Plp1NeoΔASE allele. Some of the female progeny contain the EIIa-cre transgene as well. Primers used for genotyping generate amplicons of 315 (Plp1ΔASE), 333 (Plp1), and 1505 (Plp1NeoΔASE) bp. Notice that removal of the Neo cassette was incomplete in two of the females; both ΔASE and NeoΔASE products were generated. The upper band (~400 bp) in the right lane of females is a hybrid between WT and ΔASE strands.
W4/129S6 ES clones ES901 and ES914, which contain the Plp1NeoΔASE rearrangement, were independently injected into C57BL/6J blastocysts and transplanted into pseudopregnant ICR females (Gene Targeting Facility, University of Iowa, Iowa City). [All procedures involving the use of mice were approved by the Institutional Animal Care and Use Committees at the University of Arkansas for Medical Sciences and the University of Iowa, in compliance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and the National Research Council’s Guide for the Care and Use of Laboratory Animals, and adhered to ARRIVE guidelines (Kilkenny et al. 2010).] The resulting chimeras were bred to C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME, USA) to generate female heterozygotes (Plp1+/NeoΔASE). Because the Plp1 gene is located on the X chromosome and the W4 ES cells are genotypically male, only female progeny will carry the rearranged allele. Germline transmission was assessed by PCR and Southern blot analyses similar to that described for the ES clones. Removal of the Neo gene cassette was achieved by breeding Plp1NeoΔASE mice with EIIa-Cre transgenic mice (The Jackson Laboratory), which expresses Cre recombinase under control of the adenovirus EIIa promoter early in embryogenesis (Lakso et al. 1996). Genomic DNA was isolated from tail biopsies (0.5 cm) of mice, and genotyping performed using sense (WT2; 5′-GGGCTGCCACATCCTTATTA-3′) and antisense (WT2′; 5′-TGTGTTCACCGTGCAACTTT-3′) primers. The amplicon from the WT (Plp1) allele is 333 bp, while a 315 bp product is generated from the rearranged allele without Neo (Plp1ΔASE); the Plp1ΔASE allele contains a single loxP site in lieu of the ASE. Failure to excise the Neo gene cassette from the Plp1NeoΔASE allele would result in a 1505 bp amplicon.
RNA extraction
Mice were anesthetized with isoflurane, decapitated, and brains rapidly removed. Total RNA was isolated immediately thereafter from the brains, separately, using the TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. RNA was resuspended in DEPC treated water, and samples aliquoted and stored at −70°C until further use.
Ribonuclease protection assay (RPA)
RPA analysis was performed as described previously (Li et al. 2002b) with the following modifications. To generate a plasmid from which to synthesize the Plp1 riboprobe, Plp1 sequence from the mouse cDNA, pRT5-7 (LeVine et al. 1990), was amplified by PCR and cloned into the BamHI and EcoRI sites of pGEM-7Zf(−) using an exon 2 sense (5′-CGGGATCCTGTTAGAGTGTTGTGCTAGA TG-3′; incorporated BamHI site underlined) and exon 3A antisense (5′-GGAATT CAGATGGTGGTCTTGTAGTCG-3′; incorporated EcoRI site underlined) primer pair. PCR conditions included 40 cycles of amplification [denaturation at 94°C for 30 sec, annealing at 54.5°C for 30 sec, extension at 68°C for 20 sec]. The resulting plasmid, termed E2-3A, was linearized with BamHI and an antisense RNA probe generated by in vitro transcription in the presence of [α-32P]UTP (NEN Life Science Products, Boston, MA) with the MAXIscript® T7 Kit (Applied Biosystems, Foster City, CA, USA). The antisense 32P-riboprobe was 371 nucleotides (nt) in length, of which 319 nt were complementary to Plp1 mRNA sequences. Thus, hybridization of the riboprobe to Plp/Dm20 transcripts, and subsequent digestion with RNase A and RNase T1 would result in the protection of 319 nt. In addition, a 304 nt full-length antisense riboprobe for endogenous mouse β-actin sequences was produced with the pTRI-β-actin-Mouse template (Applied Biosystems), of which, 245 nt can be protected.
Full-length riboprobes were excised from 6% polyacrylamide/8 M urea gels and eluted in 350 μL of Elution Buffer (5 M ammonium acetate, 1 mM EDTA, 0.1% SDS) at 37°C for 3 h. RPAs were performed with the RPA III Kit (Applied Biosystems) according to the manufacturer’s recommendations. In brief, 25 μg of total RNA from brain of mice at P9 to P390 of age, or 40 μg from younger animals (P2 through P7), was hybridized to ~8 × 105 cpm of 32P-labeled riboprobes, overnight at 43.5°C. Afterwards, the reactions were treated with a mixture of RNase A/T1 and protected fragments fractionated on a 6% polyacrylamide/8 M urea gel. Dried gels were initially analyzed on a 445 SI PhosphorImager with ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA, USA) and then subjected to autoradiography. Results are presented as the mean ratio of Plp/Dm20 combined to β-actin (signal strength), with a minimum of 3 animals, per timepoint, per genotype.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Assessment of the relative levels of Plp/Dm20 mRNA (combined) was also determined by real-time qRT-PCR analysis using the TaqMan Gene Expression Assay (Applied Biosystems). Single-stranded cDNAs were synthesized from total RNA (1.0 μg) using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) in a final reaction volume of 20 μL per manufacturer’s specifications. Conditions for RT consisted of three (single) incubation steps: 25°C for 5 min, 42°C for 30 min, and 85°C for 5 min. cDNAs were utilized immediately or stored at −70°C until needed. Quantitative PCR (qPCR) was carried out in a final reaction volume of 25 μL in iQ Supermix (Bio-Rad) containing 1 μL (demyelination/remyelination study) or 2 μL (developmental study) of the RT reaction mixture (cDNAs), 900 nM of primers, and 250 nM of probe. Primer/probe sets were obtained from Applied Biosystems including one for β-actin (catalog No. 4352933E), which served as the reference gene. A custom primer/probe set was designed for Plp1 [sense: 5′-CAAGACCTCTGCCAGTATAGG-3′; antisense: 5′-CAGCAATAAACAGGTGGAAGG-3′; probe: 6FAM-TGCCAGAATGTATGGTGTTCTCCCAT-MGBNFQ] which recognizes all splice variants (Plp and Dm20). qPCR was run (in triplicate) for 40 cycles (95°C for 15 s, 60°C for 1 min) in an iQ5 real-time PCR detection system (Bio-Rad) following a hot start (95°C for 10 min). Results for the developmental study were interpreted using the 2−ΔΔCT method (Schmittgen and Livak 2008), and are reported as the fold difference relative to a uniform control (qRT-PCR product from pooled brain mRNA of P21 WT mice). mRNA levels with the cuprizone demyelination/remyelination study were calculated instead from a standard curve using the same uniform control (P21 WT mice), as previously described (Pereira et al. 2011). The ratio of Plp1 (Plp and Dm20 combined) to β-actin mRNA was calculated for each sample, for standardization. Results are presented as the percent value of corrected Plp1 in cuprizone-treated mice relative to the average amount in untreated mice across all timepoints (within a genotype), which was arbitrarily set at 100%.
Western blot analysis
Preparation of whole brain lysates and Western blot analysis was performed as described previously (Li et al. 2009) with the following minor modifications. Proteins were denatured in Gel Loading Buffer (31.25mM Tris pH 6.8, 2% SDS, 5% glycerol, 0.05 mg/mL bromophenol blue, 0.785% β-mercaptoethanol) for 10 min at 55°C prior to fractionation on an SDS-PAGE gel (10% polyacrylamide), and subsequently transferred to a nitrocellulose membrane (Optitran BA-S85, Schleicher & Schuell, Florham Park, NJ, USA) per the manufacturer’s recommendations. Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline with Tween-20 (TBST; 20 mM Tris, pH 7.6, 150 mM NaCl, 0.1% Tween-20) for 1 h at 22°C, washed three times with TBST (15 min each), and subsequently incubated with primary mouse monoclonal antibodies. The anti-PLP/DM20 antibody (Abcam, Cambridge, MA, USA; catalog No. ab9311) was diluted 1 : 1000 in TBST plus 3% BSA and membranes were incubated overnight at 4°C. The anti-β-actin antibody (Abcam, catalog No. ab49900) was diluted 1 : 25,000 in TBST plus 5% non-fat dry milk and membranes were incubated for 1 h at 22°C. Blots were washed three times (5 min each) with TBST at 22°C and then incubated with secondary antibody (Jackson Immunoresearch, West Grove, PA, USA) for 1 h. The anti-mouse secondary antibody was diluted 1 : 10000 in TBST plus 5% non-fat dry milk. Immunoreactive bands were visualized with the ECL Plus Western Blotting Detection System (GE Healthcare Biosciences, Piscataway, NJ, USA) and analyzed on a Storm 840 PhosphorImager with ImageQuant TL (GE Healthcare Biosciences).
Cuprizone-induced demyelination
Plp1ΔASE mice were crossed to C57BL/6J mice for seven generations prior to initiation of cuprizone-induced demyelination to enrich for the C57BL/6J genetic background. Animals were provided food and water available, ad libitum, throughout the study. Beginning at 8 weeks of age, male Plp1ΔASE/Y and Plp1+/Y littermates were fed 0.2% cuprizone [bis(cyclohexanone)oxaldihydrazone; Sigma-Aldrich Inc., St. Louis, MO, USA] in milled rodent chow for 6 weeks. Untreated (control) mice were fed milled chow (without cuprizone) during the same time period, after which both groups were returned to normal chow (pellets) for 1 to 3 weeks. Mice were anesthetized with isoflurane and brains quickly removed at 0, 1, 2, 3, 4, 5, 7, and 9 week’s post-initiation of the study (cuprizone feeding). Brains were placed in a Petri dish (on ice) containing PBS and subsequently dissected retaining the portion of tissue containing most of the corpus callosum – coronal sections at −0.22 and −0.82 mm from the bregma, followed by a horizontal cut at the midline just below the fornix, and retention of the dorsal piece. Total RNA was extracted from the tissue block and qRT-PCR analysis performed. Three animals per group (± cuprizone treatment/genotype) were investigated for every timepoint.
Statistical analysis
Statistical significance for differences in the levels of β-gal activity between transfected Plp1-lacZ constructs was determined by one-way ANOVA, with Tukey’s post hoc analysis using the SAS 9.2 Software package (SAS, Cary, NC, USA). Bonferroni’s post hoc analysis yielded similar interpretations (data not shown). Statistical significance for differences in Plp1 gene expression between WT and ΔASE mice (RPA and qRT-PCR analyses) was determined by one-way ANOVA using the Generalized Linear Model (GLM) procedure from the SAS 9.2 Software package.
RESULTS
The ASE element is critical for high levels of Plp1-lacZ expression in oligodendroglial cell lines
As illustrated in Fig. 2a, the mouse Plp1 gene contains seven major exons, with a relatively large first intron. A variety of Plp1-lacZ constructs have been generated to map regulatory elements within Plp1 intron 1 DNA that are operative in N20.1 oligodendroglial cells. These constructs, depicted in Fig. 1, contain Plp1 genomic DNA [2.4 kb 5′-flanking DNA, exon 1, and first 37 bp of exon 2] which is used to drive expression of a lacZ reporter gene cassette. The PLP(+)Z construct also includes all of Plp1 intron 1 DNA, while PLPΔ809-5807 retains the intron except for positions 809-5807 based on numbering the intron from 1 to 8140 (Wight and Dobretsova 1997), and PLP(−)Z is missing the intron altogether. Similar to previous results (Dobretsova and Wight 1999), transfection analysis in N20.1 cells resulted in high levels of β-gal activity for the PLP(+)Z and PLP(−)Z constructs, and a much lower level for PLPΔ809-5807 [compare PLP(+)Z at 194% and PLPΔ809-5807 at 17% to PLP(−)Z, which was arbitrarily set at 100% for analysis in N20.1 cells]. Reinstatement of Plp1 intron positions 1093-1177 at the deletion-junction site of PLPΔ809-5807 was able to rescue β-activity back to level with PLP(+)Z [181% for PLPΔ809-5807 + ASE], suggesting that the ASE is a potent positive regulatory element. PLPΔASE was constructed to test whether the ASE is the sole positive regulatory element within the intron.
Fig. 1.
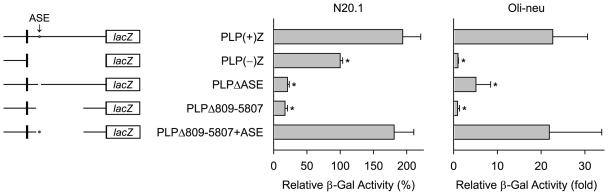
Transfection analysis of Plp-lacZ constructs in N20.1 and Oli-neu cells. Left: Schematic depiction of the Plp1-lacZ constructs used in this study (black boxes portray Plp1 exon 1 and the first 37 bp of exon 2). The star portrays the ASE sequence in Plp1 intron 1. Regions of Plp1 intron 1 DNA missing from the Plp1-lacZ constructs are indicated by the absence of a line. PLP(+)Z contains the entire intron, whereas PLP(−)Z is missing the intron altogether. Right: Transfection results represent the mean ± SD of β-gal activity (n ≥ 6) relative to PLP(−)Z, which was arbitrarily set at 100% (N20.1 cells) or 1-fold (Oli-neu cells) in every experiment. *Significant difference (p < 0.05) between PLP(+)Z and other constructs by one-way ANOVA with Tukey’s post-hoc analysis.
The PLPΔASE construct is identical to PLP(+)Z except it is missing Plp1 intron 1 positions 1084-1176 and contains a single loxP site in lieu of the ASE sequence. The loxP site was added to mimic the targeted Plp1 gene rearrangement in mice as discussed later. Deletion of just the ASE resulted in low levels of β-gal activity in N20.1 cells (21% for PLPΔASE), which were similar to that with PLPΔ809-5807. Thus, the ASE appears to be the sole positive regulatory element from Plp1 intron 1 that is operative in N20.1 cells.
To ascertain whether these findings are restricted merely to N20.1 cells, transfection analysis was also performed in Oli-neu cells. The Oli-neu cell line was derived by immortalizing enriched cultures of primary oligodendrocytes from mouse with the t-neu oncogene (Jung et al. 1995), and appears to be at a slightly later developmental stage than the N20.1 cell line (Pereira et al. 2011). Since the range of β-gal activity was so great between some of the constructs, transfection results with Oli-neu cells are reported as fold difference relative to PLP(−)Z, which was arbitrarily set at 1. As shown in Fig. 1, the level of β-gal activity with PLP(+)Z was 23-fold higher than that for PLP(−)Z in Oli-neu cells. Deletion of Plp1 intron 1 positions 809 to 5807 resulted in low levels of β-gal activity (0.9-fold for PLPΔ809-5807), similar to that for PLP(−)Z, suggesting that a positive regulatory element(s) lies within this 5 kb segment. Insertion of intron 1 positions 1093-1177 into the deletion-junction site of PLPΔ809-5807 was able to fully restore β-gal activity (22-fold for PLPΔ809-5807 + ASE) back to the level attained with PLP(+)Z, indicating that the ASE element is also operative in Oli-neu cells. Deletion of the ASE sequence alone resulted in moderately low levels of β-gal activity. Because the level of β-gal activity with PLPΔASE (5.1-fold) was still higher than that for PLPΔ809-5907 (0.9-fold), an additional positive regulatory element is active in Oli-neu cells between intron 1 positions 809-5807. Taken together, the transfection analysis in N20.1 and Oli-neu cells suggests that the ASE is a potent regulatory element through which an increase in Plp1 gene activity can be mediated – at least in oligodendrocytes.
Generation of mice missing the ASE element from the Plp1 gene (Plp1ΔASE)
To understand the role that the ASE might play in modulating Plp1 gene expression in vivo, intron 1 positions 1084-1176 were excised from the native gene in mouse using a Cre-mediated knock-in strategy (Fig. 2). Initially, a floxed Neo gene cassette was inserted in lieu of the ASE sequence via homologous recombination in ES cells with the targeting vector. Of the 201 ES clones screened, 5 clones demonstrated correct targeting (2.5% rate for homologous recombination). Four male chimeras (F0 founders) were generated through injection of ES901 or ES914 ES clones into mouse blastocysts. Three of the chimeras [ES901/1 (80% agouti), ES901/2 (100% agouti) and ES914/2 (15% agouti)] sired pups with the agouti coat color indicating the possibility of germline transmission of the mutant PlpNeoΔASE allele. Agouti progeny from the resulting F1 generation were screened for presence of the mutant allele, which segregated in a Mendelian fashion. All F1 agouti females were heterozygous for the mutant allele (PlpNeoΔASE/+) while F1 agouti males were hemizygous for the WT gene (Plp+/Y) as expected since male founders can only transmit the rearranged gene to female progeny; the Plp1 gene is located on the X chromosome. Subsequently, F1 females heterozygous for the Plp1NeoΔASE allele were bred to EIIa-Cre transgenic mice to remove the Neo gene cassette (see Fig. 2c). Progeny positive for the Plp1ΔASE allele, which is identical to the native gene except it contains a single loxP site in lieu of the ASE (lacks 93 bp of intron 1), were backcrossed with the Cre-deleter for several more generations to ensure that the Neo cassette had been removed from all cells in the animal (data not shown).
The ASE is essentially dispensable for correct developmental regulation of Plp1 gene expression in brain
To discern the role for the ASE in vivo, in context of the native gene, RPA analysis was performed with RNA isolated from male mice harboring the rearranged (Plp1ΔASE/Y) or the WT (Plp1+/Y) gene. Total RNA was obtained from the brains of mice at various ages (P2-P390) and the overall amount of Plp/Dm20 transcripts (combined) relative to that for β-actin was determined for each animal (Fig. 3). No significant differences were noted except at P15, where the combined level of Plp/Dm20 transcripts was 30% lower in ASE-deleted mice (Plp1ΔASE/Y) compared to WT mice, however there was a tendency towards a reduction in the ASE-deleted mice during the first several weeks postnatal. Similar results were also obtained by qRT-PCR analysis, although no significant differences between the genotypes were noted (Fig. 4), which is not altogether surprising since small differences are generally lost (imperceptible) after PCR amplification. Likewise, Western blot analysis did not demonstrate any significant difference in the levels of PLP or DM20 in Plp1ΔASE/Y and Plp1+/Y mice at P15 to P35 of age (Fig. 5). These results suggest that the ASE is virtually dispensable for normal regulation of Plp1 gene expression during development. Perhaps other elements in the Plp1 gene, not present in the PLP(+)Z construct (Fig. 1), can functionally compensate for its loss.
Fig. 3.

Developmental analysis (RPA) of Plp1 gene expression in male WT (Plp1+/Y) and ΔASE (Plp1ΔASE/Y) mice. Total RNA was isolated from whole brain of mice at the indicated ages and hybridized to 32P-labeled antisense riboprobes for Plp1 (371 nt) and β-actin (304 nt). Forty micrograms of total RNA was used in the reactions with mice at P2-P7 of age, while only 25 μg was used for mice at P9-P390. Protected fragments of 319 nt (Plp1) and 245 nt (β-actin) result after digestion with RNase A and RNase T1. The Plp1 riboprobe is unable to distinguish between Plp and Dm20 mRNA species; therefore it is a measure of the combined value. The mean ratio ± SD of Plp1 to β-actin mRNA levels is plotted for each timepoint/genotype as determined by PhosphorImager analysis (n ≥ 4 for P2-P7; n ≥ 3 for P9-P390). A representative autoradiogram is shown at the top of each plot. RPA analysis showed a significant decrease (*p < 0.0025) in Plp1 gene expression (Plp and Dm20 mRNA levels combined) in ΔASE mice (30% lower) compared with WT mice at P15.
Fig. 4.

qRT-PCR analysis of Plp1 gene expression in WT and ΔASE male mice from P2 to P390. A custom designed primer/probe set for Plp1 was used, which detects all splice variants (Plp and Dm20). Results were obtained using the 2−ΔΔCT method, with β-actin as the reference gene. Results are plotted as the mean level ± SD of Plp1 (Plp and Dm20 mRNA combined) relative to that from a uniform control (pooled brain mRNA from P21 WT mice) for each timepoint/genotype (n ≥ 4 for P2-P7; n ≥ 3 for P9-P390). While there tended to be a slight reduction in the level of Plp1 gene expression in ASE-deleted (ΔASE) mice compared to WT littermates at P15 and P21, the difference was not significant.
Fig. 5.

Western blot analysis of Plp1 gene products in brain from WT and ΔASE male mice at P15-P35 of age. Protein was isolated from whole brain of mice at the indicated ages, and the relative levels of PLP and DM20 protein determined using an antibody that recognizes both isoforms. Four animals were evaluated for each timepoint/genotype. Blots were incubated first with an anti-PLP/DM20 antibody, and then stripped and incubated with an anti-β-actin antibody. Autoradiograms for PLP and DM20 are shown at the top for all timepoints; only the P15 timepoint is shown for β-actin due to space constraints. Plots represent the mean ratio ± SD of PLP or DM20 to β-actin (n = 4) for each timepoint/genotype by PhosphorImager analysis. No significant difference was noted between the levels of PLP or DM20 in WT and ΔASE mice.
Lack of the ASE does not significantly deter Plp1 gene expression during remyelination
In demyelinating diseases, such as multiple sclerosis (MS), a combination of effects can lead to a failure in remyelination including depletion of myelinating cells, deficient recruitment of OPCs, and changes in gene expression programs (reviewed in Lubetzki et al. 2005). The myelin basic protein (Mbp) gene has been shown to contain multiple regulatory modules which are recruited selectively, in different combinations, to modulate Mbp gene activity during primary myelination, myelin maintenance, and remyelination in oligodendrocytes and Schwann cells (Farhadi et al. 2003). Since a single module (M4) was deemed to be important for regulating Mbp gene expression in oligodendrocytes during remyelination, we investigated if a similar scenario might be applicable with the ASE. Relative levels of Plp1 gene activity in ASE-deleted and WT mice undergoing acute demyelination and subsequent remyelination were evaluated by qRT-PCR analysis (Fig. 6). To induce demyelination, 8 week old Plp1ΔASE/Y and Plp1+/Y mice were fed milled rodent chow containing 0.2% cuprizone for up to 6 weeks, and then switched back to normal rodent chow for up to 3 weeks. Similar to published results with this model (reviewed by Matsushima and Morell 2001), animals showed a marked decrease in Plp/Dm20 mRNA levels following cuprizone ingestion (minimal at week 2), but after 5 weeks of treatment, the levels began to rise (Fig. 6). One week after discontinuation of cuprizone treatment (week 7 in Fig. 6), Plp/Dm20 mRNA levels were back to pretreatment levels. While there was no significant difference in Plp/Dm20 mRNA levels between ASE-deleted and WT mice, there was a hint that the levels in the ASE-deleted mice may have been somewhat restrained during the recovery (remyelination) phase (see weeks 5-9 in Fig. 6).
Fig. 6.

Cuprizone-induced changes in Plp1 gene expression in WT and ΔASE mice. Mice at 8 weeks of age (week 0) were placed on a diet containing 0.2% cuprizone for up to 6 weeks (week 0-6) and then switched back to normal rodent chow for the remainder of the study. Untreated mice (not exposed to cuprizone) were also analyzed for both genotypes (WT and ΔASE). Animals were sacrificed at weekly intervals, and total RNA isolated from a defined block of tissue from the corpus callosum (see methods for more details). Real time qRT-PCR analysis was performed, and mRNA levels for Plp1 (Plp and Dm20 combined) and β-actin were determined from a standard curve generated with the same uniform control as in Fig. 4. The ratio of Plp1 (Plp and Dm20 combined) to β-actin mRNA was calculated for each sample, for standardization. Normalized values for Plp1 in untreated mice (no cuprizone) were averaged across all timepoints and set at 100% for a given genotype (WT or ΔASE). Results are plotted as the mean value ± SD of corrected Plp1 in cuprizone-treated mice relative to the average amount in untreated mice across all timepoints for a specific genotype (n = 3 for each timepoint/genotype).
DISCUSSION
Mouse transgenesis with reporter constructs has greatly aided in the identification and characterization of cis-regulatory elements for a host of genes including CD69 (Vazquez et al. 2009), interleukin 4 (Yagi et al. 2007), Mbp (Foran and Peterson 1992), type X collagen (Zheng et al. 2009), and Plp1 (Wight et al. 1993). However, as with any model, there are limitations. For instance, random integration of transgenes can lead to inadvertent position effects from the surrounding chromatin, thereby necessitating that multiple (independent) transgenic lines be scrutinized. Moreover, it is possible that additional regulatory elements, important for governing the expression of a particular gene, may not have been included in the transgene. To avoid these pitfalls, the ASE was targeted for deletion from the native (mouse) Plp1 gene in order to study its role in vivo. While incorporation of the equivalent mutation (loxP site in lieu of the ASE) in a Plp1-lacZ construct (PLPΔASE) caused a dramatic decrease in expression in transfected N20.1 and Oli-neu cells (Fig. 1), loss of the ASE from the endogenous gene had, at most, a very minimal effect on Plp1 gene activity during development; RPA analysis indicated that the combined level for Plp/Dm20 transcripts in brain was 30% lower in ASE-deleted mice (Plp1ΔASE/Y) compared to WT littermates (Plp1+/Y) at P15 (Fig. 3). Plp/Dm20 mRNA levels were similar at all other timepoints examined. Analysis by qRT-PCR did not reveal a difference in Plp/Dm20 mRNA levels between the genotypes at any timepoint, although there was a hint that the levels in the ASE-deleted mice may have been somewhat tempered at P15 and P21 (Fig. 4). However, no significant difference was detected at the protein level (Fig. 5). Thus, the ASE is dispensable for appropriate Plp1 gene regulation during development. This could be due simply to the fact that the ASE is not functional in vivo, or that other elements in the Plp1 gene can functionally compensate for its lack. Accordingly, the ASE-deleted mice did not display any abnormal phenotype out to 16 months of age. Even the neurological/behavioral deficits observed in Plp1 null mice are generally mild (for review see Rosenbluth et al. 2006).
The slight, but statistically significantly, decrease in Plp/Dm20 mRNA levels for ASE-deleted mice at P15 by RPA analysis suggests that the ASE may actually be operational in vivo. It is possible that functionally redundant regulatory element(s) may not have been included in PLP(+)Z, which would explain why removal of the ASE from the Plp1-lacZ construct [PLPΔASE] led to such a dramatic reduction in its activity in the oligodendroglial cell lines (Fig. 1). The MyoD gene encodes an essential transcription factor responsible for muscle differentiation in vertebrates and contains functionally redundant cis-acting regulatory modules – a distal regulatory region (DRR) and a core enhancer – that can compensate for one another’s loss (Chen et al. 2002; Chen and Goldhamer 2004). The HoxD gene (Zákány et al. 1997; Beckers and Duboule 1998) also exhibits functional redundancy in its transcriptional regulation. Thus, it is entirely likely that redundancy in gene regulation represents a safety mechanism, which ensures the expression of critical/important genes.
Alternatively, there could be another regulatory element present in Plp1 intron 1 DNA that can compensate for loss of the ASE during most of development, but is not active at the stage consistent with N20.1 cells. Transfection analysis in Oli-neu cells, which appear to be at a later developmental stage than N20.1 cells by virtue of attaining a much higher level of PLP(+)Z expression relative to PLP(−)Z, seems to support this premise. Even though the ASE is a very potent positive regulatory element in Oli-neu cells, it is not the only positive regulatory element in Plp1 intron 1 DNA; β-gal activity for PLPΔASE was still 5-fold higher than PLP(−)Z in Oli-neu cells (Fig. 1).
A couple of other regions in Plp1 intron 1 having enhancer-like activity were identified using a controlled transgenesis approach that targeted reporter transgenes to the hypoxanthine phosphoribosyltransferase (HPRT) locus (Tuason et al. 2008). Specifically, highly conserved regions of mouse Plp1 DNA were tested for their ability to support expression of a minimally promoted [basal heat shock protein (hsp) promoter] eGFP-lacZ reporter gene. A 1171 bp segment of intron 1 DNA (situated downstream of the ASE) led to transgene activity in mature oligodendrocytes in a temporal manner consistent with the endogenous Plp1 gene, and as such, was named the wmN1 enhancer (Tuason et al. 2008). It is plausible that this element may be responsible for the slight elevation of β-gal activity in Oli-neu cells for PLPΔASE relative to PLP(−)Z, since removal of the just the wmN1 enhancer region from PLP(+)Z led to a modest decline in [PLPΔwmN1] activity (Fig. S1). Perhaps the wmN1 enhancer can counteract for loss of the ASE in the native gene at certain times of development, but is unable to fully do so in context of Plp1-lacZ constructs in transfected Oli-neu cells due to the lack of signaling from other cell types or the immature nature of the cell line itself. The wmN1 enhancer was found to be active during the latter stages of oligodendrocyte maturation (Tuason et al. 2008) and therefore may not be optimally active in Oli-neu cells. Additionally, another element was identified further downstream in Plp1 intron 1 and named wmN2 (Tuason et al. 2008). The wmN2 enhancer was found to support relatively high levels of expression in the PNS when incorporated in the minimally promoted eGFP-lacZ transgene, while much lower levels were detected in white matter areas of the CNS. Interestingly, CNS expression of the transgene containing the wmN2 region was found to be maximal at P14, barely detectable at P30, and extinguished by P60 (Tuason et al. 2008), which happens to coincide with the timing of a maximal effect observed for Plp1 gene regulation when the ASE is absent (Fig. 3). Perhaps the wmN2 enhancer can compensate for loss of the ASE in the native Plp1 gene, but appears to be essential (much like the ASE) for attaining decent levels of Plp1-lacZ expression in Oli-neu cells (Fig. S2).
The ASE was also evaluated for enhancer activity in the controlled transgenesis study by Tuason et al. (2008). While the ASE was able to support expression of the eGFP-lacZ transgene in some (unidentified) cell types, it did not do so in oligodendrocytes, oligodendrocyte progenitor cells (OPCs) or Schwann cells. Previously, we have shown that the ASE needs be in context of an intron in order to function in N20.1 cells. It lost its ability to augment expression of a reporter gene when repositioned upstream of the mouse Plp1 promoter, or downstream of the (lacZ) reporter gene (Meng et al. 2005). Thus, it is not surprising that the ASE did not sustain expression in oligodendrocytes when placed upstream of the basal hsp promoter in the controlled transgenesis study by Tuason et al. (2008). Perhaps the ASE functions to enhance transcriptional elongation in oligodendroglial cells, which becomes immaterial when relocated to a site outside of the transcribed region.
It is conceivable that not enough sequence surrounding the ASE was removed to obliterate its activity in the ASE-deleted mice. However, in the study by Tuason et al. (2008), much larger fragments (2538 bp and 300 bp) of Plp1 genomic DNA that encompass the ASE were tested by means of enhancer trap. Neither fragment [ASE2538 and ASE300] directed transgene expression in oligodendrocytes, although transgene activity was detected in select regions of the CNS in other cell types. Hence, targeting a broader region of sequence for deletion of the ASE probably would not change the outcome of the present study, where loss of Plp1 intron 1 positions 1084-1176 caused a significant reduction in expression of PLPΔASE in N20.1 and Oli-neu cells, but had little effect on expression of the native Plp1 gene in mouse. While these seemingly contradictory results might be due a difference in chromatin structure between the two systems – transiently transfected cell lines and ASE-deleted mice – previously we have shown that the ASE is still able to augment expression of Plp1-lacZ constructs in N20.1 cells when stably transfected (Dobretsova et al. 2000). Thus, it appears that ASE activity is mediated via a factor(s) that acts as a true transcriptional activator rather than one that affects chromatin structure per se. Perhaps the disparate outcomes obtained here reflect a difference in transcription factor repertoires between the oligodendroglial cell lines and the ASE-deleted mice. Alternatively, loss of the ASE from the native Plp1 gene may be compensated for by other functionally redundant elements in the gene which are either not present in PLPΔASE, or nonfunctional/not optimally active in N20.1 and Oli-neu cells. Maybe the oligodendroglial cell lines lack, or fail to properly activate, ASE-binding factor(s) due to the absence of an important signal such as those produced by neuron-glia interactions.
Similar to the situation during development, lack of the ASE from the native gene in mouse did not significantly alter Plp1 gene expression after acute demyelination with cuprizone, although there was a hint that the levels may have been somewhat restrained in the ASE-deleted mice during the recovery (remyelination) phase (Fig. 6). Thus, the ASE is not the sole regulator of Plp1 gene expression during remyelination. Whether this is due to compensation by another element (e.g., wmN1 enhancer) or whether the ASE is not important for Plp1 gene expression during remyelination is unknown at this time.
In conclusion, the current studies demonstrate that the ASE is largely (if not entirely) dispensable for appropriate regulation of Plp1 gene expression in vivo. This could be due to a difference between the transfection and transgenic paradigms. Namely, that the ASE is functional when examined by transfection analysis in cell culture, but this effect does not translate to the animal. On the other hand, transgenic studies (Li et al. 2002b) have clearly demonstrated the necessity of Plp1 intron 1 DNA for accurate expression of Plp1-lacZ transgenes in mice. It is possible that another element in the intron (besides the ASE) may be wholly important or, alternatively, able to compensate for loss of the ASE from the native Plp1 gene. As well, other elements, not present in the Plp1-lacZ transgenes, may have functional redundancy with the ASE. Because there was a modest decrease in Plp/Dm20 mRNA levels with ASE-deleted mice at P15 (Fig. 3), and a hint of diminished levels at other times during development and during remyelination, the ASE may actually be active in vivo. Whether this is due a weak effect imparted by the ASE itself or to compensation by another element in the Plp1 gene remains to be seen.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr. Anna Dobretsova for scientific advice and discussion with respect to the project and to Dr. Glenn K. Matsushima for guidance with the cuprizone model of acute demyelination. We would also like to thank Jennifer Johnson and Gail Waggoner for help with the animal colonies. This work was supported by grants from the National Multiple Sclerosis Society (RG 2705), NIH (R01NS037821; P30NS047546; UL1TR000039), and intramural funding from the UAMS College of Medicine Research Council. The authors have no competing financial interest or further conflict of interest.
Abbreviations used
- ASE
antisilencer/enhancer
- β-gal
β-galactosidase
- ES
embryonic stem;
- floxed
flanked by loxP sites
- HPRT
hypoxanthine phosphoribosyltransferase
- HSV
herpes simplex virus
- Mbp
myelin basic protein (gene)
- MS
multiple sclerosis
- n
number of samples
- Neo
neomycin resistance
- nt
nucleotides
- OPC
oligodendrocyte progenitor cell
- P
postnatal day
- PGK
phosphoglycerate kinase
- PMD
Pelizaeus-Merzbacher disease
- PLP
myelin proteolipid protein
- Plp1
myelin proteolipid protein (gene)
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- RLU
relative light units
- RPA
ribonuclease protection assay
- SPG2
spastic paraplegia type 2
- TBST
Tris-buffered saline with Tween-20
- TK
thymidine kinase
- WT
wild-type
REFERENCES
- Beckers J, Duboule D. Genetic analysis of a conserved sequence in the HoxD complex: regulatory redundancy or limitations of the transgenic approach? Dev. Dyn. 1998;213:1–11. doi: 10.1002/(SICI)1097-0177(199809)213:1<1::AID-AJA1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Chen JC, Goldhamer DJ. The core enhancer is essential for proper timing of MyoD activation in limb buds and branchial arches. Dev. Biol. 2004;265:502–512. doi: 10.1016/j.ydbio.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Chen JC, Ramachandran R, Goldhamer DJ. Essential and redundant functions of the MyoD distal regulatory region revealed by targeted mutagenesis. Dev. Biol. 2002;245:213–223. doi: 10.1006/dbio.2002.0638. [DOI] [PubMed] [Google Scholar]
- de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol. Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobretsova A, Wight PA. Antisilencing: myelin proteolipid protein gene expression in oligodendrocytes is regulated via derepression. J. Neurochem. 1999;72:2227–2237. doi: 10.1046/j.1471-4159.1999.0722227.x. [DOI] [PubMed] [Google Scholar]
- Dobretsova A, Kokorina NA, Wight PA. Functional characterization of cis-acting DNA antisilencer region that modulates myelin proteolipid protein gene expression. J. Neurochem. 2000;90:1500–1510. doi: 10.1046/j.1471-4159.2000.0751368.x. [DOI] [PubMed] [Google Scholar]
- Dobretsova A, Kokorina NA, Wight PA. Potentiation of myelin proteolipid protein (Plp) gene expression is mediated through AP-1-like binding sites. J. Neurochem. 2004;90:1500–1510. doi: 10.1111/j.1471-4159.2004.02683.x. [DOI] [PubMed] [Google Scholar]
- Eng LF, Chao RC, Gerstl B, Pratt D, Tavastsjerna MG. The maturation of human white matter myelin: fractionation of the myelin membrane proteins. Biochemistry. 1968;7:4455–4465. doi: 10.1021/bi00852a042. [DOI] [PubMed] [Google Scholar]
- Farhadi HF, Lepage P, Forghani R, Friedman HC, Orfali W, Jasmin L, Miller W, Hudson TJ, Peterson AC. A combinatorial network of evolutionarily conserved myelin basic protein regulatory sequences confers distinct glial-specific phenotypes. J. Neurosci. 2003;23:10214–10223. doi: 10.1523/JNEUROSCI.23-32-10214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J. Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell. Mol. Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J. Neurosci. 1997;17:181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudz TI, Schneider TE, Haas TA, Macklin WB. Myelin proteolipid protein forms a complex with integrins and may participate in integrin receptor signaling in oligodendrocytes. J. Neurosci. 2003;22:7398–7407. doi: 10.1523/JNEUROSCI.22-17-07398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave K-A, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Zhang Z, Mullins C, Bessert D, Lee I, Nave K-A, Appikatla S, Skoff RP. Different proteolipid protein mutants exhibit unique metabolic defects. ASN NEURO. 2009;1(3) doi: 10.1042/AN20090028. art:e00014.doi:10.1042/AN20090028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K. PLP1-related inherited Dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazale K, Chlichlia K, Blankenfeld GV, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinctive degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi: 10.1371/journal.pbio.1000412. doi:10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl Acad. Sci. USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine SM, Wong D, Macklin WB. Developmental expression of proteolipid protein and DM-20 mRNAs and proteins in the rat brain. Dev. Neurosci. 1990;12:235–250. doi: 10.1159/000111853. [DOI] [PubMed] [Google Scholar]
- Li S, Dobretsova A, Kokorina NA, Wight PA. Repression of myelin proteolipid protein gene expression is mediated through both general and cell type-specific negative regulatory elements in nonexpressing cells. J. Neurochem. 2002a;82:159–171. doi: 10.1046/j.1471-4159.2002.00962.x. [DOI] [PubMed] [Google Scholar]
- Li S, Moore CL, Dobretsova A, Wight PA. Myelin proteolipid protein (Plp) intron 1 DNA is required to temporally regulate Plp gene expression in the brain. J. Neurochem. 2002b;83:193–201. doi: 10.1046/j.1471-4159.2002.01142.x. [DOI] [PubMed] [Google Scholar]
- Li S, Greuel BT, Meng F, Pereira GB, Pitts A, Dobretsova A, Wight PA. Leydig cells express the myelin proteolipid protein gene and incorporate a new alternatively spliced exon. Gene. 2009;436:30–36. doi: 10.1016/j.gene.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubetzki C, Williams A, Stankoff B. Promoting repair in multiple sclerosis: problems and prospects. Curr. Opin. Neurol. 2005;18:237–244. doi: 10.1097/01.wco.0000169739.83793.e0. [DOI] [PubMed] [Google Scholar]
- Macklin WB, Campagnoni CW, Deininger PL, Gardinier MV. Structure and expression of the mouse myelin proteolipid protein gene. J. Neurosci. Res. 1987;18:383–394. doi: 10.1002/jnr.490180302. [DOI] [PubMed] [Google Scholar]
- Macklin WB, Gardinier MV, Obeso ZO, King KD, Wight PA. Mutations in the myelin proteolipid protein gene alter oligodendrocyte gene expression in jimpy and jimpymsd mice. J. Neurochem. 1991;56:163–171. doi: 10.1111/j.1471-4159.1991.tb02576.x. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Zolova O, Kokorina NA, Dobretsova A, Wight PA. Characterization of an intronic enhancer that regulates myelin proteolipid protein (Plp) gene expression in oligodendrocytes. J. Neurosci. Res. 2005;82:346–356. doi: 10.1002/jnr.20640. [DOI] [PubMed] [Google Scholar]
- Nave K-A, Lai C, Bloom FE, Milner RJ. Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc. Natl Acad. Sci. USA. 1987;84:5665–5669. doi: 10.1073/pnas.84.16.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: changes in myelin composition during brain maturation. J. Neurochem. 1973;21:759–773. doi: 10.1111/j.1471-4159.1973.tb07520.x. [DOI] [PubMed] [Google Scholar]
- Pereira GB, Dobretsova A, Hamdan H, Wight PA. Expression of myelin genes: comparative analysis of Oli-neu and N20.1 oligodendroglial cell lines. J. Neurosci. Res. 2011;89:1070–1078. doi: 10.1002/jnr.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J, Nave K-A, Mierzwa A, Schiff R. Subtle myelin defects in PLP-null mice. Glia. 2006;54:172–182. doi: 10.1002/glia.20370. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A Laboratory Manual. 3rd edition Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Skoff RP, Saluja I, Bessert D, Yang X. Analyses of proteolipid protein mutants show levels of proteolipid protein regulate oligodendrocyte number and cell death in vitro and in vivo. Neurochem. Res. 2004;29:2095–2103. doi: 10.1007/s11064-004-6882-0. [DOI] [PubMed] [Google Scholar]
- Simons R, Alon N, Riordan JR. Human myelin DM-20 proteolipid protein deletion defined by cDNA sequence. Biochem. Biophys. Res. Commun. 1987;146:666–671. doi: 10.1016/0006-291x(87)90580-8. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- Tuason MC, Rastikerdar A, Kuhlmann T, Goujet-Zalc C, Zalc B, Dib S, Friedman H, Peterson A. Separate proteolipid protein/DM20 enhancers serve different lineages and stages of development. J. Neurosci. 2008;28:6895–6903. doi: 10.1523/JNEUROSCI.4579-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez BN, Laguna T, Carabana J, Krangel MS, Lauzurica P. CD69 gene is differentially regulated in T and B cells by evolutionarily conserved promoter-distal elements. J. Immunol. 2009;183:6513–6521. doi: 10.4049/jimmunol.0900839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verity AN, Bredesen D, Vonderscher C, Handley VW, Campagnoni AT. Expression of myelin protein genes and other myelin components in an oligodendrocytic cell line conditionally immortalized with a temperature-sensitive retrovirus. J. Neurochem. 1993;60:577–587. doi: 10.1111/j.1471-4159.1993.tb03188.x. [DOI] [PubMed] [Google Scholar]
- Wight PA, Dobretsova A. The first intron of the myelin proteolipid protein gene confers cell type-specific expression by a transcriptional repression mechanism in non-expressing cell types. Gene. 1997;201:111–117. doi: 10.1016/s0378-1119(97)00435-6. [DOI] [PubMed] [Google Scholar]
- Wight PA, Dobretsova A. Where, when and how much: regulation of myelin proteolipid protein gene expression. Cell. Mol. Life Sci. 2004;61:810–821. doi: 10.1007/s00018-003-3309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight PA, Duchala CS, Readhead C, Macklin WB. A myelin proteolipid protein-lacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J. Cell Biol. 1993;123:443–454. doi: 10.1083/jcb.123.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi R, Tanaka S, Motomura Y, Kubo M. Regulation of the Il4 gene is independently controlled by proximal and distal 3′ enhancers in mast cells and basophils. Mol. Cell. Biol. 2007;27:8087–8097. doi: 10.1128/MCB.00631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yool DA, Klugmann M, McLaughlin M, Vouyiouklis DA, Dimou L, Barrie JA, McCulloch MC, Nave K-A, Griffiths IR. Myelin proteolipid proteins promote the interaction of oligodendrocytes and axons. J. Neurosci. Res. 2001;63:151–164. doi: 10.1002/1097-4547(20010115)63:2<151::AID-JNR1007>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Zákány J, Gérard M, Favier B, Duboule D. Deletion of a HoxD enhancer induces transcriptional heterochrony leading to transposition of the sacrum. EMBO J. 1997;16:4393–4402. doi: 10.1093/emboj/16.14.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Keller B, Zhou G, Napierala D, Chen Y, Zabel B, Parker AE, Lee B. Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J. Bone Miner. Res. 2009;24:1022–1032. doi: 10.1359/JBMR.081249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.