

Abstract
Due to the emergence of drug-resistant strains and the cumulative toxicities associated with current therapies, demand remains for new inhibitors of HIV-1 replication. The HIV-1 matrix (MA) protein is an essential viral component with established roles in the assembly of the virus. Using virtual and surface plasmon resonance (SPR)-based screening, we describe the identification of the first small molecule to bind to the HIV-1 MA protein and to possess broad range anti-HIV properties.
Despite the successes of highly active antiretroviral therapy, current therapies for HIV-1 infection are limited by the development of multidrug-resistant virus and by significant cumulative drug toxicities. The development of new classes of antiretroviral agents with novel modes of action is therefore highly desirable and is a driving force for the pursuit of small-molecule inhibitors of the nonenzymatic viral proteins.
The HIV-1 replication cycle, similar to other retroviruses, can be divided into early (preintegration) and late (postintegration) stages. The Gag polyprotein is a structural protein that plays a central role in the late stages of viral replication. The Gag polyprotein consists of several domains, three of which are functionally conserved among retroviruses: the nucleocapsid (NC) domain; the capsid (CA) domain; and the myristoylated matrix (MA) domain. Despite the pivotal role played by Gag in the HIV-1 replication cycle, there are currently no approved drugs targeting either the full-length polyprotein or its component proteins.
The HIV-1 MA protein, encoded as the N-terminal portion of Gag, is critically involved in the late, assembly stages in the life cycle of HIV-1. MA is a 132-amino-acid structural protein that is posttranslationally myristoylated at the N-terminus. The three-dimensional structure of HIV-1 MA has been determined by nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography.1–3 MA is a structural molecule, partially globular, and composed of five α-helices. The helices α1, α2, and α3 are organized around the central and buried α4,1 whereasα5 is projected from the packed helical bundle, making the C-terminal region of the protein distinct from its globular N-terminus. In solution, myristoylated MA (myr-MA) is in dynamic equilibrium between monomeric and trimeric species, whereas nonmyristoylated MA is exclusively monomeric.2, 4, 5 Current data, based on crystallographic experiments,1 electron microscopic observations,6, 7 and docking calculations,8 point to a trimeric organization of MA monomers in the mature virion. Such organization appears to be also used by other HIV-1 structural proteins,9 in an organization defined by the Gag precursor. A recent study has indicated, however, that although the myr-MA oligomerizes in solution as a trimer and crystallizes in three dimensions as a trimer unit, myristoylated MA assembles on phosphatidylserine-cholesterol membranes as hexamer rings.10 This finding has been echoed in another study looking at oligomerization of MA on phosphatidylcholine-cholesterol-phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2], in which the authors found that MA organizes as hexamers, each one composed of a trimer of MA.11
Two main functions of MA in the assembly of HIV-1 are well established: membrane binding and envelope incorporation. Membrane binding of HIV-1 Gag is mediated by two signals in MA: the N-terminal myristic acid and the conserved basic region between amino acids 17 and 31.12–14 The myristate moiety is considered to be regulated by a mechanism termed a myristoyl switch, whereby the N-terminal myristate is sequestered in the MA globular domain, but a structural change exposes the myristate and enhances Gag membrane binding.5, 15–22 The MA basic domain is involved in specific localization of Gag to the plasma membrane, with mutations in this domain shifting the Gag localization from plasma membrane to intracellular vesicles in HeLa and T cells.23–27 The basic domain of MA has been demonstrated to interact with PI(4,5)P2, a minor phospholipid that is concentrated primarily on the cytoplasmic leaflet of the plasma membrane.20, 28–30 PI(4,5)P2 binding to HIV-1 MA is thought to serve two functions: inducing the conformational change that triggers myristate exposure and acting as a site-specific membrane anchor, allowing the targeting of Gag to the plasma membrane.
The most effective targets for the development of small-molecule inhibitors are likely to be functionally critical pockets within the viral proteins that are highly conserved between viral isolates. Therefore, to assess the suitability of the PI(4,5)P2-binding site as a target for inhibitor design, we assessed the conservation of it’s component residues. We analyzed the amino acid sequences of the matrix domains from a subset of the available HIV-1 Pr55Gag proteins from the National Center for Biotechnology Information Entrez Protein database (~20,000 sequences from a total of 50,133) using BLAST (Basic Local Alignment Search Tool),31 assessed the level of the sequence conservation at each amino acid, and then highlighted this conservation on a surface representation of the MA protein (Fig. 1). As can been seen from Figure 1, the amino acids present at the surface of the MA protein vary widely, ranging from less than 20% to complete conservation. However, the structural recess that runs across the globular head of MA and encompasses the PI(4,5)P2-binding site shows remarkable conservation (Fig. 1). Such a high level of conservation may be required for regions of viral proteins that interact with cellular components, and highlights the potential of the PI(4,5)P2-binding site as an attractive antiviral target.
Figure 1. Conservation of surface residues of the HIV-1 MA protein.
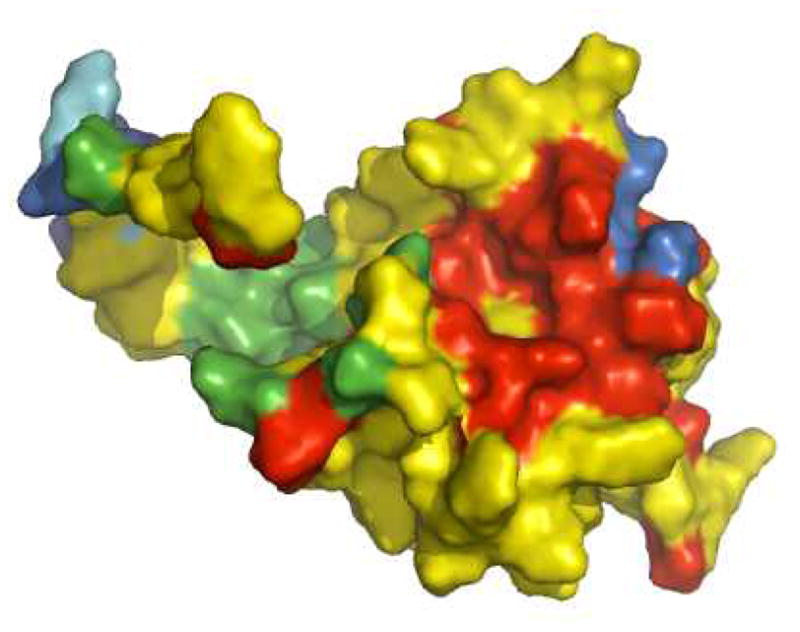
Color scheme: red = >99% conservative residue; yellow = >80%–99%; green = >50%–80%; cyan = >20%–50%; blue = 0%–20%. Residues surrounding a structural recess that serves as a binding site for PI(4,5)P2 are highly conserved as indicated by the red coloring.
We therefore undertook a structure-guided search for small, drug-like molecules that inhibit viral replication by binding to this highly conserved area on the HIV-1 MA protein. To identify potential hits, we employed a virtual screening protocol. This protocol, performed using QUANTUM software,32–34 included fast molecular docking for the generation of binding poses and molecular dynamics simulations to rank the ligand poses according to their binding affinities. The simulations were implemented using a proprietary version of the MM/Poisson Boltzman–Surface Area (MM/PB–SA) force field.35 We used the Enamine Screening Collection, which comprised approximately 1,168,625 exclusive drug-like compounds filtered according to Vernalis’ guidelines (Cambridge, UK) at the time of screening.36 To limit the necessary calculations to a reasonable number, we performed extensive clustering of the ligand library, while keeping as much of the full chemical diversity of the available library as possible (see Materials and Methods). A compound was regarded as a hit if its predicted affinity (KD) was in the range 1 to 10 μM and it displayed favorable, drug-like chemical characteristics. Docking of compounds from the Enamine Screening Collection to a static MA model yielded no compounds with a predicted KD of less than 1 μM and 29 compounds with the KD ranging from 1 to 10 μM. These 29 best docked conformers were then taken to the molecular dynamics study, where the calculated protein-ligand binding energies were refined with regard to protein and ligand flexibility. After these compounds were refined, we identified four with predicted KD of less than 1 μM and 15 more compounds with KD ranging up to 10 μM. These 19 compounds were purchased for use in downstream biophysical and biological assays.
First, we tested whether the identified compounds could interact with MA using surface plasmon resonance (SPR). The HIV-1 MA protein poses a challenge for conventional SPR assays, as the PI(4,5)P2-binding site and surrounding regions on the MA protein are enriched in lysines and arginines, residues commonly used to covalently link proteins to the sensor surface using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) chemistry. 37, 38 Therefore, in order to ensure that the PIP(4,5)P2-binding site remained intact upon attachment, we developed an immobilization method based upon the properties of the HaloTag (Promega, Madison, WI). The HaloTag is a mutant haloalkane dehalogenase designed to covalently bind to synthetic ligands (HaloTag ligands).39 We therefore created a C-terminally HaloTagged version of the HIV-1 MA protein. This construct also contained a C-terminal His-tag to allow purification via immobilized metal affinity chromatography. A HaloTag ligand, containing a terminal primary amine, was attached to the sensor surface using standard amine chemistry. The MA-HALO-H6 protein was then covalently attached to the ligand by injection over this surface. A reference surface was created in a similar manner using a H6-SUMO-HALO protein. The 19 compounds were then screened at one concentration (50 μM) over these surfaces to identify specific binders of the MA protein. Unfortunately, the majority of the compounds nonspecifically interacted with the chloroalkane HaloTag ligand. One exception, compound 14 (N2-(Phenoxyacetyl)-N-[4-(1 piperidinylcarbonyl)benzyl]glycinamide), however, was identified as being able to interact specifically with HIV-1 MA using this novel immobilization system. This analysis, shown in Figure 2, demonstrated that compound 14 interacts with HIV-1 MA with an equilibrium dissociation constant of 171 (± 50) μM.
Figure 2. Sensorgrams depicting the interaction of the compound 14 with Halotag-immobilized HIV-1LAI MA.

Compound 14 at concentrations in the range 6.25–100 μM are shown. Colored lines indicate experimental data, whereas black lines indicate fitting to a simple 1:1 binding model. Kinetic parameters for compound 14–HIV-1LAI MA interaction: ka = 2.54 (±0.8) × 103 M−1s−1; kd = 0.4 (± 0.07) s−1; KD = 171 (±50) μM.
We next sought to investigate the binding site of compound 14 by performing direct binding studies with MA mutants that were created based upon the docking model (Figure 3A). Residues Leu21, Lys27, and Asn80, were individually mutated to alanine, and the effect of the mutagenesis on the binding of compound 14 at a single concentration (100 μM) relative to wild-type MA was assessed using SPR. For comparison, and to take into account minor differences in the ligand density of the mutant surfaces, responses were normalized to the theoretical Rmax (maximum analyte binding capacity of the surface in RU), assuming a 1:1 interaction. As shown in Figure 3B, mutation of residues Leu21, Lys27 and Asn80 drastically reduced the binding of compound 14 compared with wild-type MA. This clearly demonstrates that residues within the PI(4,5)P2-binding site of HIV-1 MA are required for interaction with compound 14.
Figure 3.
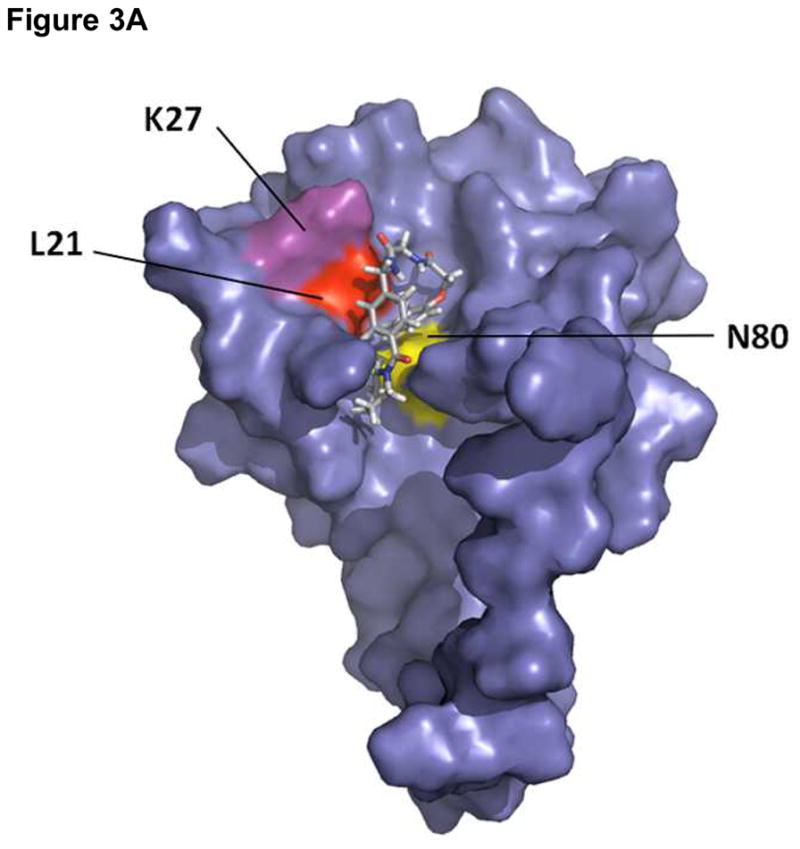

(A) Surface rendering of HIV-1 MA showing the position of residues chosen for mutation to alanine relative to the predicted binding site of compound 14 Compound 14 is shown as a stick representation (B) Sensorgrams illustrating the effect of mutation of residues in the proposed compound 14 binding site of HIV-1 on compound binding. The interaction of compound 14 at a concentration of 100 μM with wild-type and mutant versions of the MA protein was assessed using SPR. To allow comparison, responses were normalized to the theoretical Rmax, assuming a 1:1 interaction
Having identified compound 14 as a small molecule that specifically binds to the HIV-1 MA protein, and in a region overlapping the PI(4,5)P2-binding site, we next investigated whether it had the capacity to inhibit HIV-1 replication using a standardized PBMC-based anti-HIV-1 assay. As a key issue in the development of novel HIV drugs is their ability to inhibit the replication of genetically diverse isolates, especially isolates from the most globally prevalent subtypes A, B, and C, we performed the assay with a panel of HIV-1 clinical isolates and laboratory strains from different geographic locations. This panel of isolates included HIV-1 group M subtypes A, B, C, D, E, F, and G, as well as HIV-1 group O (Table 1). The panel included CCR5-tropic (R5), CXCR4-tropic (X4), and dual-tropic (R5X4) viruses. The toxicity of compound 14 was assessed in parallel. Compound 14 inhibited the replication of all the viruses tested, with half-maximal inhibitory concentrations (IC50) in the range 16 – 31 μM; a half-maximal toxicity concentration (TC50) was not reached within the concentration range evaluated (TC50 >100 μM). Moreover, compound 14 inhibited the multiply drug-resistant strain HIV-1MDR769,41 indicating a different mechanism of action from the inhibitors currently in use in the clinic. Interestingly, the effective concentration of compound 14 in the anti-HIV assays is lower than the equilibrium dissociation constant that we derived using SPR. A similar situation has been observed with the Gag-directed small molecule CAP-1, which displays low affinity for the HIV-1 capsid protein (KD = 0.8 mM), but whose half maximal inhibitory concentration (IC50) is approximately 60 μM.42
Table 1.
Therapeutic spectrum of compound 1 against HIV-1 Subtypes.
| HIV-1 isolate | Compound | IC50 (μM) | Antiviral index† (TC50/IC50) |
|---|---|---|---|
| 92UG031 Subtype A |
MTI535 | 17.8 (±2.6) | > 5.6 |
| 89BZ167 Subtype B |
MTI535 | 17.7 (±2.9) | > 5.7 |
| 92BR030 Subtype B |
MTI535 | 20.3 (±2.9) | > 4.9 |
| 92BR025 Subtype C |
MTI535 | 16.3 (±4.3) | > 6.1 |
| 93IN101 Subtype C |
MTI535 | 16.3 (±4.2) | > 6.1 |
| 92UG024 Subtype D |
MTI535 | 16.0 (±7.8) | > 6.25 |
| CMU08 Subtype E |
MTI535 | 17.7 (±4.8) | > 5.7 |
| 93BR020 Subtype F |
MTI535 | 17.3 (±4.8) | > 5.8 |
| G3 Subtype G |
MTI535 | 18.9 (±2.9) | > 5.3 |
| BaL Subtype B |
MTI535 | 27.8 (± 5.0) | > 3.6 |
| NL4-3 Subtype B |
MTI535 | 30.8 (± 0.9) | > 3.3 |
| MDR769 Subtype B |
MTI535 | 18.8 (± 3.3) | > 5.3 |
TC50 values for all compounds were determined to be >100 μM in this study. Numbers in parentheses represent one standard deviation derived from three replicate assays.
We next tested the specificity of compound 14 for HIV-1 using cellular assays and a panel of viruses from different families. Again, compound toxicity was assessed in parallel. Compound 14 was evaluated against this panel of viruses up to a high-test concentration of 100 μM and displayed no inhibitory effects on the replication of herpes simplex type 1, Dengue serotypes 1–4, influenza H1N1, respiratory syncytial virus, yellow fever, Japanese encephalitis, Vaccinia, or Chikungunya viruses. Therefore, compound 14 appears to be specific for HIV-1 (Supplemental Table 1).
In summary, we have identified a novel compound that binds to the conserved PI(4,5)P2-binding site on HIV-1 MA and exhibits broad-spectrum anti-HIV activity with IC50 values in the range of 16 – 31 μM for all isolates tested (Table 1). The broad-spectrum activity of this compound is particularly exciting, highlighting the HIV-1 MA protein as a new viral target with significant therapeutic potential. Although the dissociation constant (KD) of compound 14 is not currently within an acceptable range for clinical use, the successful improvement of the antiviral compound CAP-1 to higher potency suggests that compound 14 could also serve as a good starting point for the development of derivatives with increased affinity and efficacy. Moreover, mechanism of action studies of compound 14 should shed light upon the roles of the HIV-1 MA in the viral replication cycle.
Methods
Virtual Screening
The structures of ligands for virtual screening were taken from the Enamine Screening Collection library. The Enamine library was clustered using the Jarvis–Patrick algorithm43–45 implemented in QUANTUM (Quantum Pharmaceuticals, Moscow, Russia). The measure of dissimilarity (“distance”) between the molecules was determined by Tanimoto similarity calculated with Daylight fingerprints of the molecules (Daylight Chemical Information Systems, Inc.; Aliso Viejo, CA). The clustering parameters were chosen such that a reasonable number of clusters (~72k) was obtained. The compounds representing the cluster centroids were taken for subsequent screening. As a result, the library of 1M compounds was reduced to a library of roughly 72k cluster representatives of appropriate molecular weight. The compounds were prepared for docking by extraction from the Enamine-supplied sdf files and processing through the QUANTUM structure recovery and typization software components in batch mode.
The 2GOL (X-ray)46 and 2H3Z (NMR)47 HIV-1 MA structures from the RCSB Protein Data Bank were used for molecular modeling. The complex 2GOL was selected on the basis of its high resolution (2.2 Å) and 2H3Z was chosen because it contains a bound ligand (di-C4-phosphatidylinositol-(4,5)-bisphosphate [di-C4-PI(4,5)P2]). Prior to molecular modeling we removed the capsid protein p24 (CA) from the 2GOL structure and di-C4-PI(4,5)P2 from the 2H3Z structure. The docking area was then restricted by a box of 20 Å × 20 Å × 20 Å on the 2GOL structure and 19 Å × 20 Å × 21 Å on the 2H3Z structure. This region encompasses the di-C4-PI(4,5)P2 binding pocket of MA.48
The virtual screening procedure included two stages: (1) docking to a static protein model and (2) refinement using a dynamic protein model. These two procedures were performed using QUANTUM software utilities. Docking to a static protein model included identification of the ligand position in the binding pocket with the minimal binding energy, and its estimation. In the second stage, the binding energies for hits were refined with regard to the protein flexibility using molecular dynamics. The refinement procedure employed is a complete free energy perturbation molecular dynamics run for the whole protein-ligand complex in aqueous environment. Thus, it considers both protein and ligand flexibility. In addition, the calculation of physical-chemical properties of small molecules was performed using Q-Mol ver. 1.0.6 (Quantum Pharmaceuticals). A compound was regarded as a hit if the predicted affinity (KD) was in the range 1 to 10 μM and it displayed favorable, drug-like chemical characteristics.
Chemicals
All chemicals were purchased from Enamine Ltd. and were 95% pure or greater. Compound 14 was resynthesized by Enamine Ltd. using the following route:
Analytical details are provided in supplemental information.
Recombinant protein production and purification for surface plasmon resonance
A codon-optimized gene that codes for the HIV-1LAI MA protein fused via a small linker to a C-terminally histidine-tagged HaloTag protein (MA-HALO-H6) was synthesized and inserted into pUC57 vector by GenScript Corp. (Piscataway, NJ). This synthetic gene was flanked by NdeI and BamH1 restriction sites to facilitate subcloning in to the Escherichia coli expression vector pET11a (Novagen, EMD Millipore Corporation, Darmstadt, Germany). To provide a control protein for the surface plasmon resonance analysis, the HaloTag region of the MA-HALO-H6 gene was amplified from using primers designed to facilitate ligation-independent cloning into the vector pETHSUL.49 This vector is designed for the insertion of genes of interest in frame with an N-terminal small ubiquitin-related modifier (SUMO) tag.49 The recombinant pETHSUL plasmid was verified for the presence of HaloTag insert by restriction digestion and sequence analysis (Genewiz, Inc., South Plainfield, NJ). The resultant vector codes for a N-terminally histidine tagged SUMO-HaloTag fusion protein (H6-SUMO-HALO). The purification of both MA-HALO-H6 and H6SUMO-HALO was achieved via immobilized metal affinity chromatography using a TALON cobalt resin affinity column (ClonTech). The E. coli strain BL21 (DE3) Codon+-RIL (Stratagene, La Jolla, CA) was used for expression. Two milliliters of Luria-Bertani broth, containing 100 μg mL−1 ampicillin and 50 μg mL−1 chloramphenicol, were inoculated with a single transformed colony and allowed to grow at 37°C for 9 h. A total of 100 μL of the preculture was used to inoculate 100 mL of the autoinducing media ZYP-5052 50 containing 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol. The culture was grown at 30°C for 16 h. Cells were harvested by centrifugation at 1076 × g for 20 min at 4°C and the pellet was suspended in 30 mL PBS (Roche, Nutley, NJ) containing 2.5 mM imidazole. Cells were lysed by sonication and the supernatant clarified by centrifugation at 11,952 × g (SS-34, Sorvall RC 5C Plus) for 20 min at 4°C. The supernatant was removed and applied to a TALON cobalt resin affinity column (ClonTech), previously equilibrated with PBS (Roche). Loosely bound proteins were removed via seven-column volumes of PBS containing 7.5 mM imidazole. Tightly associated proteins were eluted in three-column volumes of PBS containing 250 mM imidazole. The eluates were pooled and immediately used for SPR analysis. Individual alanine mutations were introduced in to the wild-type MA-HALO-H6 expression vector by site-directed mutagenesis. Mutant MA-HALO-H6 proteins were purified as described above.
Surface plasmon resonance direct binding assays
Interaction analyses were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules CA). ProteOn GLH sensor chips were preconditioned with two short pulses each (10 s) of 50 mM NaOH, 100 mM HCl, and 0.5% SDS. Then the system was equilibrated with PBS-T buffer (20 mM Na-phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensorchip was activated with a mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, 1 mM HaloTag Amine (O4) ligand (Promega, Madison, WI) in 200 mM Tris buffer pH 8.0, was injected across the ligand flow strip for 5 min at a flow rate of 30 μl min−1. Excess active ester groups on the sensor surface were capped by a 5-min injection of 1 M ethanolamine HCl (pH 8.5). HIV-1LAI MA-HALO-H6, purified as described above, was then injected across the HaloTag ligand surface at a flow rate of 30 μl min−1 for 14 min, resulting in the immobilization of immobilized approximately 8,000 response units (RU). A reference surface was similarly created by immobilizing a H6-SUMO-HALO protein to the same density. In small-molecule assays, the buffering system used is particularly important owing to the low responses obtained in binding assays. Among the most commonly used buffers, PBS was found to be the most suitable for the MA small-molecule binding assays, as with a Tris-based buffer, variable and drifting baselines were observed, complicating evaluation of the data. A stock solution of the compound 14 was prepared by dissolving in 100% DMSO to a final concentration of ~10 mM. To prepare the sample for analysis, 10 μL of the compound stock solution was added to 20 μL of 100% DMSO and this was made to a final volume of 1 mL by addition of sample preparation buffer (PBS, pH 7.4). Preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Two-fold dilutions were then prepared in running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected across the MA-HALO-H6 and H6-SUMO-HALO surfaces a flow rate of 100 μL min−1, for a 1-min association phase, followed by a 5-min dissociation phase using the “one shot kinetics” capability of the Proteon instrument.51 Specific regeneration of the surfaces between injections was not needed owing to the nature of the interaction. Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses of a buffer injection and responses from the reference flow cell were subtracted to account for nonspecific binding and injection artifacts. Experimental data were fitted to a simple 1:1 binding model. The average kinetic parameters (association [ka] and dissociation [kd] rates) generated from 6 data sets were used to define the equilibrium dissociation constant (KD).
Anti-HIV efficacy evaluation in human peripheral blood mononuclear cells
HIV-1 infection of human peripheral blood mononuclear cells (PBMC) was performed as described previously.52 Briefly, fresh PBMCs, seronegative for HIV and hepatitis B virus, were isolated from blood samples of the screened donors (Biological Specialty Corp., Colmar, PA) by using lymphocyte separation medium (LSM; density, 1.078 ± 0.002 g/mL; Cellgro; Mediatech, Inc., Manassas, VA) by following the manufacturer’s instructions. Cells were stimulated by incubation in 4 μg/mL phytohemagglutinin (PHA; Sigma) for 48 to 72 h. Mitogenic stimulation was maintained by the addition of 20 U/mL recombinant human interleukin-2 (rhIL-2; R&D Systems, Inc., Minneapolis, MN) to the culture medium. PHA-stimulated PBMCs from at least two donors were pooled, diluted in fresh medium, and added to 96-well plates at 5 × 104 cells/well. Cells were infected (final multiplicity of infection [MOI] of ≅ 0.1) in the presence of nine different concentrations of compound 14 (triplicate wells/concentration) and incubated for 7 days. To determine the level of virus inhibition, cell-free supernatant samples were collected for analysis of reverse transcriptase activity.53 Following removal of supernatant samples, compound cytotoxicity was measured by the addition of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; CellTiter 96 reagent; Promega) by following the manufacturer’s instructions.
Viral isolates were obtained from the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, as follows: HIV-1 group M isolates 92UG031 (subtype A, CCR5-tropic), 92BR030 (subtype B, CCR5-tropic), 92BR025 (subtype C, CCR5-tropic), 92UG024 (subtype D, CXCR4-tropic), and 93BR020 (subtype F, CCR5/CXCR4 dual-tropic) from the UNAIDS Network for HIV Isolation and Characterization 54; HIV-1 group M isolate 89BZ167 (subtype B, CXCR4-tropic; also referred to as “89BZ_167,” “89_BZ167,” “BZ167,” or “ GS 010”) from Dr. Nelson Michael 55–58; HIV-1 group M isolate 93IN101 (subtype C, CCR5-tropic) from Dr. Robert Bollinger and the UNAIDS Network for HIV Isolation and Characterization 54; HIV-1 group M isolate CMU08 (subtype E, CXCR4-tropic) from Dr. Kenrad Nelson and the UNAIDS Network for HIV Isolation and Characterization 54; HIV-1 group M isolate G3 (subtype G, CCR5-tropic) from Alash’le Abimiku 59; HIV-1 group O isolate BCF02 (CCR5-tropic) from Sentob Saragosti, Françoise Brun-Vézinet, and François Simon 60; HIV-1 clinical isolate MDR769 (presumed Group M, Subtype B) from Dr. Thomas C. Merigan.41
Supplementary Material
Acknowledgments
This work was supported in part by NIH/NIAID grants 1R03AI078790-01A1 (Cocklin, PI) and 1R21AI087388-01A1 (Cocklin, PI). We thank Diana Winters (Academic Publishing Services, Drexel University College of Medicine) for proofreading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Proc Natl Acad Sci USA. 1996;93:3099. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. J Mol Biol. 1994;244:198. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 3.Verli H, Calazans A, Brindeiro R, Tanuri A, Guimaraes JA. J Mol Graphics Modell. 2007;26:62. doi: 10.1016/j.jmgm.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Matthews S, Barlow P, Boyd J, Barton G, Russell R, Mills H, Cunningham M, Meyers N, Burns N, Clark N, et al. Nature. 1994;370:666. doi: 10.1038/370666a0. [DOI] [PubMed] [Google Scholar]
- 5.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Proc Natl Acad Sci USA. 2004;101:517. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nermut MV, Hockley DJ, Bron P, Thomas D, Zhang WH, Jones IM. J Struct Biol. 1998;123:143. doi: 10.1006/jsbi.1998.4024. [DOI] [PubMed] [Google Scholar]
- 7.Nermut MV, Hockley DJ, Jowett JB, Jones IM, Garreau M, Thomas D. Virology. 1994;198:288. doi: 10.1006/viro.1994.1032. [DOI] [PubMed] [Google Scholar]
- 8.Forster MJ, Mulloy B, Nermut MV. J Mol Biol. 2000;298:841. doi: 10.1006/jmbi.2000.3715. [DOI] [PubMed] [Google Scholar]
- 9.Huseby D, Barklis RL, Alfadhli A, Barklis E. J Biol Chem. 2005;280:17664. doi: 10.1074/jbc.M412325200. [DOI] [PubMed] [Google Scholar]
- 10.Alfadhli A, Huseby D, Kapit E, Colman D, Barklis E. J Virol. 2007;81:1472. doi: 10.1128/JVI.02122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alfadhli A, Barklis RL, Barklis E. Virology. 2009 doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryant M, Ratner L. Proc Natl Acad Sci USA. 1990;87:523. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottlinger HG, Sodroski JG, Haseltine WA. Proc Natl Acad Sci USA. 1989;86:5781. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W, Parent LJ, Wills JW, Resh MD. J Virol. 1994;68:2556. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hermida-Matsumoto L, Resh MD. J Virol. 1999;73:1902. doi: 10.1128/jvi.73.3.1902-1908.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono A, Freed EO. J Virol. 1999;73:4136. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paillart JC, Gottlinger HG. J Virol. 1999;73:2604. doi: 10.1128/jvi.73.4.2604-2612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Resh MD. Proc Natl Acad Sci USA. 2004;101:417. doi: 10.1073/pnas.0308043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed EO, Summers MF. J Mol Biol. 2007;366:574. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Proc Natl Acad Sci USA. 2006;103:11364. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spearman P, Horton R, Ratner L, Kuli-Zade I. J Virol. 1997;71:6582. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W, Resh MD. J Virol. 1996;70:8540. doi: 10.1128/jvi.70.12.8540-8548.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. J Virol. 1994;68:5311. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermida-Matsumoto L, Resh MD. J Virol. 2000;74:8670. doi: 10.1128/jvi.74.18.8670-8679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ono A, Freed EO. J Virol. 2004;78:1552. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ono A, Orenstein JM, Freed EO. J Virol. 2000;74:2855. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan X, Yu X, Lee TH, Essex M. J Virol. 1993;67:6387. doi: 10.1128/jvi.67.11.6387-6394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. J Virol. 2008;82:2405. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. Biochemistry. 2006;45:4077. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- 30.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Proc Natl Acad Sci USA. 2004;101:14889. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A. PLoS currents. 2011;3:RRN1253. doi: 10.1371/currents.RRN1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Bioorg Med Chem Lett. 2010;20:5411. doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Timakhov RA, Fedichev PO, Vinnik AA, Testa JR, Favorova OO. Acta naturae. 2011;3:47. [PMC free article] [PubMed] [Google Scholar]
- 35.Fedichev PO, Getmantsev EG, Menshikov LI. J Comput Chem. 2011;32:1368. doi: 10.1002/jcc.21719. [DOI] [PubMed] [Google Scholar]
- 36.Baurin N, Baker R, Richardson C, Chen I, Foloppe N, Potter A, Jordan A, Roughley S, Parratt M, Greaney P, Morley D, Hubbard RE. J Chem Inf Comput Sci. 2004;44:643. doi: 10.1021/ci034260m. [DOI] [PubMed] [Google Scholar]
- 37.Johnsson B, Lofas S, Lindquist G. Anal Biochem. 1991;198:268. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- 38.Johnsson B, Lofas S, Lindquist G, Edstrom A, Muller Hillgren RM, Hansson A. J Mol Recognit. 1995;8:125. doi: 10.1002/jmr.300080122. [DOI] [PubMed] [Google Scholar]
- 39.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. ACS chemical biology. 2008;3:373. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 40.Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. Anal Biochem. 2006;358:281. doi: 10.1016/j.ab.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Palmer S, Shafer RW, Merigan TC. AIDS. 1999;13:661. doi: 10.1097/00002030-199904160-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. J Mol Biol. 2003;327:1013. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 43.Jarvis RA, Patrick EA. IEEE Transactions on Computers. 1973;22:1025. [Google Scholar]
- 44.Li W. J Chem Inf Model. 2006;46:1919. doi: 10.1021/ci0600859. [DOI] [PubMed] [Google Scholar]
- 45.Willett P. Similarity and Clustering in Chemical Information Systems. Letchworth, Hertfordshire, England: Research Studies Press; 1987. [Google Scholar]
- 46.Kelly BN, Howard BR, Wang H, Robinson H, Sundquist WI, Hill CP. Biochemistry. 2006;45:11257. doi: 10.1021/bi060927x. [DOI] [PubMed] [Google Scholar]
- 47.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Proc Natl Acad Sci USA. 2006;103:11364. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haffar O, Dubrovsky L, Lowe R, Berro R, Kashanchi F, Godden J, Vanpouille C, Bajorath E, Bukrinsky M. J Virol. 2005;79:13028. doi: 10.1128/JVI.79.20.13028-13036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weeks SD, Drinker M, Loll PJ. Protein Expr Purif. 2007;53:40. doi: 10.1016/j.pep.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Studier FW. Protein Expr Purif. 2005;41:207. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. Anal Biochem. 2006;358:281. doi: 10.1016/j.ab.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB., 3rd J Virol. 2012;86:8472. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buckheit RW, Jr, Swanstrom R. AIDS Res Hum Retroviruses. 1991;7:295. doi: 10.1089/aid.1991.7.295. [DOI] [PubMed] [Google Scholar]
- 54.Gao F, Yue L, Craig S, Thornton CL, Robertson DL, McCutchan FE, Bradac JA, Sharp PM, Hahn BH. AIDS Res Hum Retroviruses. 1994;10:1359. doi: 10.1089/aid.1994.10.1359. [DOI] [PubMed] [Google Scholar]
- 55.Brown BK, Darden JM, Tovanabutra S, Oblander T, Frost J, Sanders-Buell E, de Souza MS, Birx DL, McCutchan FE, Polonis VR. J Virol. 2005;79:6089. doi: 10.1128/JVI.79.10.6089-6101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jagodzinski LL, Wiggins DL, McManis JL, Emery S, Overbaugh J, Robb M, Bodrug S, Michael NL. J Clin Microbiol. 2000;38:1247. doi: 10.1128/jcm.38.3.1247-1249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michael NL, Herman SA, Kwok S, Dreyer K, Wang J, Christopherson C, Spadoro JP, Young KKY, Polonis V, McCutchan FE, Carr J, Mascola JR, Jagodzinsi LL, Robb ML. J Clin Microbiol. 1999;37:2557. doi: 10.1128/jcm.37.8.2557-2563.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vahey M, Nau ME, Barrick S, Cooley JD, Sawyer R, Sleeker AA, Vickerman P, Bloor S, Larder B, Michael NL, Wegner SA. J Clin Microbiol. 1999;37:2533. doi: 10.1128/jcm.37.8.2533-2537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abimiku AG, Stern TL, Zwandor A, Markham PD, Calef C, Kyari S, Saxinger WC, Gallo RC, Robertguroff M, Reitz MS. Aids Research and Human Retroviruses. 1994;10:1581. doi: 10.1089/aid.1994.10.1581. [DOI] [PubMed] [Google Scholar]
- 60.Loussertajaka I, Chaix ML, Korber B, Letourneur F, Gomas E, Allen E, Ly TD, Brunvezinet F, Simon F, Saragosti S. J Virol. 1995;69:5640. doi: 10.1128/jvi.69.9.5640-5649.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.