

Abstract
Neisseria meningitides is a gram-negative diplococcus bacterium and is the main causative agent of meningitis and other meningococcal diseases. Alanine aminopeptidase from N. meningitides (NmAPN) belongs to the family of metallo-exopeptidase enzymes, which catalyze the removal of amino acids from the N-terminus of peptides and proteins, and are found among all the kingdoms of life. NmAPN is suggested to be mostly responsible for proteolysis and nutrition delivery, similar to the orthologs from other bacteria.
To explore the possibility of NmAPN being a potential drug target for inhibition and development of novel therapeutic agents, the specificity of the S1 and S1′ binding sites were explored using an integrated approach. Initially, an extensive library consisting of almost 100 fluorogenic substrates derived from both natural and unnatural amino acids, were used to obtain a detailed substrate fingerprint of the S1 pocket of NmAPN. A broad substrate tolerance of NmAPN was revealed, with bulky basic and hydrophobic ligands being the most favored substrates. Additionally, the potency of a set of organophosphorus inhibitors of neutral aminopeptidases, amino acid and dipeptide analogues was determined. Inhibition constants in the nanomolar range, determined for phosphinic dipeptides, proves the positive increase in inhibition impact of the P1′ ligand elongation. The results were further verified via molecular modeling and docking of canonical aminopeptidase phosphinic dipeptide inhibitors in the NmAPN active site. These studies present comprehensive characterization of interactions responsible for specific ligand binding. This knowledge provides invaluable insight into understanding of the enzyme and development of novel NmAPN inhibitors.
Keywords: M1 aminopeptidase, Neisseria meningitidis, fluorogenic substrates, organophosphorus inhibitors, S1 and S1′ binding sites specificity
1. Introduction
Meningitis is the inflammation of the membranes (the meninges) lining the brain and spinal cord. It is primarily caused by an infection of various microorganisms such us bacteria, parasites, fungi, and viruses [1]. Bacterial meningitis is a particularly dangerous disease as it can result in brain damage and eventually, if not treated promptly or left untreated, can lead to death. It mostly threatens children during the first year of life more than at any other age [2]. Three types of bacteria are the leading cause of bacterial meningitis: Haemophilus influenzae type b (Hib), Neisseria meningitidis, or Streptococcus pneumoniae. The routine immunization of infants in many countries has reduced the occurrence of severe Hib cases [2–4], leaving N. meningitides and S. pneumoniae as the most common sources of bacterial meningitis worldwide [5–7]. Despite the development of many antibiotics and vaccines, meningitis continues to have unacceptably high morbidity and mortality rates. The World Health Organization estimates that worldwide annual burden of meningococcal disease is 500,000 cases and leads to 50,000 deaths [8]. The incidences are estimated to be ten times greater in less-developed countries than in the Western world (2–5/100,000) [9].
There is a pressing need for the identification of novel drug targets to improve our understanding, prevention, and treatment of bacterial diseases. Control of highly specialized proteases of pathogenic microorganisms is considered a future therapeutic intervention, alternative to the action of the traditional broad-spectrum antibiotics, for which an increasing resistance is observed in the clinical practice [10]. Enzymes drug targets in the bacteria seem to be relatively easier to alter than mammalian ones. The latter are frequently implicated in complex proteolytic networks and their inhibition effects are often either ambiguous or redundant [11]. Proteases of single cell organisms function in a less complicated environment, are more bioavailable and predictable targets, and their elimination is usually lethal to the microorganism.
Several zinc-dependent proteases, known to be etiological factors of diseases, are targeted for designing new directed antibiotics [12]. This includes a neutral aminopeptidase from N. meningitides belonging to the M1 family [13]. This enzyme (E.C. 3.4.11.2, alanine aminopeptidase, aminopeptidase N, APN) is a conserved, zinc-dependent exopeptidase involved in biological functions in both eukaryotic and prokaryotic cells [14,15]. APN’s activity has been found in mammals, plants and single cell organisms. APN is known to act on various peptide substrates with broad substrate specificity. In humans, the most significant physiological role of APN is associated with angiogenesis, immunological responses and signal transduction, whereas pathogenic activity is connected with cancer malignancy and metastasis [16–18]. Orthologous enzymes in single-celled organisms are mostly responsible for proteolysis and nutrition delivery[19,20].
In this work, we present substrate specificity profiling of the alanine aminopeptidase from human pathogen N. meningitidis (NmAPN). In particular, we identified a fingerprint of substrate specificity in the S1 pocket using natural and unnatural amino acids attached to a 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. This approach has recently been applied to identify in detail the ligand preferences of aminopeptidases from different sources [21–24]. Organophosphorous inhibitors, including those, which were recently validated as antimalarial agents of M1 and M17 aminopeptidases from Plasmodium falciparum [19,25], were also screened. To visualize the binding mode of phosphinic pseudodipeptides substrates to the S1 and S1′ pockets of the active site of NmAPN, we used molecular docking. The docking results provided structural insights into the active site’s substrate/inhibitor binding and selectivity.
2. Materials and Methods
2.1. General
To examine the S1 pocket requirements of the alanine aminopeptidase from Neisseria meningitides, a 98-membered library containing natural and unnatural amino acids was used (for the full list of compounds and their structures see Table S1 in Supplementary Information). Among the fluorogenic substrates, 82 have been described earlier [23]. 16 new compounds (L-Tyr(Bzl)-ACC, L-Pip-ACC, L-Cit-ACC, L-Glu(Bzl)-ACC, L-hSer-ACC, L-Tle-ACC, L-Chg-ACC, 6-OBzl-L-Nle-ACC, L-Abu(Bth)-ACC, L-Arg(NO2)-ACC, L-Agp-ACC, L-hSer(Bzl)-ACC, 4-F-L-Phe-ACC, penta-F-L-Phe-ACC, L-Asp(Bzl)-ACC, 3-pyridyl-L-Ala-ACC) were synthesized and characterized according to the protocol describe previously [21]. They were purified on an HPLC using a Waters M600 solvent delivery system equipped with a Waters M2489 detector module with preparative Waters Spherisorb S10ODS2 column. All substrates were at least of 95 % purity, which was confirmed by analytical chromatography on a Waters Spherisorb S5ODS2 column. The solvent composition system was as follows: stream A (water with 0.1 % of trifluoroacetic acid, TFA] and stream B (acetonitrile/water 80%/20% (v/v) with 0.1% of TFA). The appropriate inhibitors were available from previous studies [26–28]. Gene cloning, protein expression in E. coli and purification of Nm APN were performed using the procedure described elsewhere [13].
2.2. Kinetic studies
2.2.1. Preliminary screening
Library screening was performed in 96 well plate format working at two wavelengths: excitation at 355 nm and emission at 460 nm. Neisseria meningitides alanine aminopeptidase was assayed in a 50 mM Tris-HCl buffer, at pH = 7.5. The enzyme was preincubated at 37 °C for 30 min before being added to the wells containing substrates. Final screening concentration was 3 μM (a substrate) and 1.37 nM (the enzyme). Total assay time was generally 15–30 min, and the linear portion of the progress curve was used to calculate velocity, which is proportional to kcat/Km. All experiments were repeated at least three times. Analysis of the results was based on total relative fluorescence units (RFU) for each substrate, setting the highest value to 100% and adjusting the other results accordingly.
2.2.2. Kinetic parameters determination
NmAPN was preincubated for 30 min in the buffer as described above. After incubation the enzyme was added to the wells containing selected substrates. The release of the fluorophore was monitored continuously for at least 15 minutes at an enzyme concentration of 1.13 nM (the total volume of 100 μL). The linear portion of the progress curve was used to calculate velocity of hydrolysis. Values of kcat, Km and kcat/Km were calculated from the reaction velocity to substrate concentration plot using the Michaels-Menten equation (V = Vmax × [S]/(Km + [S])) and Vmax = [E] × kcat with help of GraphPad Prism program (V – velocity of hydrolysis, Vmax – maximum enzyme velocity, Km – the Michaelis-Menten constant, [S] – substrate concentration and [E] – enzyme concentration). Each substrate was assayed at eight different concentrations. The experiments were repeated at least three times and the average as well as the standard deviation values were calculated. The concentration of DMSO in the assay was less than 1% (v/v).
2.2.3. Inhibitors
The phosphorus containing inhibitors were screened against NmAPN at 37 °C in the assay buffer as described above. The enzyme was preincubated for 30 min at 37 °C with an inhibitor before adding the substrate (L-Arg-ACC, final concentration 20 μM) to the wells. The conditions were as follows: total volume 100 μL, eight different inhibitor concentrations, and enzyme concentration 0.85 nM. The release of the ACC fluorophore was monitored. The linear portion of the progress curve was used to calculate velocity. IC50 values were calculated from the reaction velocity to inhibitor concentration plot fitted with a non-linear regression method implemented in GraphPad Prism program. Each experiment was repeated at least three times and the results are presented as the average with standard deviation. The competitive binding mode was evidenced from the Lineweaver-Burk plot. Ki values were calculated using Cheng-Prusoff equation: Ki = IC50/[1 + (S/Km)] [29], where IC50 was the concentration of the inhibitor that effected with 50% inhibition, S was the concentration of the substrate used, and Km was the Michaelis constant determined for enzyme/substrate pair. The concentration of DMSO was less than 1% (v/v).
2.3. Molecular modeling
Molecular modeling studies were performed using Discovery Studio (Accelrys, Inc.) program package. Crystal structure of the native aminopeptidase N from N. meningitidis (pdb id. 2GTQ) [13] with protons added (assuming the protonation state of pH 7) was used as the starting point. The structure of inhibitor 4 was copied from the crystal structure of hPheP(CH2)Phe-PfAPN complex (pdb id. 3EBI) [30] after superimposition of key active site residues of both proteins. Starting structure of inhibitor 5 was obtained by appropriate modification of compound 4, in above-mentioned complex. Minimizations were done with CHARMM force field, using Smart Minimizer algorithm, up to RMS gradient 0.1 Å. The structure of all residues forming the enzyme active site and the inhibitor were optimized, while all other protein residues were kept fixed. Molecular surfaces were obtained using Soft method and colored according to interpolated charge.
3. Results and discussion
3.1. Fluorogenic substrates library
A set of 98 fluorogenic substrates was used to investigate the specificity of the Neisseria meningitides APN active site, in particular the S1 binding pocket. Apart from the previously used 82 substrates [23], the library complexity was increased by 16 novel compounds. The compounds were synthesized, purified and characterized using the approach analogous to the one previously described [21]. The library contained amino acids of defined stereochemistry and diverse side chain structures. This included the members of the natural L analogues, with the exception of cysteine, and their D counterparts. In addition, a selection of the L amino acids that had modified side chains (with the functionalities protected, substituted at the aromatic rings, higher homologues, etc.), was tested. The library also included a set of non-natural residues that comprised several alkyl, arylalkyl, cycloalkyl, heterocyclic and heteroaromatic derivatives. Thus, the members of the collection differed in size and functionality (for the full list of compounds see Table S1 in Supplementary Information). This structural variety would indicate a range of possible interactions in the S1 pocket and would allow us to distinguish even subtle differences when compared with the specificity of related aminopeptidases. Recombinant fully-functional NmAPN was expressed in Escherichia coli, isolated and purified as described previously [13]. To compare the rate of hydrolysis of the fluorogenic substrates in optimal conditions, an initial library screening was performed. It identified compounds that are best cleaved and revealed their preliminary kinetic parameters. The lowest measured Michaelis constant (Km) values were in the 30 μM range, such that the final substrate concentration, equal for each individual compounds, was adjusted to 3 μM (approximately 10-fold below the lowest Km) and the enzyme concentration to 1.37 nM. In these conditions, the cleavage of substrates, recorded as an increase of the fluorescence signal proportional to kcat/Km values (kcat – turnover number, kcat/Km – catalytic efficiency of the enzyme), and was monitored in independent triplicate experiments. The plots summarizing the obtained average rates of hydrolysis are presented in Fig. 1 and 2 as the percentage referenced to the fastest cleavage rate.
Fig. 1.
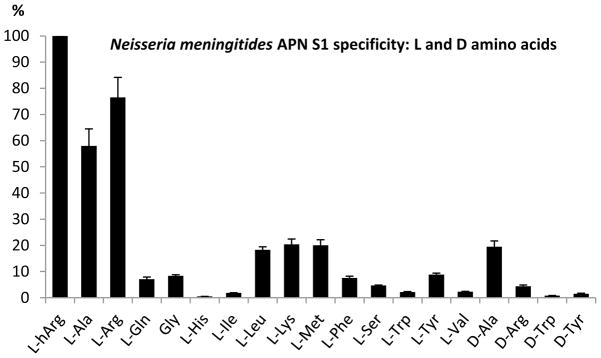
Substrate specificity of NmAPN for ACC derived natural L-amino acids and D-derivatives. Y-axis represents the rate of hydrolysis, proportional to the kcat/Km value, expressed as the percentage of the best substrate cleavage (L-hArg-ACC, 100%). Error bars represent standard deviation. ACC derived L-Asn, L-Asp, L-Glu, L-Pro, L-Thr (the L series) and D-Asp, D-Asn, D-Glu, D-Gln, D-His, D-Leu, D-Lys, D-Met, D-Phe, D-Pro, D-Ser, D-Thr, D-Val (the D series) were not hydrolyzed.
Fig. 2.

Substrate specificity of NmAPN for ACC non-natural amino acids. Substrates bearing a linear, non-substituted side chain are colored blue, hydrophobic, branched aliphatic and cycloaliphatic – red, heteroatom-containing aliphatic and cycloaliphatic – green, aromatic and heteroaromatic – orange. Y-axis represents the rate of hydrolysis, proportional to the kcat/Km value, expressed as the percentage of the best substrate cleavage (L-hArg-ACC, 100%). Error bars represent standard deviation. ACC derived pyrrolidin-2-yl-L-Ala, 2S,3S-Apns, L-Dab, L-Tic, β-Ala, 6-Ahx, L-Phg, 4-I-L-Phe, 4(1-carboxymethoxy)-L-Phe, L-Dab(Z), 4-guanidino-L-Phe, L-Tyr(Bzl), L-Cit, L-Tle, L-Chg, L-Agp were not hydrolyzed.
Among tested substrates, ACC derived L-homoarginine (L-hArg) appeared to be the best substrate for NmAPN. As far as the natural amino acid analogues are considered (Fig. 1), L-arginine and L-alanine are clearly preferred, being hydrolyzed at 76.5% and 58.0% of L-hArg rate, respectively. Other proteinogenic amino acids derivatives were hydrolyzed at a considerably lower rate (< 20% for hydrophobic/basic L-Leu, L-Lys and L-Met). Charged and hydrophilic compounds (L-Asp, L-Asn, L-Glu, L-His, L-Thr, etc.) were not processed. It is worth to note that two ACC compounds of the D series were also hydrolyzed by the enzyme, namely D-Ala-ACC (25 % of the L-Ala-ACC cleavage rate) and D-Arg-ACC (6% of the L-Arg-ACC cleavage rate).
NmAPN tolerates many different side chains of an unnatural amino acids in the S1 pocket (Fig. 2). The best substrates (38.5–74.0% of L-hArg-ACC hydrolysis rate) can be characterized as containing bulky hydrophobic residues, with an additional heteroatom functionalization, namely: L-arginine(NO2), L-homotyrosine, L-2-amino-3-(2′-benzothiazolyl)butyric acid (L-Abu(Bth)), L-homophenylalanine and L-glutamic acid benzyl ester. Most of them are aromatic (Fig. 2, highlighted orange). The remaining ACC substrates of the same class of compounds are hydrolyzed with lesser efficiency, which indicates much lower specificity.
Interestingly, linear aliphatic chains of a small to medium size (L-Abu, L-Nva, L-allylGly, containing 2–3 carbon atoms in the side chain, Fig. 2, highlighted blue) are processed at the range of 20–40% efficiency of the optimal substrate velocity. More extended hydrophobic amino acids that are alkyl (both linear and branched, Fig. 2, highlighted red), arylalkyl or heteroarylalkyl substituted, are also tolerated by the enzyme to some extent. In contrast, hydrophilic substrates that have an additional functionalized heteroatom moiety at a 2–3 carbon atoms side chain are generally poorly cleaved in comparison to the reference compound. Also, the sterically constrained phenylglycine, cyclohexylglycine, and cyclic (L-Pro, L-Tic) ACC derivatives as well as β-amino acids are poor substrates for NmAPN.
Whole library screening revealed a broad preference of substrates of NmAPN. To obtain precise values of the kinetic parameters of cleavage, kcat, Km and kcat/Km were determined for almost 40 compounds (selected data presented in Table 1, all measured constants in Table S2 in Supplementary Information). The lowest Michaelis constant values were found for long and basic L-homoarginine (27.7 ± 2.3 μM), medium size aliphatic L-norvaline (28.8 ± 0.8 μM) and hydrophobic heteroaromatic L-Abu(Bth) (30.3 ± 4.8 μM). Interestingly, each of these three substrates belongs to a different group of substrates as shown in Table 1. These results demonstrate that the length and type of the side chain have a strong influence on a substrate recognition and binding. Among non-substituted and non-branched alkyl substituents (group A) nPr residue of L-Nva seems to be optimal (Fig. 3). Km values of shorter analogues are 6–7 fold higher, namely 174.7 ± 1.6 μM for L-Ala and 161.6 ± 1.1 μM for L-Abu. Further elongation of L-Nva by a methylene group to L-Nle has a less pronounced, but still negative influence on affinity (52.8 ± 6.0 μM). Similarly deteriorating effect on affinity is observed in the case of a more rigid unsaturated allylglycine analogue.
Table 1.
The kinetic data (Km, kcat/Km, kcat) for selected ACC substrates of N. mengitides aminopeptidase N (each compound was formally assigned to one of four groups in relation to its characteristic structural features), nd – not determined.
| Group | Code | Km [μM] | kcat/Km [s−1M−1] | kcat [s−1] |
|---|---|---|---|---|
| A (linear, non-substituted) | Gly | >250 | nd | nd |
| L-Ala | 174.7 ± 1.6 | 163 000 ± 3 000 | 28.4 ± 1.0 | |
| L-Abu | 161.6 ± 1.1 | 85 000 ± 7 000 | 13.8 ± 0.1 | |
| L-propargylGly | 121.3 ± 2.3 | 63 000 ± 2 000 | 7.74 ± 0.25 | |
| L-Nva | 28.8 ± 0.8 | 116 000 ± 2 000 | 3.33 ± 0.09 | |
| L-allylGly | 59.7 ± 2.7 | 82 000 ± 3 000 | 4.90 ± 0.16 | |
| L-Nle | 52.8 ± 6.0 | 52 000 ± 4 000 | 2.72 ± 0.18 | |
| L-Met | 80.7 ± 0.8 | 74 000 ± 2 000 | 5.99 ± 0.18 | |
|
| ||||
| B (hydrophobic, branched aliphatic and aromatic) | L-Leu | 85.4 ± 0.5 | 40 000 ± 1 000 | 3.45 ± 0.02 |
| L-neopentylGly | 61.0 ± 1.6 | 27 000 ± 1 000 | 1.64 ± 0.10 | |
| L-hLeu | 56.9 ± 1.8 | 46 000 ± 1 000 | 2.65 ± 0.08 | |
| L-Cha | 121.9 ± 1.0 | 31 000 ± 1 000 | 3.84 ± 0.04 | |
| L-hCha | 41.4 ± 1.3 | 77 000 ± 3 000 | 3.19 ± 0.13 | |
| L-Phe | >250 | nd | nd | |
| 4-Cl-L-Phe | 149.1 ± 2.6 | 34 000 ± 1 000 | 5.05 ± 0.11 | |
| 4-Br-L-Phe | >250 | nd | nd | |
| L-hPhe | 100.8 ± 0.6 | 129 000 ± 1 000 | 13.0 ± 0.1 | |
| L-Igl | 48.9 ± 2.2 | 35 000 ± 2 000 | 1.72 ± 0.08 | |
| L-2-Nal | 50.7 ± 8.9 | 51 000 ± 7 000 | 2.6 ± 0.27 | |
|
| ||||
| C (aliphatic, heteroatom terminated) | L-hSer | 79.2 ± 6.9 | 79 000 ± 5 000 | 6.23 ± 0.67 |
| 3-OH-L-Pro | 57.2 ± 2.7 | 21 000 ± 2 000 | 1.19 ± 0.07 | |
| L-Gln | >250 | nd | nd | |
| L-Lys | 88.7 ± 1.2 | 69 000 ± 2 000 | 6.15 ± 0.31 | |
| L-Arg | 64.1 ± 1.2 | 205 000 ± 7 000 | 13.1 ± 0.6 | |
| L-Arg(NO2) | 60.4 ± 4.8 | 120 000 ± 2 000 | 7.25 ± 0.20 | |
| L-hArg | 27.7 ± 2.3 | 312 000 ± 23 000 | 8.63 ± 0.59 | |
|
| ||||
| D (heteroatom containing aromatic and heteroaromatic) | L-Tyr | 46.4 ± 2.5 | 36 000 ± 2 000 | 1.66 ± 0.05 |
| L-hTyr | 47.4 ± 1.6 | 270 000 ± 11 000 | 12.7 ± 0.6 | |
| L-hSer(Bzl) | 146.8 ± 7.9 | 28 000 ± 1 000 | 4.16 ± 0.13 | |
| L-Asp(Bzl) | 117.4 ± 4.0 | 31 000 ± 1 000 | 3.64 ± 0.05 | |
| L-Glu(Bzl) | 50.6 ± 2.7 | 111 000 ± 5 000 | 5.60 ± 0.28 | |
| 6-OBzl-L-Nle | 59.9 ± 2.8 | 54 000 ± 1 000 | 3.24 ± 0.03 | |
| L-Abu(Bth) | 30.3 ± 4.8 | 173 000 ± 28 000 | 5.25 ± 0.85 | |
Fig. 3.
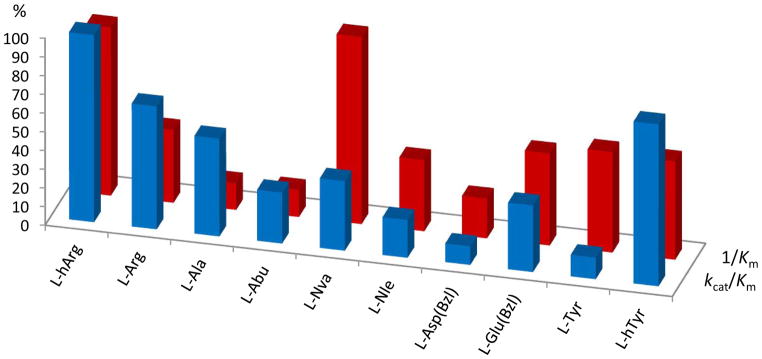
The kinetic parameters of selected homologous substrates, referenced to kcat/Km of L-hArg (100%, blue) and 1/Km of L-Nva (100%, red). Among alkyl derivatives (L-Ala, L-Abu, L-Nva, L-Nle), medium size nPr side chain of L-Nva is the most preferentially bound in the S1 cavity, although L-Ala-ACC is processed at a higher hydrolysis rate. Other examples show a significance of the P1 residue structure extension: CH2 elongated analogs (homo) are generally better recognized and faster cleaved than their natural counterpart.
Mixed results are observed for hydrophobic, branched alkyl, cycloalkyl or arylalkyl substituted substrates (group B). From one side, substrates elongation by insertion of two carbon atoms between the amino acid backbone and the bulky terminal appears to be beneficial as illustrated by the following substrate pairs: L-Leu versus L-hLeu (85.4 μM and 56.9, respectively), L-Cha versus L-hCha (121.9 ± 1.0 μM and 41.4 ± 1.3 μM, respectively), L-Phe versus L-hPhe (>250 μM and 100.8 ± 0.6 μM, respectively), and others (Fig. 3). From the other side, sterically constrained derivatives (L-neopentylGly, L-Igl, L-2-Nal) also exhibit Km at the range of 50–60 μM. Substitution on the aromatic portion of L-Phe by a neutral group (methyl, halogen, nitro groups) has no significant influence on its binding affinity to the S1 pocket.
As heteroatom functionalities vary in the studied library of the fluorogenic substrates, the impact of heteroatom substitution in the substrate was also tested. Among linear derivatives terminated with different groups (group C), extended basic L-homarginine is clearly the best. Other substrates of a related character are also accepted. Several compounds with good affinity are found for heteroatom containing modified aromatic and related residues (group D). This includes L-Tyr and L-hTyr ACC derivatives, terminated with a p-phenol residue (46.4 ± 2.5 μM, 47.4 ± 1.6 μM, respectively), benzyl ester of L-glutamic acid (50.6 ± 2.7 μM) and heteroaromatic L-Abu(Bth).
In conclusion, it seems that the heteroatom donors of a hydrogen bond (or positively charged moieties) are preferred at the terminus of a hydrophobic substituent, whereas acceptors are well tolerated in the core.
The results of kcat/Km measurements (Table 1) were found to correlate well with the preliminary profile, which was obtained through the full library screening. Similarly, as indicated above, L-hArg (312 000 ± 23 000 M−1s−1), L-hTyr (270 000 ± 11 000 M−1s−1), L-Arg (205 000 ± 7 000 M−1s−1) and L-Abu(Bht) (173 000 ± 28 000 M−1s−1) substrates were processed with the highest rate of hydrolysis. Apparently, long and basic or bulky, hydrophobic and heteroatom modified amino acids are preferred by the NmAPN. L-Ala-ACC, which holds a small non-branched substituent exhibits a comparable value of kcat/Km (163 000 ± 3 000 M−1s−1, approximately 50% of the calculated maximum of L-hArg-ACC). In this case a visibly lower binding affinity is compensated by the highest kcat value (28.4 ± 1.0 s−1).
The turnover rates for other substrates are at least two-fold lower. Such values are characteristic for hydrophobic and basic residues of ACC substrates, namely: L-Abu-ACC (13.8 ± 0.1 s−1), L-Arg-ACC (13.1 ± 0.6 s−1), L-hPhe-ACC (13.0 ± 0.1 s−1) and L-hTyr-ACC (12.7 ± 0.6 s−1). The rest of the compounds show a significantly lower kcat.
An interesting variation in the relationship between the catalytic efficiency and the substrate structure can be observed between orthologous APNs from different species. Three mammalian (human, porcine and rodent) and one protozoan (a functionally active recombinant Plasmodium falciparum, PfAPN) enzyme have been kinetically profiled [21,24]. Data from current and previous studies are compared in Fig. 4. The overall specificity of NmAPN is clearly narrower, when compared with pig and protozoan enzymes. Both pig and protozoan aminopeptidases equally accept a particularly wide range of hydrophobic and basic amino acids substrates. The human enzyme has the highest specificity towards hydrophobic substrates; with methionine being digested the fastest. In contrary basic residues of L-Arg and L-hArg ACC derivatives, which are preferentially hydrolyzed by NmAPN, are much poorer substrates and are cleaved slower by the human enzyme. Such a disparity between the S1 specificity profiles of NmAPN versus the human APN is a promising observation from an inhibitor design perspective.
Fig. 4.
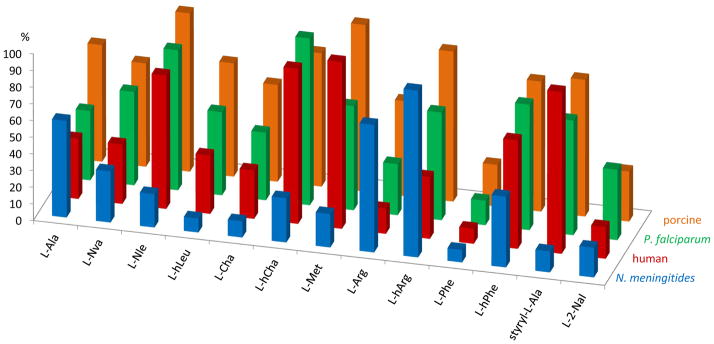
Comparison of the specificity profiles of the M1 family of alanine aminopeptidases enzymes for selected ACC derived amino acid substrates. The rate of hydrolysis, proportional to the kcat/Km value, expressed as the percentage of the best substrate cleavage (100%) is presented for N. meningitidis, P. falciparum [24], porcine and human APNs [21].
3.2. Organophosphorus inhibitors
A set of phosphorus-containing amino acid and dipeptide analogues (Fig. 5) was screened for inhibitory activity against NmAPN. It contains compounds, which are generally considered transition state inhibitors as they closely mimic the structure and charge distribution of the tetrahedral intermediate formed in the amide bond hydrolysis. The compounds were shown to regulate the activity of other metalloproteases [32–34]. The studied group included phosphonic derivative of arginine (1), an analogue of the fasted cleaved natural amino acid substrate Arg-ACC. All other five are derivatives of homophenyloalanine: α-aminophosphonic acid (2) [26], α-amino-H-phosphinic acid (3) [27] and three phosphinic pseudodipeptide analogues (4–6) [27,28]. Previously, this hydrophobic amino acid (hPhe) appeared to bear a P1 substituent privileged for inhibition of metalloaminopeptidases [27]. It also corresponds to the structure of a good ACC substrate (kcat/Km = 129 000 ± 1 000 s−1M−1). Although Arg-ACC is hydrolyzed with higher efficiently (kcat/Km = 205 000 ± 7 000 s−1M−1), the synthesis of arginine-based phosphinic compounds would be much more problematic. Having the phenylethyl P1 residue conserved within the whole set 2–6 allows to evaluate the influence of phosphorus moiety and P1′ elongation on affinity towards the novel enzyme target. Hydrophobic portions of phenylalanine (4) and tyrosine (5) were selected as P1′ elongation in the classical phosphinic dipeptide scaffold. The latter amino acid was also applied as a fragment of the so-called extended transition state analogue (P-C-N containing backbone of 6).
Fig. 5.

Structures of tested organophosphorous inhibitors.
Compounds 1–3 and 6 appeared to be good competitive inhibitors of NmAPN with inhibition constant in the range of 1–3 μM (Table 2). ArgP (1) was found to be slightly less potent than hPheP (2) although the kinetic constants of the corresponding substrates were ranked in the reversed order. Interestingly, there is no essential difference in activity between the two structural amino acid analogues hPhePH and hPheP. It seems to be evident that two oxygen atoms of a phosphorus moiety forms complex with zinc atom. However, the kinetic results indicate that there is no additional energy gained upon binding of the third phosphonate oxygen atom. We hypothesize that it is due to the lack of any specific interactions in the active site. Likely, the bulky hydrophobic side-chain group of homophenylalanine is well accommodated in the S1 binding pocket. Similar inhibition constants have been previously reported for human (0.82 μM [21]) and porcine kidney APNs (6.1 μM [21] and 15.9 μM [26]).
Table 2.
Inhibition of the NmAPN by selected organophosphorus compounds, derivatives of arginine (1) and homophenylalanine (2–6).
| Inhibitor | IC50 [μM] | Ki [μM] |
|---|---|---|
| 1, ArgP | 1.91 ± 0.18 | 1.46 ± 0.14 |
| 2, hPheP | 1.66 ± 0.07 | 1.26 ± 0.05 |
| 3, hPhePH | 1.91 ± 0.05 | 1.45 ± 0.04 |
| 4, hPheP(CH2)Phe | 0.397 ± 0.024 | 0.302 ± 0.018 |
| 5, hPheP(CH2)Tyr | 0.070 ± 0.007 | 0.054 ± 0.005 |
| 6, hPheP(CH2NH)Tyr | 3.29 ± 0.15 | 2.51 ± 0.11 |
Compounds 4–6 have fragments, which are designed to interact with the S1′ pocket of the active site. In the case of hPheP(CH2NH)Tyr (Compound 6, Table 2) the addition of the fragment appeared ineffective as the backbone elongation distorts the overall binding mode in the S1′ region. This is evident by a slight decrease in activity (approximately two-fold) compared to the parent amino acid hPhePH and hPheP.
The backbone extensions to the P1′ region are observed to be the most successful for phosphinic pseudodipeptides (Compounds 4 and 5, Fig. 5). The hPheP(CH2)Phe and hPheP(CH2)Tyr were found to act as reversible, competitive and tight-binding inhibitors of NmAPN. The inhibition constants were measured at the nanomolar range, namely Ki = 302 ± 18 nM for hPheP(CH2)Phe and 54 ± 5 nM for hPheP(CH2)Tyr. Interestingly, the binding affinities turned out to be virtually identical to those exhibited by the same compounds towards the porcine kidney ortholog (276 nM and 36 nM, respectively, Table 3) [27]. An order of magnitude difference in potency between compounds 4 and 5 can be explained by the structural modeling using homology model of APN based on human leukotriene A4 hydrolase [27]. This model shows that compound 5, in contrast to compound 4, forms a hydrogen bond between phenolic tyrosine OH and carboxylate of Glu413 making it much tighter binder. Surprisingly, the inhibition activity displayed by compounds 4 and 5 toward PfAPN is reversed. The phenylalanine analogue (4) binds well whereas the tyrosine binding (5) is kinetically less favored (Table 3) [19,30]. It altogether suggests that NmAPN forms a specific interaction involving inhibitor OH group similar to that suggested for porcine enzyme, whereas in Pf enzyme aromatic ring substitution leads to some distortions. Thus, the subtle differences in the S1′ binding site architecture are factors that can affect the activity of closely related structural analogues.
Table 3.
Comparison of the inhibition constants Ki measured for phosphinic dipeptide inhibitors hPheP(CH2)Phe (4) and hPheP(CH2)Tyr (5) towards APNs from different organisms.
3.3. Molecular modeling
To explain these nuances and comment on the observed variations in the binding specificity of hPheP(CH2)Phe and hPheP(CH2)Tyr, the architecture of the NmAPN active site with docked ligands was studied. Structures of the inhibitor-enzyme complexes were modeled using the crystal structure of native NmAPN [13] as the starting point and the complex of P. falciparum APN with inhibitor 4 as the template [30]. Although several crystal structures of bacterial (E. coli) aminopeptidase N were recently published [35–39], the protozoan enzyme seemed to be better suited for our modeling studies. First, it is the only representative of the APNs that has been structurally characterized in the complex with the studied hPheP(CH2)Phe inhibitor. Second, the Pf and NmAPN active sites are very similar to each other and all the major catalytic residues are highly conserved. A sequence alignment analysis shows that E. coli exhibits 47% sequence identity and 62% similarity with N. meningitidis, whereas P. falciparum has 35% identity and 55% similarity (Fig. 6). Such a close relationship between listed representatives is somewhat unexpected since eukaryotic Plasmodium is evolutionary distant to bacterial N. meningitidis. From the phylogenetic perspective, a much better alignment agreement should be expected between two eukaryotic representatives (Pf and mammalian APN). However, the degree of sequence identity between the Pf and human enzyme is 31% in the active site area and only 13% in the overall sequence.
Fig. 6.

Multiple sequence alignment of three alanine aminopeptidases from bacteria (E. coli, N. meningitidis) and protozoan (P. falciparum, truncated to residues 195–1085, lacking the N-terminal extension, which contains 3 asparagine-rich regions and a putative transmembrane domain). Sequence identities are highlighted in red and similarities are shown as red letters. The key active site residues involved in the coordination of zinc atoms and in the catalytic mechanism are highlighted in orange. The residues, which are located in the S1 pocket and can control its specificity, are shown in green. The residues equivalent to Ser289 of Nm (involved in binding of the Tyr P1′ residue) are highlighted in blue (see the text for further discussion).
In general, the modeled binding mode of compound 4 to NmAPN is analogous to that observed for P. falciparum enzyme (Fig. 7A). The phosphinate group is involved in a bidentate zinc ion complexation. It also accepts a hydrogen bond from the hydroxyl group of Tyr377, which indicates the significant role the residue plays in the catalysis, namely stabilization of the transition state [35]. The terminal amino moiety is bound by two charge-assisted hydrogen bonds with the Glu260 and Glu117, while the carboxylate is exposed to the solvent and forms only one interaction with the Gly257 amide proton. The aromatic portion of hPhe fragment is located in a deep narrow S1 cavity of a neutral character (formed by Tyr372, Met256, Gln115, Asn255, Gln818, Phe254 and Met103 residues). The P1′ benzyl side chain lays on the surface shaped by Val270 and Phe271 residues.
Fig. 7.


The modeled structures of the complexes of phosphinate inhibitors 4 and 5 with N. meningitidis APN (panel A and B, respectively). The enzyme molecular surface is colored according to the interpolated charge (red – negative, grey – neutral and blue – positive).
The Met256 residue directly precedes the characteristic GAMEN motif and, when disordered, can partially block the S1 pocket. Conformational ordering of E. coli Met260 (equivalent to N. meningitides Met256) was suggested to accompany accommodation of bulkier P1 residues [35]. In the present studies, this assumption can explain the fast cleavage of small aliphatic substrate derivatives (that use a limited size of the S1 pocket) but also a preferential binding of extended heteroatom-modified ones (that uses the whole available cavity room). Potential P1-S1 interactions may include electrostatic and ionic binding of 2,3- and 2,4-disubstituted ligands (similarly to e.g. bestatine [35] and diamino compounds [39,40]) in the glutamate-rich anionic site formed by Glu117, Glu260 and Glu316. Furthermore, a set of characteristic amide side chains is located in the S1 periphery. Apparently, Asn255 and Gln818 are responsible for superior binding of guanidine-terminated ligands as evidenced here, in analogy to the same role of Asn373 (Asn369 of Nm) and Gln821 postulated previously for EcAPN [35,37].
The mode of binding modeled for inhibitor 5 (Fig. 7B) was found to be nearly the same as described above for hPheP(CH2)Phe. Both the backbone and the side chains are located in a manner analogous to 4. Nevertheless, in the case of hPheP(CH2)Tyr, an additional specific hydrogen bond between Ser289 hydroxyl and compound 5 hydroxyl was recognized. This represents the crucial difference in the pattern of interactions between phenylalanine and tyrosine compounds. This most likely is the key to the discrimination factor of their affinities to NmLAP (Ki = 302 nM versus Ki = 53.6 nM, respectively). In the eukaryotic enzymes (both P. falciparum and mammalian orthologs), the position corresponding to the flexible side chain of Ser289 is occupied by a more sterically constrained threonine (Thr297 for Pf, Fig. 6). As the effect, the binding of hPheP(CH2)Tyr by plasmodia APN does not cause any energy gain compared to hPheP(CH2)Phe. On the contrary, slight distortions may be responsible for a decrease in activity (Ki = 79 nM versus Ki = 232 nM, respectively) [19,30]. However, in porcine kidney APN the role of the hydrogen bond counterpart for phenol OH is taken by a neighboring carboxylate (Glu413, see above) and the presence of the hydroxyl is again favorable [27]. In E. coli the parallel position in the sequence alignment is mutated to the residue of a rather different character – Arg293.
4. Conclusions
APN is a multifunctional enzyme that is essential to many biological functions in mammals and other organisms [14–19]. Therefore, it might be an attractive target for drug design and discovery. Understanding of both, the enzyme specificity and selective inhibition is an important step into the better understanding of the enzyme function and role it might play in bacterial meningitis progression. We have presented comprehensive studies of the NmAPN specificity. The fluorogenic substrate screening revealed ligands that bind specifically to the S1 pocket. The best fit into the narrow, cylindrical cavity of the active site, was observed for bulky hydrophobic (in majority, aromatic) P1 side chains that preferentially contain additional functions (Fig. 8). Heteroatoms are well accepted at the distance of 2–3 atoms from the carbon α adjacent to phosphorus. They likely form hydrogen bonds with the glutamate residues (Glu117, Glu260 and Glu316), typically binding the N-terminus of the substrates. The other functionality can be designed to explore the distal part of the S1 pocket containing amide residues of Asn255, Asn269 and Gln818.
Fig. 8.

A summary of favorable features of the NmAPN ligand structure identified in the current study, which should enhance the binding affinity of potential inhibitors (as shown based on the phosphinic dipeptide scaffold), FG – a heteroatom containing functional group.
As evidenced by studies on organophosphorus inhibitors and by molecular modeling, potent inhibitors of NmAPN should be definitely extended to the P1′ fragment. Phosphinic dipeptides, transition state analogues following this requirement, appeared to be tight, competitive inhibitors. Aromatic residues seem to be favorably bound in a shallow hydrophobic S1′ cleft. Their terminal part can be further modified, for example by a moiety directed to Ser289 (Fig. 8) and/or to other neighboring residues.
Supplementary Material
Acknowledgments
Artur Mucha is supported by the Polish Ministry of Science and Higher Education Grant N N302 159937. The Drag laboratory is supported by the Foundation for Polish Science and the State for Scientific Research Grant N N401 042838 in Poland. Marcin Poręba and Anna Byzia are supported by European Union Human Capital National Cohesion Strategy. Bogusław Nocek, Rory Muligan and Andrzej Joachimiak are supported by a grant from the National Institutes of Health (GM094585). The use of software resources (including the Accelrys programs) of the Wroclaw Centre for Networking and Supercomputing, Poland, is kindly acknowledged.
Footnotes
Dedicated to Professor Henri-Jean CRISTAU on the occasion of his 70th birthday.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sáez-Llorens X, McCracken GH., Jr Bacterial meningitis in children. Lancet. 2003;361:2139–2148. doi: 10.1016/S0140-6736(03)13693-8. [DOI] [PubMed] [Google Scholar]
- 2.Mace SE. Acute bacterial meningitis. Emerg Med Clin North Am. 2008;26:281–317. doi: 10.1016/j.emc.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Wenger JD, Hightower AW, Facklam RR, Gaventa S, Broome CV. Bacterial meningitis in the United States, 1986: report of a multistate surveillance study. The Bacterial Meningitis Study Group. J Infect Dis. 1990;162:1316–1322. doi: 10.1093/infdis/162.6.1316. [DOI] [PubMed] [Google Scholar]
- 4.Pickering LK, Baker CJ, Long SS, McMillan JA, editors. Red Book: 2006 Report of the Committee on Infectious Diseases. 27. Elk Grove Village: American Academy of Pediatrics; 2006. Haemophilus influenzae infections; pp. 310–318. [Google Scholar]
- 5.Schuchat A, Robinson K, Wenger JD, Harrison LH, Farley M, Reingold AL, Lefkowitz L, Perkins BA. Active Surveillance Team. Bacterial meningitis in the United States in 1995. N Engl J Med. 1997;337:970–976. doi: 10.1056/NEJM199710023371404. [DOI] [PubMed] [Google Scholar]
- 6.Johri S, Gorthi SP, Anand AC. Meningococcal meningitis. MJAFI. 2005;61:369–374. doi: 10.1016/S0377-1237(05)80071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison LH, Trotter CL, Ramsey ME. Global epidemiology of meningococcal disease. Vaccine. 2009;275:B51–B63. doi: 10.1016/j.vaccine.2009.04.063. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization. Meningococcal Position Paper. [Accessed on May 28, 2012];Weekly Epidemiological Record No 40. 2002 77:329–340. Available at: http://www.who.int/immunization/wer7740meningococcal_Oct02_position_paper.pdf. [Google Scholar]
- 9.Chaudhuri A, Martinez-Martin P, Kennedy PG, Andrew Seaton R, Portegies P, Bojar M, Steiner I. EFNS guideline on the management of community-acquired bacterial meningitis: report of an EFNS Task Force on acute bacterial meningitis in older children and adults. Eur J Neurol. 2008;15:649–659. doi: 10.1111/j.1468-1331.2008.02193.x. [DOI] [PubMed] [Google Scholar]
- 10.Supuran CT, Scozzafava A, Clare BW. Bacterial protease inhibitors. Med Res Rev. 2002;22:329–372. doi: 10.1002/med.10007. [DOI] [PubMed] [Google Scholar]
- 11.Drag M, Salvesen GS. Emerging principles in protease- based drug discovery. Nat Rev Drug Discov. 2010;9:690–701. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Supuran CT, Mastrolorenzo A. Bacterial zinc proteases and their inhibition. Curr Enz Inhib. 2011;7:2–23. [Google Scholar]
- 13.Nocek B, Mulligan R, Bargassa M, Collart F, Joachimiak A. Crystal structure of aminopeptidase N from human pathogen Neisseria meningitides. Proteins. 2008;70:273–279. doi: 10.1002/prot.21276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauvois B, Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4112) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev. 2006;26:88–130. doi: 10.1002/med.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luan Y, Xu W. The structure and main functions of aminopeptidase N. Curr Med Chem. 2007;14:639–647. doi: 10.2174/092986707780059571. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Xu W. Aminopeptidase N (APN/CD13) as a target for anti-cancer agent design. Curr Med Chem. 2008;15:2850–2865. doi: 10.2174/092986708786242840. [DOI] [PubMed] [Google Scholar]
- 17.Wickström M, Larsson R, Nygren P, Gullbo J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011;102:501–508. doi: 10.1111/j.1349-7006.2010.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mina-Osorio P. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med. 2008;14:361–371. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skinner-Adams TS, Stack CM, Trenholme KR, Brown CL, Grembecka J, Lowther J, Mucha A, Drag M, Kafarski P, McGowan S, Whisstock JC, Gardiner DL, Dalton JP. Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials. Trends Biochem Sci. 2010;35:53–61. doi: 10.1016/j.tibs.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Gonzales T, Robert-Baudouy J. Bacterial aminopeptidases: properties and functions. FEMS Microbiol Rev. 1996;18:319–344. doi: 10.1111/j.1574-6976.1996.tb00247.x. [DOI] [PubMed] [Google Scholar]
- 21.Drag M, Bogyo M, Ellman JA, Salvesen GS. Aminopeptidase fingerprints. An integrated approach for identification of good substrates and optimal inhibitors. J Biol Chem. 2010;285:3310–3318. doi: 10.1074/jbc.M109.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poreba M, Gajda A, Picha J, Jiracek J, Marschner A, Klein CD, Salvesen GS, Drag M. S1 pocket fingerprints of human and bacterial methionine aminopeptidases determined using fluorogenic libraries of substrates and phosphorus based inhibitors. Biochimie. 2012;94:704–710. doi: 10.1016/j.biochi.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Zervoudi E, Papakyriakou A, Georgiadou D, Evnouchidou I, Gajda A, Poreba M, Salvesen GS, Drag M, Hattori A, Swevers L, Vourloumis D, Stratikos E. Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem J. 2011;435:411–420. doi: 10.1042/BJ20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poreba M, McGowan S, Skinner-Adams TS, Trenholme KR, Gardiner DL, Whisstock JC, To J, Salvesen GS, Dalton JP, Drag M. Fingerprinting the substrate specificity of M1 and M17 aminopeptidases of human malaria, Plasmodium falciparum. PLoS One. 2012;7:e31938. doi: 10.1371/journal.pone.0031938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skinner-Adams TS, Lowther J, Teuscher F, Stack CM, Grembecka J, Mucha A, Kafarski P, Trenholme KR, Dalton JP, Gardiner DL. Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds. J Med Chem. 2007;50:6024–6031. doi: 10.1021/jm070733v. [DOI] [PubMed] [Google Scholar]
- 26.Drag M, Grembecka J, Pawełczak M, Kafarski P. alpha-Aminoalkylphosphonates as a tool in experimental optimisation of P1 side chain shape of potential inhibitors in S1 pocket of leucine- and neutral aminopeptidases. Eur J Med Chem. 2005;40:764–771. doi: 10.1016/j.ejmech.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Grembecka J, Mucha A, Cierpicki T, Kafarski P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J Med Chem. 2003;46:2641–2655. doi: 10.1021/jm030795v. [DOI] [PubMed] [Google Scholar]
- 28.Dziełak A, Pawełczak M, Mucha A. A three-component Mannich-type condensation leading to phosphinic dipeptides – extended transition state analogue inhibitors of aminopeptidases. Tetrahedron Lett. 2011;52:3141–3145. [Google Scholar]
- 29.Schneider EL, Craik CS. Positional scanning synthetic combinatorial libraries for substrate profiling. Methods Mol Biol. 2009;539:59–78. doi: 10.1007/978-1-60327-003-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGowan S, Porter CJ, Lowther J, Stack CM, Golding SJ, Skinner-Adams TS, Trenholme KR, Teuscher F, Donnelly SM, Grembecka J, Mucha A, Kafarski P, Degori R, Buckle AM, Gardiner DL, Whisstock JC, Dalton JP. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc Natl Acad Sci U S A. 2009;106:2537–2542. doi: 10.1073/pnas.0807398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner AJ. Membrane alanyl aminopeptidase. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Elsevier; London: 2004. pp. 289–294. [Google Scholar]
- 32.Collinsova M, Jiracek J. Phosphinic acid compounds in biochemistry, biology and medicine. Curr Med Chem. 2000;7:629–647. doi: 10.2174/0929867003374831. [DOI] [PubMed] [Google Scholar]
- 33.Yiotakis A, Georgiadis D, Matziari M, Makaritis A, Dive V. Phosphinic peptides: Synthetic approaches and biochemical evaluation as Zn-metalloprotease inhibitors. Curr Org Chem. 2004;8:1135–1158. [Google Scholar]
- 34.Mucha A, Kafarski P, Berlicki Ł. Remarkable potential of the alpha-aminophosphonate/phosphinate structural motif in medicinal chemistry. J Med Chem. 2011;54:5955–5980. doi: 10.1021/jm200587f. [DOI] [PubMed] [Google Scholar]
- 35.Ito K, Nakajima Y, Onohara Y, Takeo M, Nakashima K, Matsubara F, Ito T, Yoshimoto T. Crystal structure of aminopeptidase N (proteobacteria alanyl aminopeptidase) from Escherichia coli and conformational change of methionine 260 involved in substrate recognition. J Biol Chem. 2006;281:33664–33676. doi: 10.1074/jbc.M605203200. [DOI] [PubMed] [Google Scholar]
- 36.Addlagatta A, Gay L, Matthews BW. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc Natl Acad Sci U S A. 2006;103:13339–13344. doi: 10.1073/pnas.0606167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Addlagatta A, Gay L, Matthews BW. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry. 2008;47:5303–5311. doi: 10.1021/bi7022333. [DOI] [PubMed] [Google Scholar]
- 38.Fournie-Zaluski MC, Poras H, Roques BP, Nakajima Y, Ito K, Yoshimoto T. Structure of aminopeptidase N from Escherichia coli complexed with the transition-state analogue aminophosphinic inhibitor PL250. Acta Crystallogr D Biol Crystallogr. 2009;65:814–822. doi: 10.1107/S090744490901779X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumpena R, Kishor C, Ganji RJ, Addlagatta A. Discovery of α, β - and α,γ, -diamino acid scaffolds for the inhibition of M1 family aminopeptidases. ChemMedChem. 2011;6:1971–1976. doi: 10.1002/cmdc.201100298. [DOI] [PubMed] [Google Scholar]
- 40.Lejczak B, Kafarski P, Zygmunt J. Inhibition of aminopeptidases by aminophosphonates. Biochemistry. 1989;28:3549–3555. doi: 10.1021/bi00434a060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.