

Abstract
Hyperhomocysteinemia is an independent risk factor for both acute and chronic neurological disorders but little is known about the underlying mechanisms by which elevated homocysteine can promote neuronal cell death. We recently established a role for NMDA receptor mediated activation of extracellular signal-regulated kinase-mitogen activated protein kinase (ERK-MAPK) in homocysteine-induced neuronal cell death. In the present study we examined the involvement of the stress-induced MAPK, p38 in homocysteine-induced neuronal cell death and further explored the relationship between the two MAPKs, ERK and p38, in triggering cell death. Homocysteine mediated NMDA receptor stimulation and subsequent Ca2+ influx led to a biphasic activation of p38 MAPK characterized by an initial rapid but transient activation followed by a delayed and more prolonged response. Selective inhibition of the delayed p38 MAPK activity was sufficient to attenuate homocysteine-induced neuronal cell death. Using pharmacological and RNAi approaches we further demonstrated that both the initial and delayed activation of p38 MAPK is downstream of, and dependent on activation of ERK MAPK. Our findings highlight a novel interplay between ERK and p38 MAPK in homocysteine-NMDA receptor induced neuronal cell death.
Keywords: Homocysteine, ERK MAPK, p38 MAPK, NMDA, crosstalk, neuronal cell death
INTRODUCTION
Hyperhomocysteinemia, a common metabolic disorder of the methionine cycle, is an independent risk factor for multiple neurodegenerative disorders such as cerebral stroke, age-associated dementia, Alzheimer’s disease, Parkinson’s disease, cerebral atrophy and seizures (Sacco et al. 1998; Miller 1999; Seshadri et al. 2002; Obeid and Herrmann 2006; Zoccolella et al. 2006). In vivo studies using an animal model Parkinson’s disease have shown that elevated levels of homocysteine increases the vulnerability of neurons to dysfunction and death in these animals (Duan et al. 2002). Studies using cultured neurons have also shown that elevated homocysteine can sensitize neurons to increased cellular injury in response to excitotoxic or oxidative insult, amyloid β-peptide, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Kruman et al. 2000; Duan et al. 2002; Kruman et al. 2002). Additionally, prolonged exposure to increased levels of homocysteine alone has been shown to induce cell death in cultured neurons (Lipton et al. 1997; Kruman et al. 2000; Ho et al. 2002; Mattson and Shea 2003; Poddar and Paul 2009). Despite the general recognition of the possible role of homocysteine in the etiology of multiple neurodegenerative disorders, relatively little is known about the underlying molecular mechanisms involved in homocysteine induced neuronal death.
It has been shown that oxidative injury and neuronal cell death associated with elevated levels of extracellular homocysteine involves stimulation of the N-methyl-D-aspartate (NMDA) subtype of ionotropic glutamate receptors (Lipton et al. 1997; Kruman et al. 2000; Kruman et al. 2002; Jara-Prado et al. 2003; Mattson and Shea 2003; Poddar and Paul 2009). Since over activation of NMDA receptors is known to be involved in glutamate-mediated excitotoxic cell death, it has been assumed that homocysteine activates NMDA receptors analogous to glutamate, limiting studies in this area. Contrary to this notion our recent findings (Poddar and Paul 2009) suggest that the effects of homocysteine and glutamate on NMDA receptor activation are quite different. Glutamate-mediated excitotoxic cell death has been primarily attributed to activation of NR1/NR2B subunit containing NMDA receptors (NR2B-NMDAR), (Hardingham et al. 2002; Li et al. 2002; Riccio and Ginty 2002; Kim et al. 2005; Liu et al. 2007; Zhang et al. 2007). In contrast, our findings show that homocysteine-mediated neuronal cell death involves stimulation of NR1/NR2A containing NMDAR (NR2A-NMDAR) (Poddar and Paul 2009), the pool of NMDAR that is generally thought to be involved in cell survival (Hetman and Kharebava 2006; Liu et al. 2007). Our findings further show that homocysteine-NMDAR induced neuronal cell death involves sustained activation of extracellular signal-regulated kinase (ERK MAPK), which also differs from glutamate-NMDAR mediated transient activation of ERK MAPK (Paul et al. 2003; Mao et al. 2004). These findings raise the possibility that homocysteine induced neurotoxicity involves unique signaling pathways that are different from glutamate-NMDAR mediated excitotoxic cell death.
The aim of this study was to determine the relative contribution of p38 stress activated kinase (Ono and Han 2000; Cuadrado and Nebreda 2010), another member of the MAPK family that is up regulated following NMDAR stimulation (Waxman and Lynch 2005; Poddar et al. 2010), in mediating homocysteine dependent neuronal cell death. Our results show that homocysteine leads to biphasic activation of p38 MAPK, where the initial rise is rapid but transient and the delayed increase is more sustained. We also show that homocysteine-NMDAR mediated sustained activation ERK MAPK follows a two-tier pattern. Both the initial and delayed activation of p38 MAPK are dependent on ERK MAPK activity. This novel interplay between ERK and p38 MAPK facilitates homocysteine-induced neuronal cell death.
EXPERIMENTAL PROCEDURES
Materials and reagents
All reagents required for tissue culture were obtained from Invitrogen (Carlsbad, CA, USA). L-homocysteine thiolactone, L-Glutamate, cytosine D-arabinofuranoside, glycine, EGTA and Hoechst 33342 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-phospho-ERK 1/2 (Thr202/Tyr204) monoclonal antibody (p-ERK), anti-phospho-p38 (Thr180/Tyr182) polyclonal antibody (p-p38), anti-p38 polyclonal antibody, anti-microtubule associated protein 2 (MAP2) antibody, and anti-rabbit and anti-mouse horse-radish-peroxidase conjugated secondary antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-ERK2 polyclonal antibody (ERK) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and anti-glial fibrillary acidic protein (GFAP) antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-rabbit-Alexa 488-conjugated antibody and anti-mouse-Cy3 conjugated antibodies were obtained from Molecular Probes (Eugene, OR, USA). A complete list of the pharmacological inhibitors used in this study, their physiological action and their sources have been summarized in Table S1.
Neuron culture, L-Homocysteine preparation and stimulation
Primary cortical neuronal cultures were established from rat embryos (16–17 day of gestation) as described earlier (Poddar and Paul 2009). The purity of the neuron cultures (Paul et al. 2003; Poddar and Paul 2009; Poddar et al. 2010) was determined by immunocytochemical staining with anti-MAP-2 (neuronal marker) and anti-GFAP (astrocyte marker) antibodies (Fig. S1; 95.63 ± 0.67%). For all experiments L-homocysteine was prepared fresh as described earlier (Poddar et al. 2001). Neurons (12–14 days in vitro) were treated with L-homocysteine in Hank’s balanced salt solution containing 50 μM of glycine (Lipton et al. 1997) at concentrations specified in each experiment. Pharmacological inhibitors were added either 20 min before exposure to L-homocysteine (pretreatment) or 30 min after the onset of L-homocysteine treatment (post 30 min). The Institutional Animal Care and Use Committee of University of New Mexico, Health Sciences Center approved the animal procedures used in this study.
Immunoblotting
At the specified time points, following treatment with L-homocysteine neurons were harvested and cell lysates were resolved by SDS-PAGE (8%), and subjected to immunoblotting as described earlier (Poddar and Paul 2009). Antibody concentrations were used according to manufacturer’s protocol. Signals from immune complexes developed with West Pico supersignal chemiluminescence reagents (Pierce Biotechnology, USA) were detected on X-ray films and densitometric analysis of the images was performed using the Image J software.
Caspase-3 activity assay
Caspase-3 activity was determined by monitoring the cleavage of a specific caspase substrate, DEVD-pNA, using the ApoAlert Caspase 3 colorimetric assay kit (CLONTECH). Following treatment with L-homocysteine in the presence or absence of p38 MAPK inhibitor SB203580, equal number of neurons were washed with phosphate buffered saline (pH 7.4), harvested in the cell lysis buffer (kit) and centrifuged for 10 min at 10,000 rpm and 4°C. The supernatants were incubated at 37°C for 2 hr with the substrate according to manufacturer’s protocol. Subsequent development of color was analyzed in a microplate reader at 405 nm wavelength.
Cell death assay
Neuronal cultures treated with L-homocysteine in the presence or absence of selective inhibitors for 18 hours were fixed with 4% paraformaldehyde and stained with Hoechst 33342 dye as previously described (Poddar and Paul 2009). To assess the percentage of pyknotic cells, a total of 1,500 nuclei were counted for each set of experiments (n = 3) using fluorescent microscopy.
RNA-interference using lentiviral approaches
Lentiviral particles containing short hairpin RNA (sh RNA) targeted against three different regions of ERK2 and pooled (Santa Cruz Biotechnology; Santa Cruz, CA, USA) were transduced into neurons according to manufacturer’s protocol. Briefly, 10-day old neurons (25 × 103 cells/well) were transduced with the lentiviral particles (multiplicity of infection of 10) in the presence of 8 μg/ml of Polybrene (Santa Cruz Biotechnology; Santa Cruz, CA, USA) in its original medium for 6 hr. Following infection the virus-containing medium was removed and the cells were maintained in its original medium for another four days. One set of neurons was transduced with lentivirus containing scrambled RNA following similar protocol. Infection with control copGFP containing lentiviral particles (coding insert copGFP-shRNA) under similar conditions was used to monitor transduction efficiency. CopGFP expressing cells (green positive) were counterstained with DAPI and quantitatively assessed by fluorescent microscopy. A total of 1500 cells were counted for each set of experiments. On 14th day of culture, cells were subjected to L-homocysteine treatment as specified. Suppression of ERK2 in transduced cells was evaluated by immunoblot analysis with anti-ERK antibody.
Statistical analysis
Statistical comparison was done using One-way analysis of variance (ANOVA, Bonferroni’s multiple comparison test) and differences were considered significant when p < 0.05.
RESULTS
Homocysteine-mediated regulation of p38 MAPK in neurons
We first examined the effect of homocysteine on phosphorylation and subsequent activation of p38 MAPK in neurons. Cortical neuronal cultures were treated with varying concentrations of L-homocysteine (0, 5, 50, 100, and 500 μM) for 5 min and cell lysates were analyzed by immunoblotting with anti-phospho p38 antibody. The representative immunoblot and the corresponding mean data (Fig. 1A) show a significant increase in phosphorylation of p38 MAPK following treatments with 50 μM and greater concentrations of homocysteine. Total p38 MAPK levels remain unaltered by this treatment. To determine the temporal profile of p38 MAPK activation neurons were exposed to 50 μM L-homocysteine for varying times (0, 5, 10, 20, 30 min). A rapid but transient increase in phosphorylation of p38 MAPK was observed within 2.5 – 5 min of homocysteine treatment, which returned to basal level by 30 min (Fig. 1B). With continued exposure to homocysteine for prolonged time periods (0, 2, 4, 6, 8 hr), a delayed increase in p38 MAPK phosphorylation was evident within 4 hr of treatment that peaked at 6 hr and declined thereafter (Fig. 1C). Taken together these findings suggest that homocysteine treatment leads to a transient initial activation of p38 MAPK within minutes, followed by a delayed and more sustained secondary activation beginning at 4 hr.
FIGURE 1.

Homocysteine induces biphasic pattern of p38 MAPK phosphorylation. Neuron cultures (A) were treated with different concentrations of L-homocysteine (L-Hcy) for 5 min, (B, C) were treated with 50 μM L-Hcy for the specified times. Equal amount of protein from each sample was processed for immunoblot analysis using anti-phospho-p38 (p-p38, upper panel) and p38 (lower panel) antibodies. Quantification of phosphorylated p38 MAPK by computer-assisted densitometry and Image J analysis is shown beside each immunoblot. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.01).
Role of NMDARs and Ca2+ in phosphorylation of p38 MAPK
To evaluate the role of NMDAR mediated Ca2+ influx in homocysteine-induced initial and secondary activation of p38 MAPK, neurons were either treated with L-homocysteine (50 μM) for 5 min in the presence of NMDAR antagonists, 5 μM of MK801 (Yun et al. 1998) or 200 μM of APV or treated with L-homocysteine for 4 hr where antagonists were added 30 min after onset of homocysteine treatment. Immunoblot analysis using anti-phospho p38 antibody shows that pre-treatment with either MK801 or APV effectively blocked homocysteine-induced phosphorylation of p38 MAPK by 5 min (Fig. 2A). Addition of MK801 or APV 30 min after onset of homocysteine treatment also blocked homocysteine induced secondary activation of p38 MAPK by 4 hr (Fig 2B). Removal of extracellular Ca2+ with EGTA (2 mM) before L-homocysteine (50 μM, 2.5 min) treatment blocked the initial phosphorylation of p38 MAPK (Fig. 2C). Addition of EGTA 30 min after the onset of L-homocysteine treatment (50 μM, 4 hr) also blocked the delayed phosphorylation of p38 MAPK at 4 hr (Fig. 2D).
FIGURE 2.

Homocysteine-induced p38 MAPK phosphorylation is dependent on NMDAR mediated Ca2+ influx. (A, C) Neuron cultures were treated with 50 μM L-homocysteine (L-Hcy) for 5 min in the absence or presence of (A) MK810 (5 μM) or APV (200 μM) and (C) EGTA (2 mM). (B, D) Neuron cultures were treated with 50 μM L-Hcy and 30 min after onset of homocysteine treatment (post 30′). (B) NMDA receptor blocker MK810 (5 μM) or APV (200 μM) and (D) EGTA (2 mM) were added, and the incubation was continued for 4 hr. Samples were analyzed using anti-phospho-p38 (upper panel) and p-38 (lower panel) antibodies. The extent of phosphorylation of p38 MAPK was quantified using computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min time point (p < 0.001); # significant difference from 5 min or 4 hr homocysteine treatment (p < 0.001).
Inhibition of p38 MAPK attenuates homocysteine-induced neuronal cell death
To clarify the role of p38 MAPK in homocysteine induced neuronal cell death we examined the effect of SB203580, a selective inhibitor of p38 MAPK on activation of caspase-3. Caspase-3 belongs to a family of cysteine proteases that play a critical role in apoptotic cell death (Cohen 1997; Thornberry and Lazebnik 1998). Neurons were treated with L-homocysteine (50 μM) in the absence or presence of SB203580 (5 μM) for 6 hr and cell lysates were then analyzed for caspase-3 activity by spectrophotometric assay. To examine the role of delayed p38 MAPK activation on caspase-3 activity, in a parallel set of experiments SB203580 was added 30 min after the onset of homocysteine treatment. Figure 3A shows that treatment with L-homocysteine alone led to considerable increase in caspase-3 activity. Both pre-incubation and delayed application of SB203580 significantly reduced homocysteine-induced increase in caspase-3 activity, although reduction in caspase-3 activity is slightly higher when pre-incubated with SB203580 as compared to the delayed treatment. To evaluate cell viability, in the next set of experiments neurons were either pre-incubated with SB203580 or the inhibitor was applied 30 min after the onset of L-homocysteine treatment (50 μM) and 18 hr later cells were processed for Hoechst DNA staining (Fig. 3B and C). As previously reported (Poddar and Paul 2009) treatment with L-homocysteine alone led to significant increase in neuronal cell death (17.3% ± 1.35 for control vs. 44.65% ± 1.6 for homocysteine). Application of SB203580 either prior to or 30 min after the onset of homocysteine treatment led to significant reduction in homocysteine-induced neuronal cell death (44.65% ± 1.6 for homocysteine alone vs. 25.18% ± 0.93 for pre-incubation and 29.06% ± 0.83 for delayed treatment). Taken together these findings suggest that the delayed activation of p38 MAPK plays a predominant role in homocysteine-NMDAR induced neuronal death.
FIGURE 3.
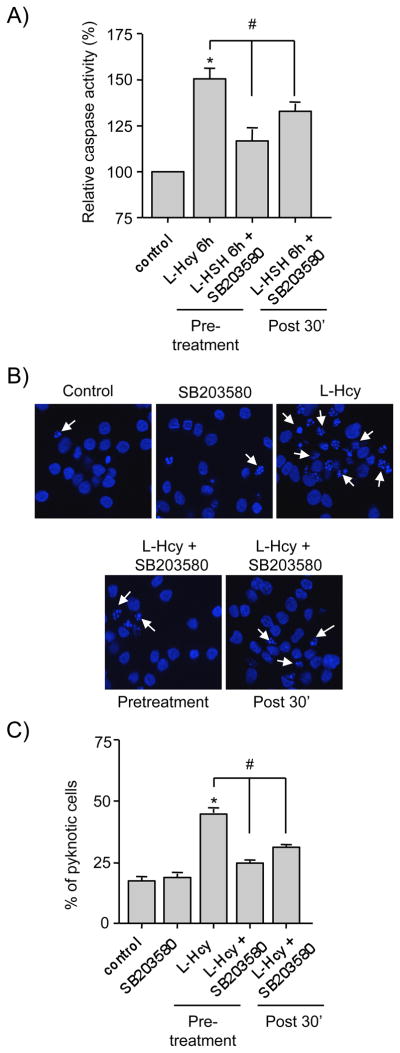
Roles of initial and delayed p38 MAPK activation in homocysteine-induced neuronal cell death. (A) Neuron cultures were treated with L-homocysteine (L-Hcy) for 6 hr and then processed for caspase-3 activity assay. In some experiments SB203580 (5 μM) was added either prior to (pretreatment) or 30 min after (post 30′) the onset of homocysteine treatment and caspase-3 activity was measured after 6 hr. * Significant difference from control (p < 0.05); # significant difference from 6 hr homocysteine treatment (p < 0.05). (B, C) Neuron cultures were treated with L-Hcy (50 μM) for 18 hr and then processed for Hoechst DNA staining to measure the extent cell death. In some experiments SB203580 (5 μM) was added either prior to (pretreatment) or 30 min after (post 30′) the onset of homocysteine treatment. (B) Representative photomicrograph demonstrating the extent of cell death. Examples of pyknotic DNA are indicated with arrows. (C) Quantitative analysis of percentage of neurons with pyknotic nuclei is represented as mean ± s.e.m. (n = 1500 cells/condition from 3 experiments). * Significant difference from control (p < 0.001); # significant difference from 18 hr homocysteine treatment (p < 0.05).
Homocysteine-induced activation of p38 MAPK temporally correlates with activation of ERK MAPK in neurons
We have previously demonstrated a role of ERK MAPK in homocysteine-NMDAR induced neuronal cell death (Poddar and Paul 2009). However, our current findings support a role for p38 MAPK in homocysteine-NMDAR induced neuronal cell death. Taken together these findings strongly suggest a central role of both ERK and p38 MAPK pathways in homocysteine induced neuronal cell death. To explore a possible correlation between the p38 and ERK MAPK activation, neurons were treated with L-homocysteine (50 μM) for varying time periods (0 min, 2.5 min, 2 hr, 4 hr, 6 hr and 8 hr). Figure 4A shows that ERK remained phosphorylated throughout the time course examined. However the increase in phosphorylation of ERK MAPK occurs in two phases. As previously reported a rapid initial increase occurs by 2.5 min (Poddar and Paul 2009). This is followed by a delayed larger increase by 4 hr that reaches maximum levels at 6 hr. Total ERK2 MAPK levels remained unaltered by this treatment. Both the initial rapid increase and the secondary delayed phase correlate with the temporal profile of phosphorylation of p38 MAPK.
FIGURE 4.

Delayed activation of ERK MAPK facilitates homocysteine-NMDAR induced neuronal cell death. Neuron cultures were treated with (A) 50 μM L-homocysteine (L-Hcy) for the specified time periods, (B, C) 50 μM L-Hcy and the following inhibitors were added 30 min after (post 30′) the onset of homocysteine treatment: (B) MK810 (5 μM) or APV (200 μM) and (C) EGTA (2 mM) and the incubation was continued for 4 hr. Protein extracts were analyzed by immunoblotting with an antibody against phosphorylated ERK (p-ERK, upper panel in A–C) and blots were re-probed with anti-ERK antibody (lower panels, A-C). The extent of phosphorylation of ERK MAPK was quantified using computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.001); # significant difference from (A) 2.5 min or (B, C) 4 hr homocysteine treatment (p < 0.01). (D) Neuronal cultures were either pre-incubated with MEK1/2 inhibitor, PD98059 (15 μM) before treatment with L-Hcy (50 μM) or the inhibitor was added 30 min after the onset of homocysteine treatment (post 30′) and the treatment was continued for 18 hr. Representative photomicrograph demonstrating the extent of cell death by Hoechst DNA staining. Quantitative analysis of the percentage of neurons with pyknotic nuclei is represented as mean ± s.e.m. (n = 1500 cells/condition from 3 experiments). * Significant difference from untreated control (p < 0.001); # significant difference from 18 hr homocysteine treatment (p < 0.01).
In our earlier study we demonstrated that the initial activation of ERK MAPK is NMDAR dependent (Poddar and Paul 2009). To determine whether the delayed activation of ERK MAPK is also NMDAR-dependent neuronal cultures were treated with L-homocysteine (50 μM, 4 hr) and NMDAR antagonists, MK801 (5 μM) or APV (200 μM) were applied 30 min after the onset of homocysteine treatment. Figure 4B shows that both MK801 and APV inhibited the phosphorylation of ERK MAPK that is normally observed at 4 hr. Addition of EGTA (2 mM) 30 min after the onset of homocysteine treatment (50 μM, 4 hr) also blocked the phosphorylation of ERK MAPK at 4 hr (Fig. 4C), suggesting a role of extracellular Ca2+ in homocysteine-NMDAR mediated delayed larger increase in ERK MAPK activity.
We next evaluated the role of the delayed activation of ERK MAPK in neuronal cell death using PD98059 (15 μM), a selective inhibitor of MEK1/2, the upstream kinase of ERK MAPK. Neurons were either pre-incubated with PD98059 or the inhibitor was added 30 min after onset of homocysteine treatment. Cell viability was assessed 18 hr later by Hoechst DNA staining. The representative photomicrograph and the corresponding mean data (Fig. 4D and E) shows that application of PD98059 either prior to or 30 min after the onset of homocysteine treatment led to significant reduction in homocysteine-induced neuronal cell death (17.3% ± 1.35 for control; 44.65% ± 1.6 for homocysteine; 23.34% ± 2.05 for pre-incubation with PD98059 and 25.29% ± 0.96 for delayed application of PD98059). Taken together these findings demonstrate that inhibiting the delayed larger increase in ERK MAPK phosphorylation is sufficient to reduce homocysteine-induced neuronal cell death.
Homocysteine-induced initial activation of p38 MAPK is dependent on ERK MAPK activity
To test the hypothesis that a possible crosstalk between ERK and p38 MAPK regulates homocysteine-induced neuronal cell death we evaluated the effects of pharmacological inhibitors of ERK and p38 MAPK on their phosphorylation. For these experiments neurons were treated with 50 μM L-homocysteine for 5 min in the absence or presence of selective inhibitors of MEK1/2 (PD98059, 15 μM; U0126, 10 μM) or p38 MAPK (SB203580, 5 μM; SB202190, 7.5 μM; SB239063, 10 μM). Inhibitor concentrations were based on earlier studies that have demonstrated that at these concentrations the pharmacological compounds can inhibit the respective pathways with no effect on the other pathway (Alessi et al. 1995; Favata et al. 1998; Davies et al. 2000; Legos et al. 2002). Immunoblot analysis used anti-phospho-p38 antibody for neurons treated with ERK MAPK inhibitors and anti-phospho-ERK antibodies for neurons treated with p38 MAPK inhibitors. Interestingly, pre-treatment with PD98059 or U0126 inhibited phosphorylation of p38 MAPK (Fig 5A) while pre-treatment with SB203580, SB202190 or SB239063 had no effect on phosphorylation of ERK MAPK (Fig. 5B), suggesting a role of ERK MAPK or its upstream kinase MEK1/2 in regulating p38 MAPK activity.
FIGURE 5.

Homocysteine-mediated initial p38 MAPK phosphorylation is downstream of ERK MAPK. (A, B) Neuron cultures were treated with L-homocysteine (L-Hcy, 50 μM, 5 min) in the absence or presence of (A) MEK1/2 inhibitors PD98059 (15 μM) or U0126 (20 μM) and (B) p38 MAPK inhibitors SB203580 (5 μM), SB202190 (7.5 μM) or SB239063 (10 μM). (C, D, E, F) Neuron cultures were treated with glutamate (50 μM, 5 min) in the absence or presence of (C, D) MK801 (5 μM), (E) PD98059 (15 μM) and (F) SB203580 (5 μM). Samples were analyzed with anti-p-p38 antibody (upper panel, A, C, E) or anti-p-ERK antibody (upper panel, B, D, F). Total p38 or ERK was analyzed ith either anti-p38 antibody (lower panel, A, C, E) or anti-ERK antibody (lower panel, B, D, F). Quantification of phosphorylated p38 and ERK MAPK was done by computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.001); # significant difference from 5 min homocysteine treatment (p < 0.001).
To determine whether the crosstalk between ERK and p38 MAPKs is unique to homocysteine-induced NMDAR signaling, in subsequent studies we examined the effect of glutamate, a well-known NMDAR agonist, on phosphorylation of ERK and p38 MAPKs. In initial experiments neurons were treated with glutamate (50 μM, 5 min) in the absence or presence of NMDAR inhibitor, MK801 (5 μM) and immunoblot analysis was performed to evaluate the phosphorylation of p38 and ERK MAPK. Results show that glutamate-induced p38 and ERK MAPK phosphorylation were both dependent on NMDAR stimulation (Fig. 5C and 5D). In subsequent experiments neurons were treated with glutamate (50 μM, 5 min) in the absence or presence of PD98059 or SB203580. Pre-treatment with PD98059 or SB203580 had no effect on the glutamate-induced phosphorylation of the p38 or ERK MAPK respectively (Fig. 5E and Fig. 5F), suggesting that the two pathways are independent of each other. Taken together the above findings suggest that crosstalk between ERK and p38 MAPKs is novel to homocysteine-induced NMDAR signaling pathway.
To directly test the role of ERK MAPK in the regulation of the p38 MAPK activity neurons were treated with L-homocysteine (50 μM, 5 min) in the presence of FR180204 (4 μM), a selective inhibitor of the ATP-dependent kinase activity of ERK (Ohori et al. 2005). The results show that FR180204 completely inhibited the phosphorylation of p38 MAPK (Fig. 6A), but had little effect on the phosphorylation of ERK (Fig. 6B). This suggests that homocysteine-induced initial phosphorylation of p38 MAPK is downstream of ERK MAPK activity.
FIGURE 6.

Homocysteine-induced initial p38 MAPK phosphorylation is dependent on ERK MAPK. (A, B) Neuron cultures were treated with 50 μM L-homocysteine (L-Hcy, 5 min) in the absence or presence of FR180204 (4 μM) and cell lysates were analyzed with (A) anti-phospho-p38 (upper panel) and anti-p38 (lower panel) antibodies or (B) anti-phospho-ERK (upper panel) and anti-ERK (lower panel) antibodies. Quantification of phosphorylated p38 and ERK was done by computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.001); # significant difference from 5 min homocysteine treatment (p < 0.001).
Homocysteine-induced delayed activation of p38 MAPK is dependent of ERK MAPK activity
For these experiments selective inhibitors of MEK1/2 (PD98059 or U0126), or p38 MAPK (SB203580, SB202190, and SB239063) were applied to neurons 30 min after the onset of L-homocysteine treatment (50 μM, 4 hr). Immunoblot analysis showed that the delayed phosphorylation of p38 MAPK at 4 hr was completely blocked by PD98059 or U0126 (Fig. 7A) whereas the phosphorylation of ERK MAPK at 4 hr remained unaltered in the presence of SB203580, SB202190 or SB239063 (Fig. 7B).
FIGURE 7.

Homocysteine-induced secondary p38 MAPK phosphorylation is downstream of ERK MAPK. (A, B) Neuron cultures were treated with 50 μM L-homocysteine (L-Hcy) for 4 hr. 30 min after (post 30′) the onset of homocysteine treatment (A) PD98059 (15 μM) or U0126 (20 μM) and (B) SB203580 (5 μM), SB202190 (7.5 μM) or SB239063 (10 μM) was added and the incubation was continued for the specified time period. Immunoblot analysis was performed using (A) anti-phospho-p38 (upper panel) and anti-p38 (lower panel) antibodies or (B) anti-phospho-ERK (upper panel) and anti-ERK (lower panel) antibodies. Quantification of phosphorylated p38 and ERK MAPK was done by computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.001); # significant difference from 4 hr homocysteine treatment (p < 0.001).
To evaluate a direct role of ERK MAPK in homocysteine induced delayed p38 MAPK activation the selective inhibitor of ERK MAPK activity, FR180204 was added to the neurons 30 min after onset of L-homocysteine treatment (50 μM, 4 hr). Figure 8A shows that inhibition of ERK MAPK activity blocked the delayed phosphorylation of p38 MAPK at 4 hr. FR180204 treatment did not alter the homocysteine-induced phosphorylation of ERK MAPK (Fig. 8B). To further confirm the role of ERK MAPK in phosphorylation and subsequent activation of p38 MAPK, we used lentiviral vectors to suppress ERK2 expression in neurons. In initial experiments, neuronal infection efficiency was established to be 58 ± 4%, using control lentiviral particles containing a green fluorescent protein (copGFP, coding insert copGFP-shRNA) (Fig. 8C). Neurons were then infected with ERK2-shRNA containing lentiviral particles, to suppress the expression of ERK2, followed by treatment with L-homocysteine (50 μM, 4 hr). Figure 8D shows that ERK2 expression was greatly reduced in these neurons (45.4 ± 2%) and was accompanied by significant reduction in homocysteine-induced activation of p38 MAPK (38.5 ± 2.3%). These findings confirm a direct role of ERK MAPK in homocysteine-induced regulation of p38 MAPK.
FIGURE 8.

Homocysteine-induced secondary p38 MAPK phosphorylation is dependent on ERK MAPK. (A, B) Neuron cultures were treated with 50 μM L-homocysteine (L-Hcy) for 4 hr in the absence or presence of FR180204 (4 μM) and samples were processed for immunoblot analysis using (A) anti-phospho-p38 (upper panel) and anti-p38 (lower panel) antibodies or (B) anti-phospho-ERK (upper panel) and anti-ERK (lower panel) antibodies. (C) Neuron cultures were transduced with lentiviral particles containing control COP-GFP insert (green). 4 days later the cells were counterstained with DAPI (blue) to test the efficiency of infection. (D) Neuron cultures transduced with or without lentiviral particles containing ERK2 shRNA or scrambled RNA were treated with 50 μM L-Hcy for 4 hr. Samples were processed for immunoblot analysis using anti-phospho-p38 (upper panel), anti-ERK (middle panel) and anti-p38 (lower panel) antibodies. (A, B, D) Quantification of phosphorylated p38 and ERK was done by computer-assisted densitometry and Image J analysis. Values are mean ± s.e.m. (n=3). * Significant difference from 0 min (p < 0.001); # significant difference from 4 hr homocysteine treatment (p < 0.001).
DISCUSSION
The current study provides the first evidence that homocysteine-NMDAR induced activation of p38 MAPK plays a crucial role in neuronal cell death. Activation of p38 MAPK shows a biphasic response, characterized by an initial rapid but transient induction followed by a delayed and more prolonged secondary increase. Although the earlier peak of p38 MAPK activation is part of homocysteine–NMDAR induced downstream signaling the later peak is primarily involved in mediating homocysteine-induced neuronal cell death. The time and duration of the secondary activation of p38 MAPK correlates with that of ERK MAPK activation, which also plays a role in facilitating homocysteine-induced neuronal cell death. Another key finding of the present study is that activation of p38 MAPK is downstream of and dependent on ERK MAPK activity establishing a novel interaction between the two MAPK pathways that facilitate homocysteine-induced neuronal cell death. These results suggest that homocysteine-NMDAR induced neuronal cell death involves a unique cascade of events that is different from downstream signaling pathways triggered by other NMDAR agonists (Fig. 9).
FIGURE 9.

Schematic representation of the crosstalk between ERK and p38 MAPKs in homocysteine-NMDAR induced neuronal cell death. (A) Homocysteine-NMDAR stimulation leads to sustained two-tier activation of ERK MAPK and biphasic activation of p38 MAPK where p38 MAPK activation is downstream and dependent on ERK MAPK. (B) Glutamate-NMDAR stimulation leads to transient activation of ERK and p38 MAPKs that are independent of each other.
ERK and p38 MAPKs are ubiquitously expressed signaling proteins that regulate neuronal functions as diverse as cell survival and cell death (Shaul and Seger 2007; Cuadrado and Nebreda 2010). They are serine/threonine kinases that are activated by dual phosphorylation of the regulatory tyrosine and threonine residues in the catalytic domain (Seth et al. 1992; Raingeaud et al. 1995). A three-tiered kinase cascade regulates the activation of these kinases (Ono and Han 2000). The first member of this cascade is MAP kinase kinase kinase (MAPKK or MEKK family), which phosphorylates and activates MAP kinase kinase (MEK 1/2 for ERK; MEK 3/6 for p38) that in turn phosphorylates and activates ERK and p38 MAPKs. Generally, transient activation of ERK MAPK by neurotrophic factors and neurotransmitters plays an important role in neuronal survival and long-term potentiation (Boulton et al. 1991; Xia et al. 1995; Segal and Greenberg 1996). In contrast, p38 MAPK is activated in response to a multitude of stress stimuli and inflammatory cytokines and can contribute to neuronal cell death (Raingeaud et al. 1995; Zhu et al. 2000; Daniels et al. 2001; Zhu et al. 2003; Choi et al. 2004; Waxman and Lynch 2005; Poddar et al. 2010). Both ERK and p38 MAPKs are activated following NMDAR stimulation by multiple agonists (Sato et al. 2001; Paul et al. 2003; Rakhit et al. 2005; Waxman and Lynch 2005; Poddar and Paul 2009; Poddar et al. 2010). Glutamate is one such agonist that leads to transient activation of both ERK and p38 MAPKs, where activation of ERK MAPK promotes neuronal cell survival and p38 MAPK promotes injury (Kim et al. 2005; Waxman and Lynch 2005; Balazs 2006; Poddar et al. 2010). In contrast, the present study demonstrates that homocysteine-NMDAR mediated sustained activation of ERK as well as p38 MAPKs facilitate neuronal cell death.
The opposing effects of ERK MAPK on neuronal survival and death following exposure to glutamate and homocysteine could be attributed to the differential activation of the NMDAR subunits. Glutamate leads to sequential activation of both the stimulatory and inhibitory (NR2B-NMDAR) pathways to limit the duration of ERK activation (Chandler et al. 2001; Paul et al. 2003; Kim et al. 2005; Poddar and Paul 2009). In contrast, homocysteine activates only the stimulatory pathway (NR2A-NMDAR) resulting in prolonged activation of ERK MAPK (Poddar and Paul 2009). The current study also emphasizes the difference between the two NMDAR agonist, glutamate and homocysteine by demonstrating that glutamate-NMDAR mediated activation of ERK and p38 MAPK pathways are parallel and independent of each other, while homocysteine-NMDAR mediated activation of p38 MAPK is downstream of and dependent on ERK MAPK activity. Taken together the study suggests that the view that p38 promotes apoptosis whereas ERK opposes apoptosis in neurons is not generalizable, and detailed analysis of these two pathways could be useful in other models of neurotoxicity.
The pharmacological inhibition of p38 MAPK activity using PD98059 and U0126 suggests an interaction between the ERK and p38 MAPK pathways but does not clarify whether p38 MAPK is activated directly by ERK MAPK or by its upstream kinase MEK1/2. However the ability of FR180204 to block p38 MAPK activity confirms a direct role of ERK MAPK in regulating p38 MAPK activity. This interpretation is based on earlier findings demonstrating that FR180204 is a potent ATP-competitive inhibitor of ERK1/2 activity that does not interfere with the MEK-dependent phosphorylation of ERK1/2 (Ohori et al. 2005). This is also consistent with our findings that suppression of ERK2 expression using shRNA approach significantly reduces homocysteine-induced phosphorylation of p38 MAPK. The direct regulatory role of ERK MAPK in p38 MAPK activation, as demonstrated in the present study, reveals a previously unidentified cellular mechanism for neuronal cell death. There are some instances where a crosstalk between two MAPKs has been suggested in non-neuronal cells. In contrast to our findings, these studies show a reciprocal phosphorylation of MAPKs where activation of one MAPK pathway leads to inactivation of the other MAPKs (Houliston et al. 2001; Xiao et al. 2002; Wang et al. 2006; Junttila et al. 2008). Additionally, these studies have emphasized the opposing effects of ERK and p38 MAPK on cell survival and cell death.
The precise mechanism by which ERK MAPK regulates the activity of p38 MAPK is yet to be determined. There is a growing body of evidence indicating that activation of ERK MAPK following stimulation of NR2A-NMDARs induces trafficking of AMPA receptor subunits to the surface. While inhibition of ERK MAPK following stimulation of NR2B-NMDARs inhibits AMPA receptor surface insertion (Ehlers 2000; Zhu et al. 2002; Kim et al. 2005; Keifer et al. 2007). Additional studies have also described a role of AMPA receptors in up-regulation of p38 MAPK activity (Krapivinsky et al. 2004). This raises the possibility that ERK-mediated trafficking of AMPA receptors may play a role in homocysteine-induced p38 MAPK activation and is an important topic for future studies.
Our study shows a sustained but two-tier pattern of ERK MAPK activation by homocysteine. A biphasic activation of p38 MAPK is also observed, where the initial phase is transient while the secondary phase is more prolonged. It is difficult to explain why the initial ERK MAPK activation remains sustained while p38 MAPK activation is transient. A probable explanation is that a phosphatase selective for p38 MAPK is activated as the cell’s protective response to inhibit the pro-apoptotic p38 MAPK pathway. Such selective activation of dual specificity phosphatases that inhibits either ERK or p38 MAPK resulting in preferential activation of the other MAPK has been reported in earlier studies (Muda et al. 1996; Xiao et al. 2002; Wang et al. 2006; Junttila et al. 2008). Regardless of this differential pattern of the initial ERK and p38 MAPK activation, it is the delayed larger increase in ERK MAPK activity and the secondary activation of p38 MAPK that is predominantly responsible for homocysteine-dependent neuronal cell death. Concerning this delayed large increase in ERK MAPK activity, evidences from earlier studies show that prolonged homocysteine exposure leads to a gradual increase in intracellular Ca2+, mitochondrial dysfunction and reactive oxygen species (ROS) generation (Kruman et al. 2000; Zieminska et al. 2006; Ganapathy et al. 2011b; Ganapathy et al. 2011a). ROS mediated activation of ERK MAPK has also been shown to play a detrimental role in oxidative neuronal injury (Stanciu et al. 2000; Stanciu and DeFranco 2002; Luo et al. 2007; Chen et al. 2009; Tuerxun et al. 2010). Thus it appears that homocysteine-NMDAR mediated intracellular Ca2+ accumulation over time may lead to mitochondrial dysfunction and subsequent ROS generation. This in turn could lead to the delayed large increase in ERK MAPK activity.
In conclusion the present study provides evidence for a novel signaling mechanism of neuronal injury involving a crosstalk between ERK and p38 MAPKs in response to elevated levels of homocysteine. Further evaluation of this molecular pathway is necessary to establish a role of hyperhomocysteinemia in the progression of neurological disorders.
Supplementary Material
Acknowledgments
We thank CW Shuttleworth for his helpful comments in preparing the manuscript. We also thank Alexandria Chen and Richard Fortenberry for technical assistance. This work was supported by the National Institutes of Health grants R21 NS065343 (Poddar, R); RO1 NS059962 (Paul, S); P20 RR15636 (Liu, KJ); and New Mexico Trust Fund, UNM HSC RAC (Poddar, R).
Abbreviations
- NMDA
N-methyl-D-aspartate
- ERK MAPK
Extracellular-signal regulated kinase
Footnotes
The authors have no conflict of interest.
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. The Journal of biological chemistry. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Balazs R. Trophic effect of glutamate. Current topics in medicinal chemistry. 2006;6:961–968. doi: 10.2174/156802606777323700. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl D-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. The Journal of biological chemistry. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Yin J, Luo Y, Huang S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. The international journal of biochemistry & cell biology. 2009;41:1284–1295. doi: 10.1016/j.biocel.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Choi WS, Eom DS, Han BS, Kim WK, Han BH, Choi EJ, Oh TH, Markelonis GJ, Cho JW, Oh YJ. Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. The Journal of biological chemistry. 2004;279:20451–20460. doi: 10.1074/jbc.M311164200. [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. The Biochemical journal. 1997;326 (Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. The Biochemical journal. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Daniels WM, Hendricks J, Salie R, Taljaard JJ. The role of the MAP-kinase superfamily in beta-amyloid toxicity. Metabolic brain disease. 2001;16:175–185. doi: 10.1023/a:1012541011123. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. The Biochemical journal. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG, Kruman, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson’s disease. Journal of neurochemistry. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. The Journal of biological chemistry. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Ganapathy PS, White RE, Ha Y, Bozard BR, McNeil PL, Caldwell RW, Kumar S, Black SM, Smith SB. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Investigative ophthalmology & visual science. 2011a;52:5515–5524. doi: 10.1167/iovs.10-6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy PS, Perry RL, Tawfik A, Smith RM, Perry E, Roon P, Bozard BR, Ha Y, Smith SB. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Investigative ophthalmology & visual science. 2011b;52:5551–5558. doi: 10.1167/iovs.11-7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature neuroscience. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Current topics in medicinal chemistry. 2006;6:787–799. doi: 10.2174/156802606777057553. [DOI] [PubMed] [Google Scholar]
- Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002;70:694–702. doi: 10.1002/jnr.10416. [DOI] [PubMed] [Google Scholar]
- Houliston RA, Pearson JD, Wheeler-Jones CP. Agonist-specific cross talk between ERKs and p38(mapk) regulates PGI(2) synthesis in endothelium. Am J Physiol Cell Physiol. 2001;281:C1266–1276. doi: 10.1152/ajpcell.2001.281.4.C1266. [DOI] [PubMed] [Google Scholar]
- Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A. Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res. 2003;5:237–243. doi: 10.1007/BF03033381. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. Faseb J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Keifer J, Zheng ZQ, Zhu D. MAPK signaling pathways mediate AMPA receptor trafficking in an in vitro model of classical conditioning. Journal of neurophysiology. 2007;97:2067–2074. doi: 10.1152/jn.01154.2006. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563–574. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Kruman, Culmsee C, Chan SL, Kruman Y, Guo Z, Penix L, Mattson MP. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legos JJ, McLaughlin B, Skaper SD, Strijbos PJ, Parsons AA, Aizenman E, Herin GA, Barone FC, Erhardt JA. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. European journal of pharmacology. 2002;447:37–42. doi: 10.1016/s0014-2999(02)01890-3. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nature neuroscience. 2002;5:833–834. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Kintner DB, Shull GE, Sun D. ERK1/2-p90RSK-mediated phosphorylation of Na+/H+ exchanger isoform 1. A role in ischemic neuronal death. The Journal of biological chemistry. 2007;282:28274–28284. doi: 10.1074/jbc.M702373200. [DOI] [PubMed] [Google Scholar]
- Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. The European journal of neuroscience. 2004;19:1207–1216. doi: 10.1111/j.1460-9568.2004.03223.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends in neurosciences. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Miller JW. Homocysteine and Alzheimer’s disease. Nutr Rev. 1999;57:126–129. doi: 10.1111/j.1753-4887.1999.tb06936.x. [DOI] [PubMed] [Google Scholar]
- Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. The Journal of biological chemistry. 1996;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M, Miyake H, Fujii T. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochemical and biophysical research communications. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- Ono K, Han J. The p38 signal transduction pathway: activation and function. Cellular signalling. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nature neuroscience. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Poddar R, Paul S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. Journal of neurochemistry. 2009;110:1095–1106. doi: 10.1111/j.1471-4159.2009.06207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Deb I, Mukherjee S, Paul S. NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. Journal of neurochemistry. 2010 doi: 10.1111/j.1471-4159.2010.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poddar R, Sivasubramanian N, DiBello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. 2001;103:2717–2723. doi: 10.1161/01.cir.103.22.2717. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. The Journal of biological chemistry. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rakhit S, Clark CJ, O’Shaughnessy CT, Morris BJ. N-methyl-D-aspartate and brain-derived neurotrophic factor induce distinct profiles of extracellular signal-regulated kinase, mitogen- and stress-activated kinase, and ribosomal s6 kinase phosphorylation in cortical neurons. Molecular pharmacology. 2005;67:1158–1165. doi: 10.1124/mol.104.005447. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ginty DD. What a privilege to reside at the synapse: NMDA receptor signaling to CREB. Nature neuroscience. 2002;5:389–390. doi: 10.1038/nn0502-389. [DOI] [PubMed] [Google Scholar]
- Sacco RL, Roberts JK, Jacobs BS. Homocysteine as a risk factor for ischemic stroke: an epidemiological story in evolution. Neuroepidemiology. 1998;17:167–173. doi: 10.1159/000026169. [DOI] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S. NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci. 2001;21:3797–3805. doi: 10.1523/JNEUROSCI.21-11-03797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Seth A, Gonzalez FA, Gupta S, Raden DL, Davis RJ. Signal transduction within the nucleus by mitogen-activated protein kinase. The Journal of biological chemistry. 1992;267:24796–24804. [PubMed] [Google Scholar]
- Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochimica et biophysica acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. The Journal of biological chemistry. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. The Journal of biological chemistry. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science (New York, NY. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Tuerxun T, Numakawa T, Adachi N, Kumamaru E, Kitazawa H, Kudo M, Kunugi H. SA4503, a sigma-1 receptor agonist, prevents cultured cortical neurons from oxidative stress-induced cell death via suppression of MAPK pathway activation and glutamate receptor expression. Neuroscience letters. 2010;469:303–308. doi: 10.1016/j.neulet.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang H, Tachado SD, Capo-Aponte JE, Bildin VN, Koziel H, Reinach PS. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Investigative ophthalmology & visual science. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science (New York, NY. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. The Journal of biological chemistry. 2002;277:14884–14893. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]
- Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-D-aspartate receptor-induced activation of p21ras. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Lee HG, Chao M, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA. Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxidants & redox signaling. 2003;5:571–576. doi: 10.1089/152308603770310220. [DOI] [PubMed] [Google Scholar]
- Zieminska E, Matyja E, Kozlowska H, Stafiej A, Lazarewicz JW. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: role of calcium and mitochondrial alterations. Neurochemistry international. 2006;48:491–497. doi: 10.1016/j.neuint.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Zoccolella S, Martino D, Defazio G, Lamberti P, Livrea P. Hyperhomocysteinemia in movement disorders: Current evidence and hypotheses. Curr Vasc Pharmacol. 2006;4:237–243. doi: 10.2174/157016106777698414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.