

Abstract
Cholesteryl ester transfer protein (CETP) mediates the net transfer of cholesteryl esters from atheroprotective high-density lipoproteins to atherogenic low-density lipoproteins or very-low-density lipoproteins. Inhibition of CETP raises high-density lipoprotein cholesterol (good cholesterol) levels and reduces low-density lipoprotein cholesterol (bad cholesterol) levels, making it a promising drug target for the prevention and treatment of coronary heart disease. Although the crystal structure of CETP has been determined, the molecular mechanism mediating cholesteryl esters transfer is still unknown, even the structural features of CETP in a physiological environment remain elusive. We performed molecular dynamics simulations to explore the structural features of CETP in an aqueous solution. Results show that the distal portion flexibility of N-terminal β-barrel domain is considerably greater in solution than in crystal; conversely, the flexibility of helix X is slightly less. During the simulations the distal end of C-terminal β-barrel domain expanded while the hydrophilic surface increasing more than the hydrophobic surface. In addition a new surface pore was generated in this domain. This surface pore and all cavities in CETP are stable. These results suggest that the formation of a continuous tunnel within CETP by connecting cavities is permitted in solution.
Keywords: CETP, MD simulations, structural flexibility, cavity, hydrophobicity, salt bridge
INTRODUCTION
Cholesteryl ester transfer protein (CETP) mediates the net transfer of cholesteryl esters (CEs) from atheroprotective high-density lipoprotein (HDL) to atherogenic low-density lipoprotein (LDL) and very-low-density lipoprotein (VLDL) with the reciprocal transfer of triglycerides.1 Low levels of HDL cholesterol (good cholesterol) and elevated levels of LDL cholesterol (bad cholesterol) in plasma are two strong risk factors for coronary heart disease.2,3 Inhibition of CETP effectively raises HDL cholesterol levels and reduces LDL cholesterol levels in humans.4,5 Therefore, CETP is considered a promising drug-design target for the prevention and treatment of coronary heart disease.6,7
CETP is a ~53 kDa (before posttranslational modification) plasma glycoprotein containing 476 amino acids.8 X-ray crystallography has provided an atomic resolution structure of CETP (PDB entry 2OBD).9 The crystal structure reveals a banana shaped conformation composed of N- and C-terminal β-barrels, a central β-sheet between the two β-barrels, and an amphiphilic helix “X” (Glu465-Ser476) at the C-terminus [see Fig. 1]. A ~60 Å long hydrophobic cavity traverses the core of CETP. This cavity is filled by two CEs while two openings nearby the central β-sheet domain are each plugged by a phospholipid. A more detailed description of the crystal structure is available in reference 9.
Figure 1. Schematic diagram of the simulation model.
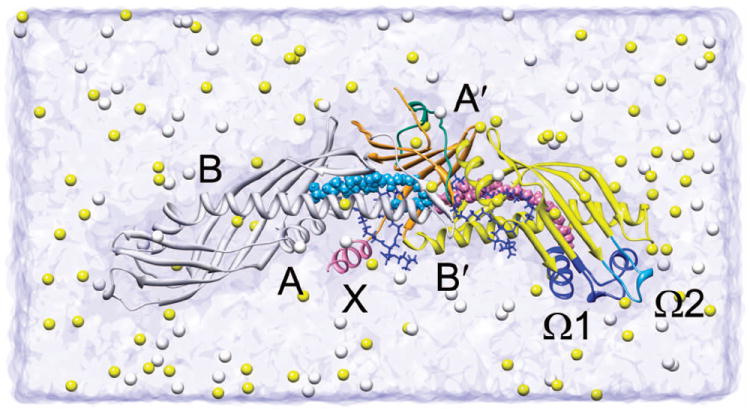
CETP is shown in ribbon representation with its N-terminal β-barrel, C-terminal β-barrel, central β-sheet and helix X colored white, yellow, orange and pink, respectively. Whenever possible, the N-terminal domain is on the left and the C-terminal domain is on the right. The flap Ω1, flap Ω2 and the central linker (Asp240-Arg259) nearby the central β-sheet are shown in blue, light blue and bluish green, respectively. Helices A, B, A′, B′ and X, and flaps Ω1 and Ω2 are labeled. The CEs are displayed as space-filling spheres. Two phospholipids are depicted as blue stick models. The Na+ (yellow) and Cl− (white) ions are shown in space-filling representation.
Three hypotheses have been proposed for the mechanism of CETP-mediated neutral lipid transfer: (1) a shuttle mechanism in which CETP acts as a carrier transporting neutral lipids between donor and acceptor lipoproteins;10 (2) a ternary complex mechanism in which CETP bridges two lipoproteins to form a transient ternary complex, with neutral lipids flowing from the donor to acceptor lipoprotein through the CETP molecule;11 and (3) a modified ternary complex mechanism involving a CETP dimer.12
Based on the analysis of CETP crystal structure, the concave surface of CETP was proposed to bind lipoprotein for neutral lipid transfer.9 Accordingly, CETP can bind only one lipoprotein at a time,9 supporting the shuttle mechanism. While recent transmission electron microscopy (TEM) studies revealed that CETP bridges HDL to LDL or VLDL to form ternary complexes, with its distal N-terminal β-barrel domain penetrating into CE core of HDL and the C-terminal β-barrel domain interacting with LDL or VLDL.13 Further studies demonstrated that internal cavities exist in the crystal structure of CETP.13,14 These results strongly suggested a tunnel mechanism for neutral lipid transfer, in which the cavities of CETP connect and form a continuous tunnel that mediates the lipid transfer between bound lipoproteins.13 The flexibility of the distal portions of β-barrel domains was suggested to facilitate the required conformational changes for the formation of continuous tunnel.13 However, in the crystallographic analysis, the distal end of N-terminal β-barrel domain shows only little flexibility.9
The crystal structure of CETP represents a static picture of CETP under crystallization conditions. The neutral lipid transfer, however, is a dynamic process under physiological conditions. Further studies of CETP structural features are required to fully elucidate the molecular mechanism of CETP-mediated lipid transfer. In particular, to predict the potential conformational changes of CETP for lipid transfer, a comprehensive knowledge of its structural flexibility is needed. To obtain a clearer understanding of the lipid transfer mechanism, especially the potential tunnel mechanism, we have to clarify whether the internal cavities in the crystallographic structure are real or merely an artifact of crystallization conditions, and explore the interactions between CETP and lipoproteins. These questions call for investigations of internal cavities and surface features of CETP.
In this paper, we performed all-atom molecular dynamics (MD) simulations to explore the structural features of CETP. Modeling focused on: structural flexibility, internal cavities, and surface hydrophobicity which are all closely related to lipid transfer function. Our simulation results provide clues for elucidating the mechanism of CETP-mediated lipid transfer.
MATERIALS AND METHODS
Preparation of simulation system
Initial coordinates of the protein with its bound CEs and phospholipids were taken from the crystal structure of CETP (PDB entry 2OBD).9 Missing hydrogen atoms were added using the AutoPSF module of VMD.15 This protein was then solvated in a cubic box of 27,662 TIP3P water molecules extending at least 15 Å from the protein-ligand complex. To neutralize the system and achieve a physiological salt concentration of 0.15 M, 86 Na+ and 71 Cl− ions were added to the water box. The resulting system consisted of ~91,000 atoms [Fig. 1].
MD simulations
All simulations were performed with the NAMD2 program.16 The CHARMM27 force field17,18 with ϕ, ψ cross-term map (CMAP) correction19 was used for the protein and ions, together with the TIP3P model for water.20 The force field for CE and phospholipid was constructed from the CHARMM27 force field and from the literature 21. The water and ions were first energy minimized and equilibrated for 0.4 ns with the protein and four lipid molecules fixed. Next, the whole system was energy minimized with the protein backbone atoms constrained (force constant of 5 kcal/mol/Å2). The system was then heated from 0 K to 310 K over 62 ps simulation. Finally, 8.5 ns MD simulation was performed in the NPT ensemble (constant number of atoms, pressure and temperature) with the constraints on backbone atoms gradually removed during the first 0.5 ns NPT simulation. To mimic physiological conditions, the temperature was kept at 310 K by using Langevin dynamics22 with a damping coefficient of 5 ps-1. And the pressure was maintained at 1 atm using the Langevin piston Nose-Hoover method,23 with a piston period of 100 fs and a decay time of 50 fs. Periodic boundary conditions and a cutoff distance of 12 Å for van der Waals interactions were applied. The Particle-Mesh Ewald method24 (grid spacing < 1 Å) was used to compute the long-range electrostatic interactions. Three replica simulations (labeled as MD1 to MD3) were carried out with different random number generator seeds to improve sampling and collect more data. The atomic coordinates were saved every 5 ps for analysis.
Analysis
All the structures from MD simulations were superimposed onto the initial structure using a least-square fit. The root mean-square deviation (RMSD) of the protein backbone atoms from the initial coordinates was used as a measure of structural differences. The solvent-accessible surface area (SASA) was calculated with a 1.4 Å probe radius. A grid size of 0.25 Å was used to measure the volume of the protein. The gyration radius of protein backbone atoms was also calculated. The root mean-square fluctuation (RMSF) of Cα atoms relative to the average structure over the last 5 ns simulations was calculated as an indicator of structural flexibility. For the analysis of salt bridges, an oxygen-nitrogen distance cutoff of 3.5 Å between charged residue side chains was employed. All above analyses were performed with VMD.
Normal mode analysis (NMA) was performed on the crystal structure and the average structure of CETP derived from the MD simulations to determine their theoretical B-factors (temperature factors). The online server (http://ignmtest.ccbb.pitt.edu/cgi-bin/anm/anm1.cgi) was used for the NMA.25 An 18 Å distance cutoff and a distance weight of 2.5 were used for interactions between Cα atoms.
The program Fpocket26 was employed to identify and characterize the volumes and existence probabilities of pockets in CETP. The minimum radius of alpha sphere was set to 3 Å. Pockets containing less than 15 alpha spheres were discarded to filter off too small pockets. 50,000 monte-carlo iterations were performed for the crystal structure and each snapshot of the MD trajectories to accurately measure the volume of pockets.
Data are presented as mean±standard deviation calculated from the three replicate simulations. All structural figures were generated with Chimera.27
RESULTS
The equilibrium structure of CETP
The CETP equilibration in the three 8.5 ns NPT simulations were verified by monitoring the time evolution of structural parameters [Fig. 2]. The RMSD of backbone atoms with respect to the initial structure rapidly increased and leveled off at ~1.55 Å after 2 ns. The volume of CETP rose to 59.4±0.1 nm3 during the first 1 ns of the simulation and then remained stable. Similarly, the SASA of CETP converged at 238.5±3.3 nm2 after an initial increase. Although the gyration radius of backbone atoms fluctuated during the MD simulations, it increased for all three simulations while the average gyration radius converged after 1 ns. Therefore, the first 3.5 ns of each simulation was considered an equilibration period while the data from the last 5 ns was used for primary analysis. The RMSD and the gyration radius of backbone atoms, and protein SASA and volume, each has the same trend over the three replicate simulations [Fig. 2], confirming the consistency of individual simulations.
Figure 2. Time evolution of CETP structural parameters over the three NPT simulations.
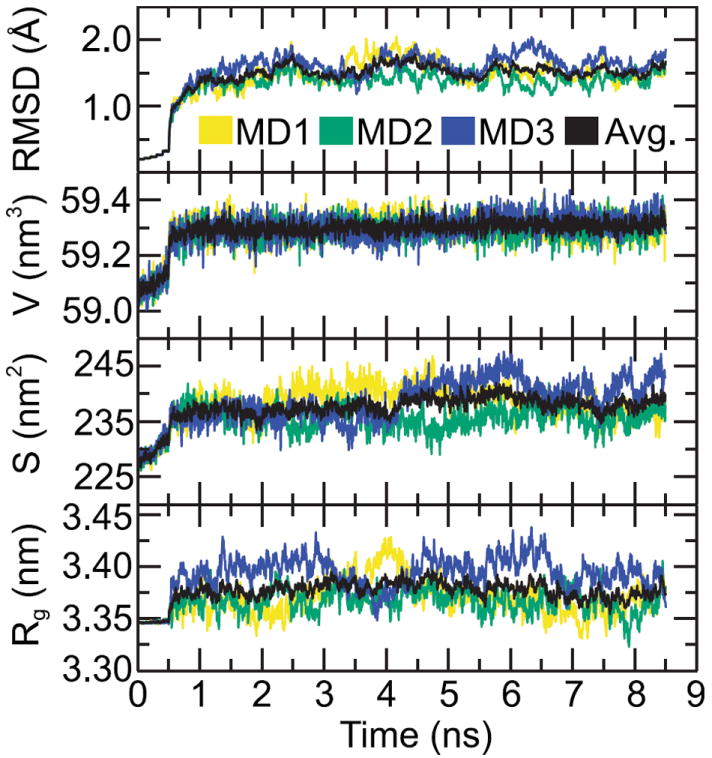
RMSD: the RMSD of backbone atoms with respect to the crystal structure; V: the volume of CETP; S: the SASA of CETP; Rg: the gyration radius of backbone atoms. Note the average value (Avg.) of each structural parameter in the three simulations converges within 2 ns.
The average structure of CETP [Fig. 3] obtained from the MD simulations shows no global conformational changes compared to the initial structure [Fig. 1], as evidenced by the low RMSD of backbone atoms. To access the individual residue displacements, the per-residue RMSD of backbone atoms of average structure relative to the initial structure is shown in Fig. 3, in which the residues with the largest RMSD are colored red. The central domain of the protein appears stable, as most residues deviate little from their initial positions. The residues with backbone RMSD greater than 1.16 Å (~20% of total residues) are mainly at the N-terminus, the distal portions of N- and C-terminal β-barrel domains, the central linker (Asp240-Arg259) nearby the central β-sheet, and the C-terminuses of helices A, B′ and X. These results demonstrate large conformational changes in these regions during the simulations.
Figure 3. Average structure of CETP obtained from the last 5 ns MD simulations.
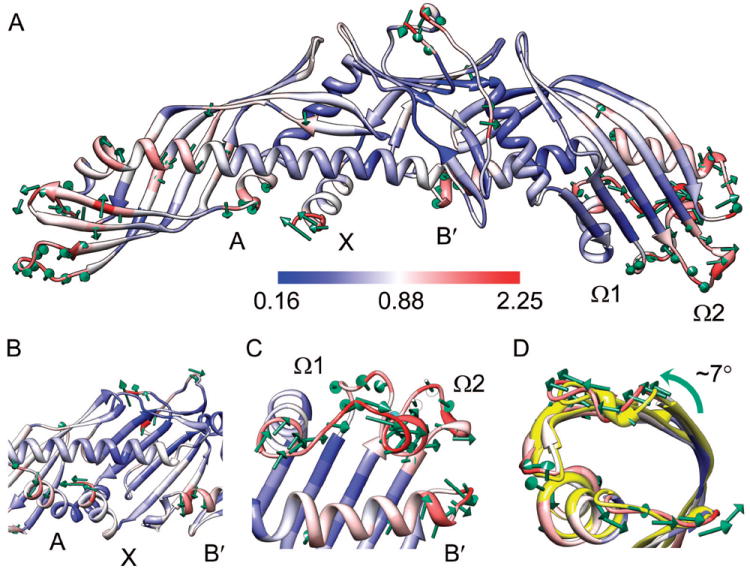
This structure is depicted as a ribbon diagram and colored according to the per-residue RMSD of backbone atoms relative to the initial structure. Green arrows indicate the movements of the backbone of the most shifted residues (top 20%) from the initial to the average positions during the simulations. To enhance clarity the lengths of arrows were scaled by a factor of 2. (A) The overall conformation of average structure. (B) The movements of the C-terminuses of helices A, X and B′. (C) The displacement of the distal end of C-terminal β-barrel domain away from the central domain of protein. (D) The rotation of the distal end of N-terminal β-barrel domain. The crystal structure (yellow) was superimposed onto the average structure using the backbone atoms.
A few key residues displayed a high degree of conformational change are shown in Fig. 3. The C-terminus of helix X moved toward the C-terminus of helix A [Fig. 3(A)]. Meanwhile, both the C-terminuses of helix A and B′ shifted away from the central cavity [Fig. 3(B)]. The distal portion of C-terminal β-barrel domain adopted a more extended conformation. The flap Ω1 (Met288-Gln320) drifted away from the protein central domain along the long axis direction of CETP [Fig. 3(C)]. Moreover, both the flap Ω2 (Phe350-Ala360) and the loop near the N-terminus of helix B′ were displaced away from the flap Ω1 [Fig. 3(A)]. In the meantime, the distal end of N-terminal β-barrel domain was rotated counterclockwise (~7°) along the long axis of N-terminal domain [Fig. 3(D)].
Structural flexibility
Structural flexibility is essential for protein biological functions28,29 and can allow the conformational changes of CETP. The RMSF of Cα atoms during the simulations were used to characterize the structural flexibility of CETP. For convenient comparison, the experimental B-factors obtained from the crystal structure9 and the theoretical B-factors from NMA were all converted to RMSF value by using Debye-Waller formula (RMSF = 3B / 8π2)1/2). In this study, the top 20% of residues with the highest Cα RMSF are considered flexible.
The distal portion of C-terminal β-barrel domain was flexible during the simulations, which agrees with crystallographic studies.9 For the C-terminal domain, the flexible residues in our simulations are mainly within residues Asp240-Leu254 (in the central linker), Asp290-Gln318 (in the flap Ω1), Pro351-Ser358 (in the flap Ω2), Lys392-Ser404 (labeled as flap Ω3) and Glu465-Ser476 (the helix X) [Fig. 4]. All these regions show elevated experimental RMSF [Fig. 4(A)]. Notably, the residues Ser439-Asp442 at the C-terminus of helix B′ are flexible in crystal and exhibited large Cα RMSF (top 30%) in our simulations, indicating that they are still relatively flexible in solution.
Figure 4. Structural fluctuation of CETP.
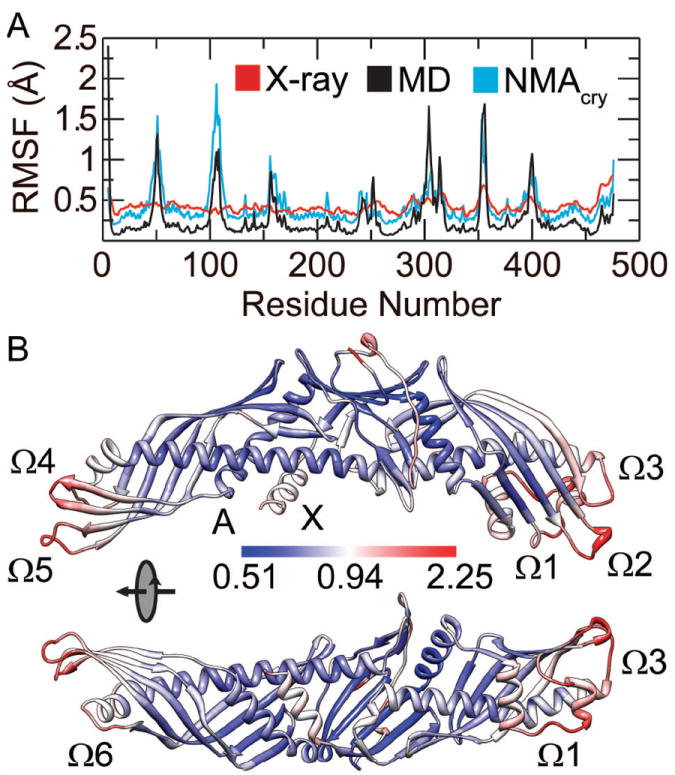
(A) Comparison of average Cα RMSF from the three MD simulations (MD) with experimental RMSF (X-ray) and theoretical RMSF obtained from the NMA of crystal structure (NMAcry). (B) The structural fluctuation of CETP during the simulations. The average structure from MD simulations is shown with residues colored according to their Cα RMSF.
Although the distal portion of N-terminal β-barrel domain shows little mobility in crystal, it is highly flexible in solution. As shown by the crystallographic study, only 4 (Glu46-Ala48, Met50) of the flexible residues are located at the distal end of N-terminal β-barrel domain.9 Contrarily, in our simulations nearly one third of the flexible residues are located here, namely Glu46-Val55 (labeled as flap Ω4), Gly100-Gln111 (labeled as flap Ω5) and Phe155-Trp162 (labeled as flap Ω6) [Fig. 4(B)]. The high flexibility of this end agrees well with the NMA of the crystal structure [Fig. 4(A)]. These results suggest that the distal end of N-terminal β-barrel domain is intrinsically flexible but becomes rigid in the crystal packing environment.
Helix X showed less flexibility in solution than in crystal. Having the highest experimental RMSF among other domains, helix X showed less fluctuation during the simulations than other mobile segments (such as the flaps Ω1 to Ω6) [Fig. 4(A)]. The reduced mobility of helix X is further confirmed by the comparison of theoretical B-factors. The theoretical B-factors of equilibrium structure obtained from the simulations are nearly the same as theoretical B-factors of the crystal structure aside from a significant decrease at the C-terminus (Supporting Information Fig. S1).
During the simulations all flexible domains underwent large conformational changes, suggesting that structural flexibility contributes to conformational changes of CETP. It is noteworthy that the C-terminus of helix A shifted away from the central cavity but was little flexible during the simulations. The likely cause for this phenomenon will be discussed in the following section.
During the simulations CEs and oleoyl tails of phospholipids generally maintained their initial conformations. The head groups of phospholipids fluctuated significantly more than the oleoyl tails and were more exposed to the aqueous solution. Notably, the head group of N-terminal phospholipid moved away from and lost its interaction with the C-cap dipole of helix B′.9 Contrarily, the head group of C-terminal phospholipid shifted towards Arg282 forming a salt bridge between them stabilizing the resulting conformation.
Role of salt bridges in the stability of CETP
Salt bridges play important roles in many aspects of protein structure and function, such as stability, domain motions and flexibility.30-32 In this study, salt bridges with occupancies more than 50% during the last 5 ns of the simulations were considered stable salt bridges. All salt bridges in the crystal structure except Asp447-Arg259 and Asp244-Lys233 were maintained during MD simulations (Supporting Information Table S1). Additionally, another three salt bridges (Asp118-Lys149, Asp214-Lys233 and Glu465-Lys29) formed and were rather stable with occupancies of 86.3%, 69.6% and 74.1%, respectively.
Although charged residues are generally evenly distributed on CETP, the stable salt bridges are distributed rather unevenly, residing mainly in the central domain of protein [Fig. 5(A)]. 12 of the 16 stable salt bridges are located in the central domain of CETP. There are 3 salt bridges in the distal half of C-terminal β-barrel domain, namely Glu291-Arg424, Asp366-Lys392 and Glu420-Arg424. The salt bridge Asp118-Lys149 formed during our simulations is the only salt bridge in the distal half of N-terminal β-barrel domain.
Figure 5. Salt bridges in CETP.
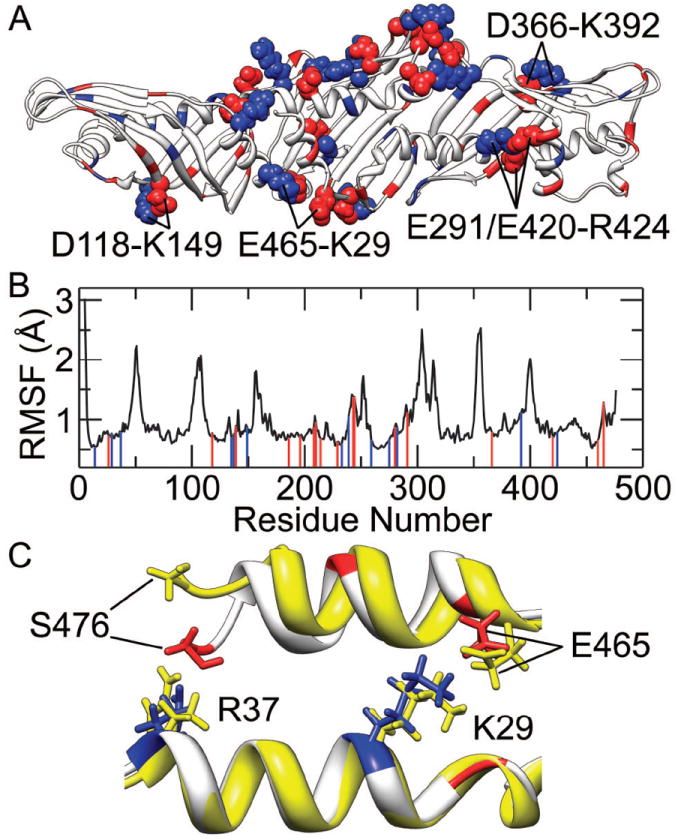
(A) Spatial distribution of charged residues and salt bridges. Positively charged residues are drawn in blue and negatively charged residues are drawn in red. Among these charged residues, the ones forming stable salt bridges during the simulations are shown in space-filling model. (B) Correlation of the salt bridges to the average Cα RMSF from the three MD simulations. Positively and negatively charged residues forming stable salt bridges are marked with blue and red lines, respectively. (C) Electrostatic interactions between the helix X and the helix A. The crystal structure (yellow) and a representative structure from the MD simulations (white) were aligned over the backbone atoms of helix A. Positively charged residues in the representative structure are colored blue while negatively charged ones and the deprotonated C-terminus (Ser476) are colored red.
The central domain of CETP, where salt bridges are mainly located, underwent few conformational changes during MD simulations and was more rigid than the distal portions of β-barrel domains. This relationship between salt bridges and structural flexibility is further illustrated in Fig. 5(B), in which nearly all flexible regions lack salt bridges. These results suggest that salt bridges expectedly contribute to the stability of the CETP central domain. In particular, the electrostatic interactions between the deprotonated (negatively charged) C-terminus (Ser476) and positively charged Arg37 should be a direct cause for the observed shift of C-terminus in helix X toward helix A [Fig. 5(C)]. Moreover, the electrostatic interactions between the C-terminus and Arg37, together with the salt bridge Glu465-Lys29 formed during the simulation, can anchor helix X more tightly to helix A. This is likely an important reason for the low flexibility of helix A and the reduced mobility of helix X in our simulations.
Surface hydrophobicity
Knowing the surface hydrophobicity of CETP is a key step for understanding the interactions of CETP with lipoproteins, which in turn helps reveal the mechanism behind CETP-mediated lipid transfer. Since exposure of hydrophobic residues to aqueous solution leads to an overall increased surface hydrophobicity,33 the SASA measurement was used to characterize CETP surface hydrophobicity.
Throughout the simulations, the SASA of both hydrophobic and hydrophilic residues increased about 8%, demonstrating that the CETP surface is generally more exposed to solvent without overall changes in surface hydrophobicity. Residues with dramatic SASA changes (at least 20.7 Å2) were identified and used to study detailed changes in surface hydrophobicity. Fig. 6 shows that these residues are mainly in distal portions of β-barrel domains and rarely in the central domain especially the central region of concave surface. The SASA of most distal portion residues in the C-terminal β-barrel domain greatly increased, however the opposite occurred for distal portion of the N-terminal β-barrel domain.
Figure 6. SASA changes of CETP during MD simulations.
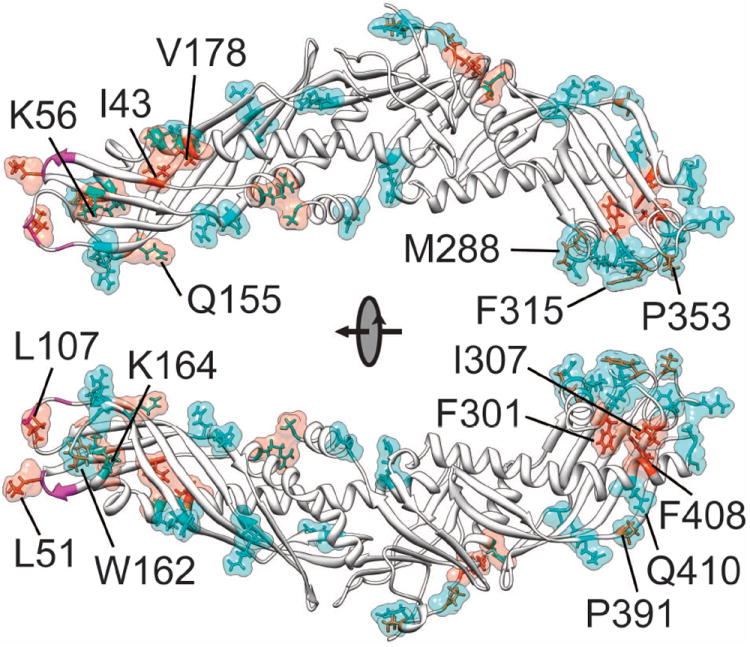
The top 10% residues with greatest SASA changes are shown in surface representation and colored cyan (SASA increase) or orange (SASA decrease). These residues are also displayed in stick models with the hydrophilic ones colored cyan and hydrophobic ones colored orange. The hydrophobic residues Ala48-Leu52, Trp105-Leu107 and Ile109 at the distal end of N-terminal β-barrel domain are highlighted in magenta.
The distal end of C-terminal β-barrel domain became more hydrophilic during the simulations. Although residues in flaps Ω1, Ω2 and Ω3 constitute about only 12% of all CETP residues, collectively their SASA increased ~15.3% which contributed to ~31% of the total SASA increase throughout the simulations. Many hydrophilic residues at this distal end, especially the ones in flap Ω1, were more exposed to solvent [Fig. 6]. The SASA of nearly all of these hydrophilic residues increased at least 30%, which is significant compared to the total SASA increase. At the same time, hydrophobic residues Phe301, Ile307 and Phe408 between the flap Ω1 and the helix B′ became more buried indicated by their decreased SASA. The increased exposure of hydrophilic residues and burying of hydrophobic residues together would increase the overall surface hydrophilicity of distal C-terminal β-barrel domain, especially in flap Ω1. Although Met288, Phe315, Pro353 and Pro391 are hydrophobic, their SASA increased leading to improved local hydrophobicity.
The overall surface hydrophobicity of the distal portion of N-terminal β-barrel domain is stable in solution. Only ~7.7% of the total SASA increase was contributed by the distal end of N-terminal β-barrel domain which accounts for ~20% of total residues. The SASA of hydrophobic residues Ile43, Leu51, Leu107 and Val178, and hydrophilic residues Gln155, Lys56 and Lys164 all decreased throughout the simulations [Fig. 6]. The burying of hydrophobic and hydrophilic residues together should lead to local but not overall changes in surface hydrophobicity. Notably from the crystal structure, the hydrophobic residues Ala48-Leu52, Trp105-Leu107 and Ile109 at the distal end of N-terminal β-barrel domain are highly exposed to solvent with average SASA of ~125.1 Å2. Unexpectedly, their total SASA decreased ~4.9% during the simulations. However the neighboring hydrophobic residue Trp162, which is buried in the crystal structure, became partially exposed to solvent which greatly increased its SASA [Fig. 6]. Consequently the distal end of N-terminal β-barrel domain remains hydrophobic in solution.
Cavities and pores in CETP
Along with the hydrophobic central cavity traversing the central β-sheet domain9 we identified three small cavities (labeled as C1, C2 and C3) and a surface pore (labeled as P1) from the crystal structure [Fig. 7(A)]. These small cavities are all completely buried in the N-terminal β-barrel and the pore P1 is partially exposed to solvent. The central cavity, internal cavities C1, C2, C3 and surface pore P1 are all highly hydrophobic, with hydrophilic residues contributing to 12.9%, 0%, 6.09%, 0% and 4.21% of their total SASA, respectively.
Figure 7. Cavities and pores in CETP.
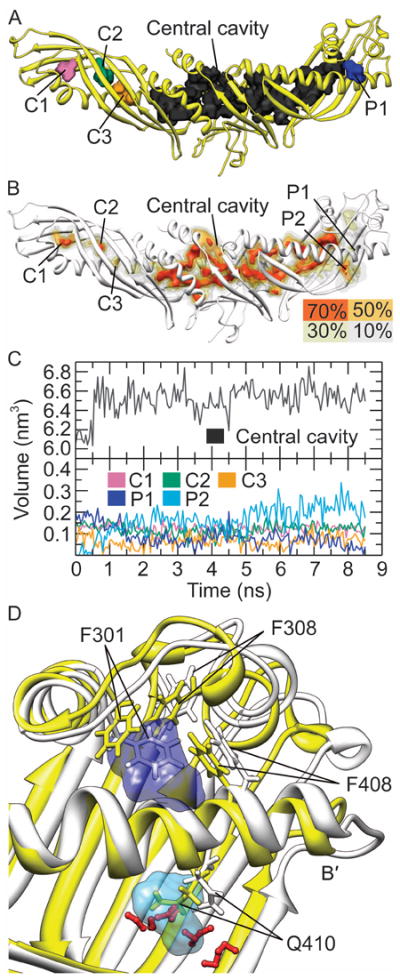
(A) The cavities and pore P1 in the crystal structure. (B) The cavities and pores during the MD simulations. The cavities and pores with existence probabilities of 70%, 50%, 30% and 10% during the last 5 ns MD simulations are all shown. (C) The volume of cavities and pores as a function of time. (D) Schematic illustration of the shrinkage of pore P1 and the formation of pore P2. The crystal structure (yellow) and a representative structure from the MD simulations (white) were aligned using the backbone atoms of the central portion of helix B′ (Ala415-Thr430). The pore P1 in the crystal structure is shown in dark blue and the pore P2 (existence probability greater than 50%) is displayed in light blue. The water molecules in the representative structure nearby the pore P2 are shown as red ball-stick models.
Throughout the simulations, the central cavity basically preserved its initial shape [Fig. 7(B)]. Cavities C1 and C2 were also rather stable, whose central parts showed existence probability higher than 70%. The existence probability of cavity C3 was low, with only a small part having an existence probability larger than 50%. All parts of pore P1 showed existence probability less than 50%, suggesting weak stability in solution. Interestingly, another surface pore (labeled as P2) was formed between the helix B′ and the flap Ω3. Contrary to pore P1 and all cavities this new surface pore is relative hydrophilic, with ~46.1% SASA comprised of hydrophilic residues.
The stability of cavities and surface pores was further confirmed by measuring their volumes [Fig. 7(C)]. The volume of the central cavity converged to 6533.7±204.2 Å3 after an initial minor increase, indicating it’s highly stable. The volume of cavity C1 was nearly unchanged throughout the simulation, converging at 121.3±36.2 Å3. Despite a certain degree of shrinkage, the cavity C2 existed at 118.3±51.6 Å3. The volume of cavity C3 decreased from 128.9 Å3 to 66.0±46.1 Å3. As expected, the pore P1 dramatically shrunk from 227.2 Å3 to only 68.5±79.2 Å3, indicating that it’s unstable in solution. In contrast, pore P2 expanded rapidly to ~193.6±81.2 Å3 becoming larger than P1 and the small cavities.
The reorientation of Phe301 side chain induced the observed shrinkage of pore P1 [Fig. 7(D)]. After all the three MD simulations, the hydrophobic side chain of Phe301 rotated inward directly occupied the previous space of pore P1. Upon the rotation of the aromatic ring, Phe301 interacted with Phe308 and Phe408 via stacking interactions, which stabilized the resulting structure. Meanwhile, the hydrophilic side chain of Gln410 moved outward and was exposed to solvent [Fig. 7(D)], confirmed by its greatly increased SASA [Fig. 6]. Its movement opened up pore P2, allowing the entry of water molecules to stabilize this partly hydrophilic pore.
DISCUSSION
The structural flexibility of CETP in our simulations shows differences from those observed in crystallographic studies. The distal end of the N-terminal β-barrel domain was flexible in our simulations but not in the crystal structure. Additionally helix X, which was determined to be the most flexible segment of CETP by crystallography,9 was less flexible than distal ends of β-barrel domains during our simulations. Also the observed flexibility of CETP in our MD simulations is consistent with NMA results and the uneven distribution of salt bridges. Salt bridges provide structural stability and are rarely located in the distal portions of β-barrel domains especially the N-terminal one. Finally helix X and helix A were anchored more tightly by electrostatic interactions. We believe that the differences in CETP flexibility between crystallographic studies and in our simulations were mostly due to different environmental conditions. A previous MD simulation study showed that the distal end of N-terminal β-barrel domain is flexible when CETP is bound to liposome surface via its concave surface.34 We can conclude that this distal end is intrinsically flexible and would remain flexible when CETP is bound to liposome.
Structural flexibility is essential for proper protein function.28,29 Helix X and flap Ω1 are flexible under crystallization conditions and were suggested to facilitate lipid transfer.9 Our MD simulations show that these domains are still flexible in solution. The distal end of the N-terminal β-barrel domain including flaps Ω4 to Ω6 was flexible throughout our simulations. Additionally, the residues Ser376-Lys379 were among the top 30% of residues which had the highest Cα RMSF in the simulations. Linker insertion mutations of human CETP at residues Ala48 (48A:EAS), Gly53 (53G:GVP), Glu165 (165Q:QGT), Ala373 (373A:D(RY)2RS) and Lys379 (379K:KGT) would markedly impair the CE transfer activity of CETP, suggesting the Ala48-Gly53 (in the flap Ω4), Gln165 (in the flap Ω6) and Ala373-Lys379 are functional regions likely responsible for lipid transfer.35 Above results indicate a fairly close relationship between flexible domains and the lipid transfer ability of CETP. This high flexibility allows for large-scale conformational changes and may play a role in the dynamic process of CETP-mediated lipid transfer.
Based on the analysis of CETP crystal structure, CETP was proposed to bind to one lipoprotein at a time via its concave surface.9 Potential conformational changes, such as the straightening of helix A and the bending of helix B around Ile190, have been suggested for CETP to accommodate lipoproteins of different sizes.9 However the central domain of CETP including the helix A and Ile190 showed little flexibility during our simulations, making it difficult for CETP to efficiently adjust its curvature in solution to accommodate different-sized lipoproteins via the suggested conformational changes.
Our simulation results indicate that the distal portions of β-barrel domains are highly flexible, which permit the hypothesized large-scale conformational changes forming a continuous tunnel by connecting internal small cavities and central cavity.13,14 The high flexibility of the distal ends of β-barrel domains also allow the proposed conformational changes at the distal ends of β-barrel domains which would open the continuous tunnel for entry and exit of CEs.13 Furthermore, internal cavities in CETP were generally stable during our MD simulations, further indicating that they are not just an artifact of crystallization conditions. It should be noted that although the surface pore P1 is not stable, a surface pore (P2) of similar size would be formed nearby. These results all support the tunnel mechanism.
The tunnel mechanism involves the interactions between the distal ends of CETP β-barrel domains and lipoproteins. The previously published TEM study shows that to facilitate neutral lipid transfer CETP penetrates ~50 Å into HDL with its N-terminal β-barrel domain, while penetrating LDL or VLDL only 20-25 Å through its distal C-terminal β-barrel domain.13 As the outer phospholipid shell of lipoproteins is 18-27 Å thick,13,36 CETP would penetrate into the surface and core of HDL but only into the surface of LDL or VLDL for binding. During our simulations the distal end of the C-terminal β-barrel domain became extended with overall hydrophilicity especially that of flap Ω1, increased. This improved hydrophilicity could assist the binding of the distal C-terminal domain to LDL or VLDL13 through hydrophilic interactions. Meanwhile, the distal end of N-terminal β-barrel domain was rotated further counterclockwise and remained hydrophobic in the simulations. By maintaining its hydrophobicity the distal N-terminal domain can interact with hydrophobic domains, or even the hydrophobic core, in lipoproteins. This is consistent with the TEM study result that CETP penetrates into the HDL core with its distal N-terminal β-barrel domain.13 This model explains why the replacement of hydrophobic Ala48 with charged residues (48A:DR(YR)2S and 48A:EAS) by linker insertion mutagenesis, located on the tip of N-terminal β-barrel domain, would partially or even completely impair the CE transfer activity of CETP.35 According to the TEM density map of the HDL-CETP complex, CETP is attached to the HDL surface at a ~45° angle and only its hydrophobic N-terminal distal end and adjacent β-sheet directly contact the hydrophobic core of HDL.13 In the distal N-terminal β-sheet the positively and negatively charged residues are spatially distributed in pairs or groups [Fig. 5(A)]. When transferring from high dielectric solvent to low dielectric HDL core, the side chains of neighboring oppositely charged residues would come closer to each other and form salt bridges for greatly increased electrostatic attractions. Given that the favorable energy of salt bridge formation in the protein interior can pay off the energetic penalty of dehydration of charged residues,37 penetrating of N-terminal domain into hydrophobic core would not be prohibited by too high energy barrier.
As the TEM result showed, CETP binds to different lipoproteins by different mechanisms,13 suggesting that additional determinants should be involved in CETP-lipoprotein binding aside from hydrophobic and hydrophilic properties.36 The N-terminal domain has a tapered configuration while the C-terminal domain adopts a more globular configuration. Penetration by the C-terminal domain would result in higher lipoprotein surface tension than penetration by the alternative end of CETP, especially for HDL which has the smallest size. This conformational difference between the two distal domains is the likely cause for lipoprotein selectivity and why the N-terminal distal end penetrates much deeper than the C-terminal end. The chemical and physical properties of lipoproteins are additional potential determining factors for selective CETP binding, such as protein composition, surface curvature, surface tension and surface charge. The binding of the CETP C-terminal domain to the LDL or VLDL surface can be modulated by lipoprotein surface charges and stabilized by the salt bridges between them. Furthermore, the charged head groups of phospholipids might be further apart due to increased lipoprotein surface curvature (for example, HDL),36 permitting CETP, especially the tapered N-terminal distal end, to deeply penetrate the lipoprotein.
The hydrophobic residue Trp162 at the distal end of N-terminal β-barrel domain is buried in the crystal structure but became partially exposed to solvent during our simulations. Considering Trp162 interacts with water when CETP is liposome-bound,34 we can conclude that Trp162 is partially exposed to solution regardless of if CETP is bound to the liposome.
The central cavity volume calculated by using Fpocket is larger than that estimated from CETP crystallography9 with at least two possible reasons for this. First, as the central cavity is opened it is difficult to precisely define its boundary. In this paper the space occupied by not only CEs but also acyl chains of phospholipids was assigned to the central cavity. This should be the primary reason for the larger central cavity volume in our studies. Second, different methods were used to calculate the central cavity volume. Since the same method was employed to estimate the central cavity volume throughout this work, the conclusion that this cavity is stable would not be changed.
Since the mutation of Asn396 to Gln through oligonucleotide-directed mutagenesis would eliminate the potential N-linked glycosylation site at Asn396 but almost does not change the CE transfer activity of CETP,38 the glycosylation at Asn396 was discarded in our MD simulations and we believe this treatment should not affect the simulation results. Due to the limitation of computational capability, in this study only three 8.5 ns NPT simulations were performed. Since multiple short trajectories are more efficient than a single long trajectory to sample conformational space,39 especially considering the maintained stability of CETP during the last 5 ns NPT simulations and agreeing results from three replicate simulations, our conclusions should not be overturned by longer calculations. In particular, the high flexibility of distal ends of β-barrel domains, the reduced flexibility of helix X, and the partially exposure of Trp162 to aqueous phase which were observed in previously published longer MD simulations34 were all captured by our shorter MD simulations. Although similar results were obtained the conclusions which can be reached in our simulations and in previous studies are different. In previous studies, CETP was simulated binding to a liposome.34 Above observations can be explained by the intrinsic properties of CETP as well as the influences of liposome binding to CETP. As CETP is simulated in solution without liposome binding in this work, we can support that the observed flexibility and exposure of Trp162 to solvent are inherent features of CETP in solution and not the result of CETP-liposome interactions. Also in previous studies the observed high flexibility of distal ends were simply attributed to the nature of loop34 while our simulation results indicated that salt bridges stabilization is an important and critical determinant of overall CETP flexibility. In our simulations the formation of salt bridge Glu465-Lys29 reduced the flexibility of helix X. Contrarily, a lack of salt bridges [Fig. 5(A)] contributed to the flexibility of not only loops in distal ends but also other domains such as the β-sheet in distal ends of β-barrels, flap Ω2 including two short helices, and the terminuses of helix B′, et al. [Fig. 4(A)].
In previous studies, the conformation of helix X was shown to rearrange and become buried inside the central cavity generating a hydrophobic pathway from the liposome surface to central cavity.34 This observed conformational change in helix X is induced by the hydrophobic patch formed by liposome phospholipids.34 In our simulation helix X maintained its conformation and only its C-terminus showed significant displacement. As helix X is amphiphilic and anchored to helix A by salt bridge Glu465-Lys29 and electrostatic interaction between its C-terminus and Arg37, it is hard for helix X to move inside the hydrophobic central cavity or undergo other forms of large-scale displacements in solution. It is unexpected that helix X will open more in this study even after a longer simulation time.
In this work we investigated the structural features of CETP in solution. The exact mechanism of neutral lipids entry and exit from the central cavity is essential to further elucidate the lipid transfer mechanism, whether through the central openings or distal ends of β-barrels. Additional research to determine how the entered neutral lipids may stabilize certain transient cavities and investigate the potential conformational changes of helix X and distal ends is required to understand lipid transfer. MD simulation studies of CETP in the presence of additional phospholipids and CE molecules or in a more hydrophobic media are urgently required.
CONCLUSION
In this paper, we performed all-atom MD simulations to characterize the structural features of CETP in solution. The simulations show that distal ends of β-barrel domains are highly flexible, though the N-terminal one initially showed little flexibility through crystallographic studies. We found that salt bridges contribute to the rigidity of protein central domain and reduce the flexibility of helix X. During the simulations the distal C-terminal β-barrel domain became more extended and generated a new surface pore. This pore and all cavities in CETP are stable. These results demonstrate that the formation of a continuous tunnel within CETP by connecting cavities is permitted in solution, in which the highly flexible distal ends of β-barrel domains may facilitate the required conformational changes. This finding corroborates the tunnel mechanism for CETP-mediated lipid transfer. In this proposed lipid transfer process distal ends of β-barrel domains, especially the N-terminal one which remains hydrophobic in solution, may mediate the binding of CETP to lipoproteins by hydrophobic and hydrophilic interactions.
Supplementary Material
Acknowledgments
We thank the Laboratory of Intelligent Information Processing and Computing (Xi’an Jiaotong University, China) for computational assistance. This work was supported by National Natural Science Foundation of China (no. 11074196), and the Cultivation Fund of the Key Scientific and Technical Innovation Project, Ministry of Education of China (no. 708082); in part by the Office of Science, Office of Basic Energy Sciences of the United States Department of Energy (contract no. DE-AC02-05CH11231) and the National Heart, Lung, And Blood Institute of the National Institutes of Health (no. R01HL115153) (G. R. and L.Z.).
Abbreviations
- CETP
cholesteryl ester transfer protein
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- VLDL
very-low-density lipoprotein
- CE
cholesteryl ester
- MD
molecular dynamics
- RMSD
root mean-square deviation
- SASA
solvent-accessible surface area
- RMSF
root mean-square fluctuation
- NMA
normal mode analysis
Footnotes
This work was performed at Xi’an Jiaotong University, People’s Republic of China.
References
- 1.Tall AR. Plasma-lipid transfer proteins. Journal of Lipid Research. 1986;27(4):361–367. [PubMed] [Google Scholar]
- 2.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease: The Framingham study. The American journal of medicine. 1977;62(5):707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 3.Camejo G, Waich S, Quintero G, Berrizbeitia ML, Lalaguna F. The affinity of low density lipoproteins for an arterial macromolecular complex A study in ischemic heart disease and controls. Atherosclerosis. 1976;24(3):341–354. doi: 10.1016/0021-9150(76)90126-x. [DOI] [PubMed] [Google Scholar]
- 4.de Grooth GJ, Kuivenhoven JA, Stalenhoef AFH, de Graaf J, Zwinderman AH, Posma JL, van Tol A, Kastelein JJP. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans - A randomized phase II dose-response study. Circulation. 2002;105(18):2159–2165. doi: 10.1161/01.cir.0000015857.31889.7b. [DOI] [PubMed] [Google Scholar]
- 5.Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: An initial multidose study of torcetrapib. Arteriosclerosis Thrombosis and Vascular Biology. 2004;24(3):490–497. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- 6.Barter PJ, Brewer HB, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein - A novel target for raising HDL and inhibiting atherosclerosis. Arteriosclerosis Thrombosis and Vascular Biology. 2003;23(2):160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 7.Barter P, Rye K-A. Cholesteryl ester transfer protein inhibition to reduce cardiovascular risk: where are we now? Trends in pharmacological sciences. 2011;32(12):694–699. doi: 10.1016/j.tips.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Drayna D, Jarnagin AS, Mclean J, Henzel W, Kohr W, Fielding C, Lawn R. Cloning and Sequencing of Human Cholesteryl Ester Transfer Protein Cdna. Nature. 1987;327(6123):632–634. doi: 10.1038/327632a0. [DOI] [PubMed] [Google Scholar]
- 9.Qiu XY, Mistry A, Ammirati MJ, Chrunyk BA, Clark RW, Cong Y, Culp JS, Danley DE, Freeman TB, Geoghegan KF, Griffor MC, Hawrylik SJ, Hayward CM, Hensley P, Hoth LR, Karam GA, Lira ME, Lloyd DB, McGrath KM, Stutzman-Engwall KJ, Subashi AK, Subashi TA, Thompson JF, Wang IK, Zhao HL, Seddon AP. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nature Structural & Molecular Biology. 2007;14(2):106–113. doi: 10.1038/nsmb1197. [DOI] [PubMed] [Google Scholar]
- 10.Barter PJ, Jones ME. Kinetic-studies of the transfer of esterified cholesterol between human-plasma low and high-density lipoproteins. Journal of Lipid Research. 1980;21(2):238–249. [PubMed] [Google Scholar]
- 11.Ihm J, Quinn DM, Busch SJ, Chataing B, Harmony JAK. Kinetics of plasma protein-catalyzed exchange of phosphatidylcholine and cholesteryl ester between plasma-lipoproteins. Journal of Lipid Research. 1982;23(9):1328–1341. [PubMed] [Google Scholar]
- 12.Tall AR. Plasma cholesteryl ester transfer protein. Journal of Lipid Research. 1993;34:1255–1274. [PubMed] [Google Scholar]
- 13.Zhang L, Yan F, Zhang S, Lei D, Charles MA, Cavigiolio G, Oda M, Krauss RM, Weisgraber KH, Rye K-A, Pownall HJ, Qiu X, Ren G. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nature Chemical Biology. 2012;8(4):342–349. doi: 10.1038/nchembio.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall J, Qiu X. Structural and biophysical insight into cholesteryl ester-transfer protein. Biochemical Society Transactions. 2011;39:1000–1005. doi: 10.1042/BST0391000. [DOI] [PubMed] [Google Scholar]
- 15.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 16.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. Journal of Computational Physics. 1999;151(1):283–312. [Google Scholar]
- 17.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 18.Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. Journal of Physical Chemistry B. 2000;104(31):7510–7515. [Google Scholar]
- 19.Mackerell A, Jr, Feig M, Brooks C., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. Journal of Computational Chemistry. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79(2):926–935. [Google Scholar]
- 21.Cournia Z, Smith JC, Ullmann GM. A molecular mechanics force field for biologically important sterols. Journal of Computational Chemistry. 2005;26(13):1383–1399. doi: 10.1002/jcc.20277. [DOI] [PubMed] [Google Scholar]
- 22.Grest GS, Kremer K. Molecular-dynamics simulation for polymers in the presence of a heat bath. Physical Review A. 1986;33(5):3628–3631. doi: 10.1103/physreva.33.3628. [DOI] [PubMed] [Google Scholar]
- 23.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation - The langevin piston method. Journal of Chemical Physics. 1995;103(11):4613–4621. [Google Scholar]
- 24.Darden T, York D, Pedersen L. Particle mesh Ewald: An N*log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 25.Eyal E, Yang LW, Bahar I. Anisotropic network model: systematic evaluation and a new web interface. Bioinformatics. 2006;22(21):2619–2627. doi: 10.1093/bioinformatics/btl448. [DOI] [PubMed] [Google Scholar]
- 26.Schmidtke P, Le Guilloux V, Maupetit J, Tuffery P. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Research. 2010;38:W582–W589. doi: 10.1093/nar/gkq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 28.Huber R, Bennett WS. Functional-significance of flexibility in proteins. Biopolymers. 1983;22(1):261–279. doi: 10.1002/bip.360220136. [DOI] [PubMed] [Google Scholar]
- 29.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33(22):6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Nussinov R. Salt bridge stability in monomeric proteins. Journal of Molecular Biology. 1999;293(5):1241–1255. doi: 10.1006/jmbi.1999.3218. [DOI] [PubMed] [Google Scholar]
- 31.Sinha N, Smith-Gill SJ. Electrostatics in protein binding and function. Current Protein & Peptide Science. 2002;3(6):601–614. doi: 10.2174/1389203023380431. [DOI] [PubMed] [Google Scholar]
- 32.Barlow DJ, Thornton JM. Ion-pairs in proteins. Journal of Molecular Biology. 1983;168(4):867–885. doi: 10.1016/s0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
- 33.Kato A, Nakai S. Hydrophobicity determined by a fluorescence probe method and its correlation with surface-properties of proteins. Biochimica Et Biophysica Acta. 1980;624(1):13–20. doi: 10.1016/0005-2795(80)90220-2. [DOI] [PubMed] [Google Scholar]
- 34.Koivuniemi A, Vuorela T, Kovanen PT, Vattulainen I, Hyvonen MT. Lipid Exchange Mechanism of the Cholesteryl Ester Transfer Protein Clarified by Atomistic and Coarse-grained Simulations. PLoS Computational Biology. 2012;8(1):e1002299. doi: 10.1371/journal.pcbi.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Deng LP, Brown ML, Agellon LB, Tall AR. Structure-Function Studies of Human Cholesteryl Ester Transfer Protein by Linker Insertion Scanning Mutagenesis. Biochemistry. 1991;30(14):3484–3490. doi: 10.1021/bi00228a019. [DOI] [PubMed] [Google Scholar]
- 36.Charles MA, Kane JP. New molecular insights into CETP structure and function: a review. Journal of Lipid Research. 2012;53(8):1451–1458. doi: 10.1194/jlr.R027011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bush J, Makhatadze GI. Statistical analysis of protein structures suggests that buried ionizable residues in proteins are hydrogen bonded or form salt bridges. Proteins-Structure Function and Bioinformatics. 2011;79(7):2027–2032. doi: 10.1002/prot.23067. [DOI] [PubMed] [Google Scholar]
- 38.Stevenson SC, Wang S, Deng LP, Tall AR. Human Plasma Cholesteryl Ester Transfer Protein Consists of a Mixture of 2 Forms Reflecting Variable Glycosylation at Asparagine-341. Biochemistry. 1993;32(19):5121–5126. doi: 10.1021/bi00070a021. [DOI] [PubMed] [Google Scholar]
- 39.Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein science : a publication of the Protein Society. 1998;7(3):649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.