

Abstract
Background. Dexamethasone (Dex) has been used to reduce inflammation in preterm infants with assistive ventilation and to prevent chronic lung diseases. However, Dex treatment results in adverse effects on the brain. Since the hippocampus contains a high density of glucocorticoid receptors (GCRs), we hypothesized that Dex affects neurogenesis in the hippocampus through inflammatory mediators. Methods. Albino Wistar rat pups first received a single dose of Dex (0.5 mg/kg) on postnatal day 1 (P1) and were sacrificed on P2, P3, P5, and P7. One group of Dex-treated pups (Dex-treated D1D2) was given mifepristone (RU486, a GCR antagonist) on P1 and sacrificed on P2. Hippocampi were isolated for western blot analysis, TUNEL, cleaved-caspase 3 staining for cell counts, and morphological assessment. Control pups received normal saline (NS). Results. Dex reduced the developmental gain in body weight, but had no effect on brain weight. In the Dex-treated D1D2 group, apoptotic cells increased in number based on TUNEL and cleaved-caspase 3 staining. Most of the apoptotic cells expressed the neural progenitor cell marker nestin. Dex-induced apoptosis in P1 pups was markedly reduced (60%) by pretreatment with RU486, indicating the involvement of GCRs. Conclusion. Early administration of Dex results in apoptosis of neural progenitor cells in the hippocampus and this is mediated through GCRs.
1. Introduction
Corticosteroids are used in preterm infants to suppress inflammation, to facilitate extubation, and/or to prevent chronic lung diseases [1–3]. However, such early dexamethasone (Dex) therapy can result in an adverse neurodevelopmental outcome [4, 5]. For example, Dex treatment reduces cerebral gray matter volume without affecting white matter and the basal ganglia, suggesting that Dex affects only certain cells in the brain [6–8].
Neurons in the dentate gyrus (DG) of the hippocampus continue to divide after term and therefore remain vulnerable to the adverse effects of steroids during the early postnatal period [9–11]. Cells in the hippocampus have a high density of glucocorticoid receptors (GCRs) [12] suggesting that they could be affected by Dex [8–13]. Dex is known to change synaptic plasticity in the hippocampus [14]. As rat and human develop over different embryonic time scales [15], rat pups on P1 correspond to the human fetus at about week 22 to 24 of gestation. The equivalence in development is reflected in brain weight, neurochemistry, electroencephalography, and synaptogenesis [16]. Therefore, rat pups can be used as an animal model for human preterm infants undergoing drug exposure [17].
Here, we studied the effects of single-dose Dex therapy and the role of GCRs on hippocampal development.
2. Materials and Methods
2.1. Animals
The study was approved by the Animal Care and Use Committee of National Cheng Kung University. Time-dated pregnant Albino Wistar rats (body weight 250–300 g) were used. Food and water were provided ad libitum. The dams were allowed to deliver naturally on gestational day 21 ± 1. Animals were kept in a ventilated room at 22 ± 2°C under a 12/12 h light/dark cycle. The day of birth was designated P0.
2.2. Study Design
In a preliminary study, each litter was divided into two groups: the Dex group received a dose of Dex phosphate (0.1, 0.2, or 0.5 mg/kg, i.p.) (Oradexon, 4 mg/mL, Organon, the Netherlands) and controls received an equal volume of normal saline (NS). Compared with the controls, changes in apoptosis were only observed with Dex 0.5 mg/kg, so this dose was used in the subsequent experiments. On day 1 (P1) pups received a single dose of 0.5 mg/kg Dex or NS. On P2, P3, P5, and P7, pups were anesthetized and euthanized with 10% chloral hydrate (W/V, 300 mg/kg, Riedel-de Haen, Germany) and then infused transcardially with NS. The body and isolated brain weights were measured to the nearest milligram. Brain tissues were processed histologically following standard protocols and cut serially at 5 μm in the coronal plane.
2.3. Immunohistochemical and Immunofluorescence Staining
Brain sections were matched to the E22 coronal plates 12 to 15 of the prenatal rat brain development atlas [18]. Sections were stained by immunohistochemical (IHC) and/or immunofluorescence (IF), TUNEL, and double-IF methods, using the protocols recommended by the manufacturers or described previously [19]. The following TUNEL detection kits and primary antibodies were used: TdT-Frag EL DNA fragmentation detection kit, ApopTag Red in situ apoptosis detection kit (Calbiochem, San Diego, CA), antibrain-derived neurotrophic factor (diluted 1 : 2000), and rabbit antiglucocorticoid (diluted 1 : 50) (Santa Cruz Biotechnology, Santa Cruz, CA), mouse antinestin (diluted 1 : 200; Santa Cruz Biotechnology), anti-OX-6 (diluted 1 : 50; AbD Serotec, Oxford, UK), and anti-NeuN (diluted 1 : 200; Chemi-Con, Temecula, CA), mouse antiactin and anti-pCNA (diluted 1 : 10000 and 1 : 1000, resp.; Millipore, Billerica, MA), and cleaved caspase 3 (diluted 1 : 100; Cell Signaling, Boston, MA). All sections were matched to the same anatomical sites for comparing cell counts between the Dex-treated and control groups. For IF staining, biotinylated anti-rabbit and anti-mouse IgG (1 : 400; Victor, Burlingame, CA) were used as the secondary antibodies; for IHC staining goat anti-rabbit IgG, H&L Chain specific Texas Red conjugate and rat anti-mouse IgG H&L Chain specific fluorescein conjugate (diluted 1 : 400; Calbiochem, San Diego, CA) were used as secondary antibodies. Sections were also double stained using the above primary antibodies and dilutions.
2.4. TUNEL Assay
The sections were subjected to TUNEL (Oncogene Research, Cambridge, MA) and IF staining. The signal was detected by the streptavidin-horseradish peroxidase conjugate and diaminobenzidine reaction.
2.5. Counts of Cells Stained for TUNEL, Cleaved-Caspase 3, and NeuN
Sections were examined by light microscopy and the images captured by a video camera coupled to a desktop computer (Eclipse 80i, Nikon, Japan). TUNEL-positive cells were identified and counted at 400x magnification. The numbers of TUNEL-positive cells in the DG, cornu ammonias 1 (CA1), CA2, and CA3 in the hippocampus were counted. For comparison with the control, sections at the same level were analyzed. Four sections were counted for each pup and the results were averaged. Cleaved-caspase 3- and NeuN-positive cells were counted at 400x magnification in four, 1 mm2 areas in the DG. Data were validated by TissueGnostics FACS-like Tissue Cytometry software (Vienna, Austria). This method was also applied to IHC, IF, and double-stained cells in subsequent experiments.
2.6. Tissue Dissection and Western Blot Analysis
The hippocampi were dissected from the pups on P2, one day after Dex or NS treatment (D1D2). Western blots were performed on cytosolic and nuclear fractions of the hippocampus homogenates as described previously [19]. Briefly, 20 μg protein homogenate, determined by the Bradford protein assay (Bio-Rad, Hercules, CA), was separated by SDS-PAGE, blotted onto nitrocellulose (Hy-Bond, Amersham, Arlington Heights, IL) and blocked with nonfat dry milk. Blots were incubated with specific primary antibody, followed by incubation with horseradish-peroxidase-conjugated secondary antibodies (Abcam, Cambridge, UK) and detected by enhanced chemiluminescence (Bio-Rad). Samples were normalized to actin and proliferating cell nuclear antigen (pCNA) proteins (EMD Millipore). Each experiment was repeated at least three times.
2.7. Mifepristone (RU486) Treatment
The GCR antagonist, RU486 (Tocris Bioscience, Ellisville, MO), was dissolved in 100 mM DMSO. Thirty minutes prior to Dex injection, P1 pups received 25 mg/kg RU486 or vehicle intraperitoneally [20]. The RU486- and DMSO-treated pups (D1D2) with or without Dex treatment were euthanized on P2, and morphological and biochemical analyses were carried out as described above.
2.8. Statistical Analysis
Quantitative results are expressed as mean ± standard error (SE). Statistical analyses were performed using one- or two-way ANOVA with a multiple comparisons posttest or the Wilcoxon signed-rank test as appropriate. P values < 0.05 were considered statistically significant. The body and brain weight results were analyzed by mixed-model ANOVA with age as the within-subject factor and Dex as the between-subject factor.
3. Results
3.1. Body and Brain Weights
Pups that had received Dex (0.5 mg/kg) showed reduced body weights. In contrast, no difference in the developmental growth of brain weight was found between the Dex and control groups (Table 1).
Table 1.
Effects of dexamethasone on rat pup body weight and brain weight.
| Age (D) | Body weight, Mean ± SE, g (N = 6) | Brain weight, Mean ± SE, g (N = 6) | ||
|---|---|---|---|---|
| NS | Dex | NS | Dex | |
| P1 group | ||||
| 1a | 7.17 ± 0.07 | 6.58 ± 0.06 | ||
| 2 | 8.13 ± 0.04 | 6.63 ± 0.03 | 0.373 ± 0.013 | 0.376 ± 0.008 |
| 3 | 9.33 ± 0.14 | 7.50 ± 0.07* | 0.387 ± 0.008 | 0.373 ± 0.006 |
| 5 | 12.45 ± 0.11 | 9.83 ± 0.16† | 0.530 ± 0.013 | 0.549 ± 0.011 |
| 7 | 16.90 ± 0.33 | 12.70 ± 0.23† | 0.699 ± 0.017 | 0.721 ± 0.007 |
N: number of pups; adays of normal saline (NS) or dexamethasone (Dex) administration to the rat pups.
In P1 group, after Dex or NS injection on day 1, the pups were sacrificed on days 2, 3, 5, and 7.
Compared pups injected with dexamethasone (Dex) and normal saline (NS) by using two-way ANOVA.
*P < 0.05; † P < 0.001.
3.2. Apoptotic Cell Death
More TUNEL-positive and cleaved-caspase 3-positive cells were found in the DG of P1 D1D2 Dex-treated group (Figures 1(b) and 1(e)), with an increase to 2.7- to 3-fold (Figures 1(c) and 1(f)) with respect to the control (Figures 1(a) and 1(d)).
Figure 1.
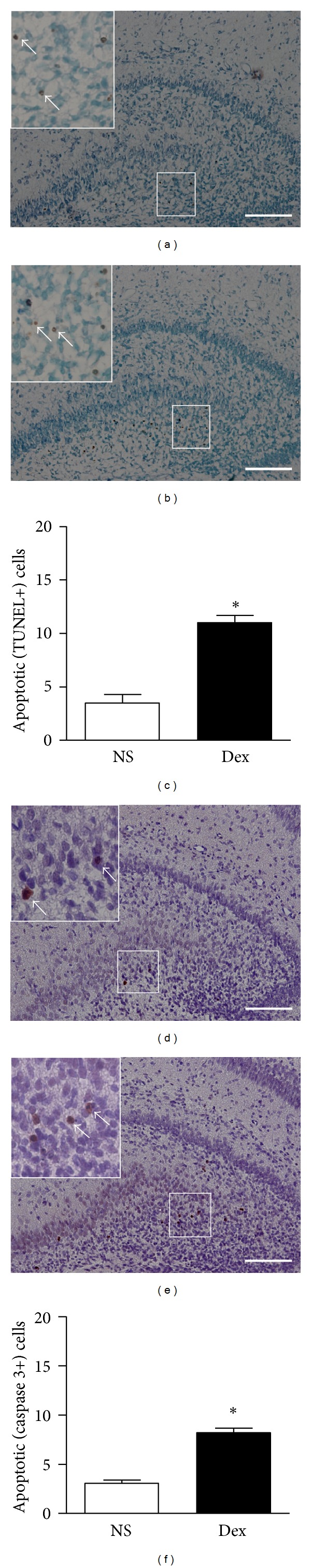
TUNEL staining of dentate gyrus (DG) P1 D1D2 pups treated with dexamethasone (Dex) or normal saline (NS) shows apoptotic cells stained brown (arrows). The Dex-treated D1D2 DG had more apoptotic cells (b) than NS control (a). High magnification photomicrographs of TUNEL-positive cells are shown in the upper left corner. TUNEL-positive cell counts revealed that the number of apoptotic cells in the Dex-treated D1D2 DG was increased when compared to that of NS control pups (n = 6, *P < 0.05) (c). Cleaved-caspase 3 staining of the DG from P1 D1D2 pups with or without Dex treatment showed apoptotic cells (arrows) ((d) and (e)). High magnification photomicrographs of cleaved-caspase 3-positive cells are shown in the upper left corner. The numbers of apoptotic cells in Dex-treated pups were increased compared to those of NS control (f) (n = 6, *P < 0.05). Magnification, 200x; scale bars, 100 μm; inset magnification, 400x. P1 D1D2: P1 pups receiving treatment on postnatal day 1 and sacrificed on day 2.
3.3. Cell Counts and Coexpression of Apoptosis and Neuronal Maturation Markers
Representative results of TUNEL, nestin, and NeuN staining are shown in Figure 2. We found about twice as many TUNEL-positive cells in the Dex-treated D1D2 group (38.1 ± 1.1) compared with the control (21.8 ± 1.2; P < 0.05) (Figure 3(a)). The Dex-treated group had a greater proportion of TUNEL-positive cells (0.75 ± 0.01) that coexpressed nestin than control (0.61 ± 0.01) (Figure 3(b)). The proportion of TUNEL-positive cells coexpressing NeuN was also greater in the Dex-treated group (0.68 ± 0.10) than in control (0.64 ± 0.01; P < 0.05) (Figure 3(c)).
Figure 2.
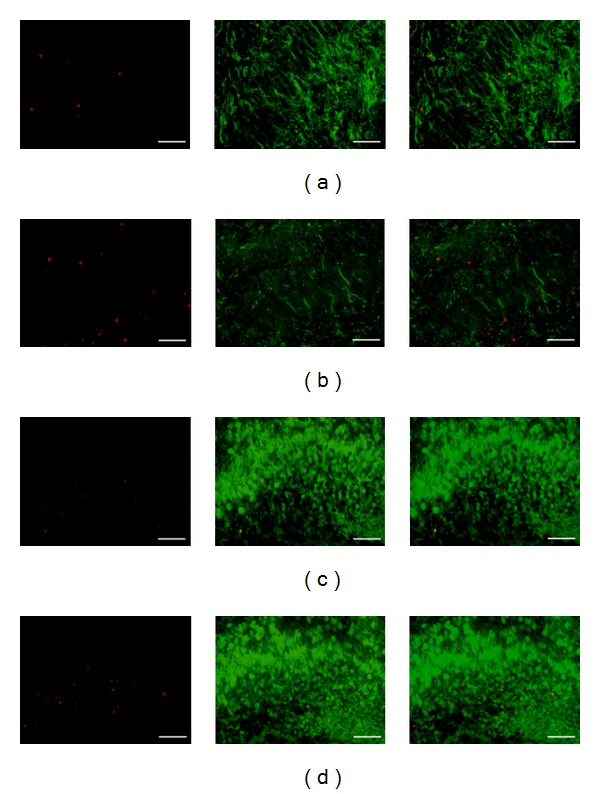
TUNEL, nestin, and NeuN expression in the hippocampus of D1D2 pups treated with normal saline (NS) or Dex. Representative double-IF micrographs demonstrate TUNEL (red) and nestin (green) staining ((a) and (b)), and TUNEL (red) and NeuN (green) staining ((c) and (d)). Cells coexpressing two proteins are merged and show as yellow or orange. Magnification, 400x, scale bars, 50 μm. Dex-treated D1D2: P1 pups receiving dexamethasone on postnatal day 1 and sacrificed on day 2.
Figure 3.
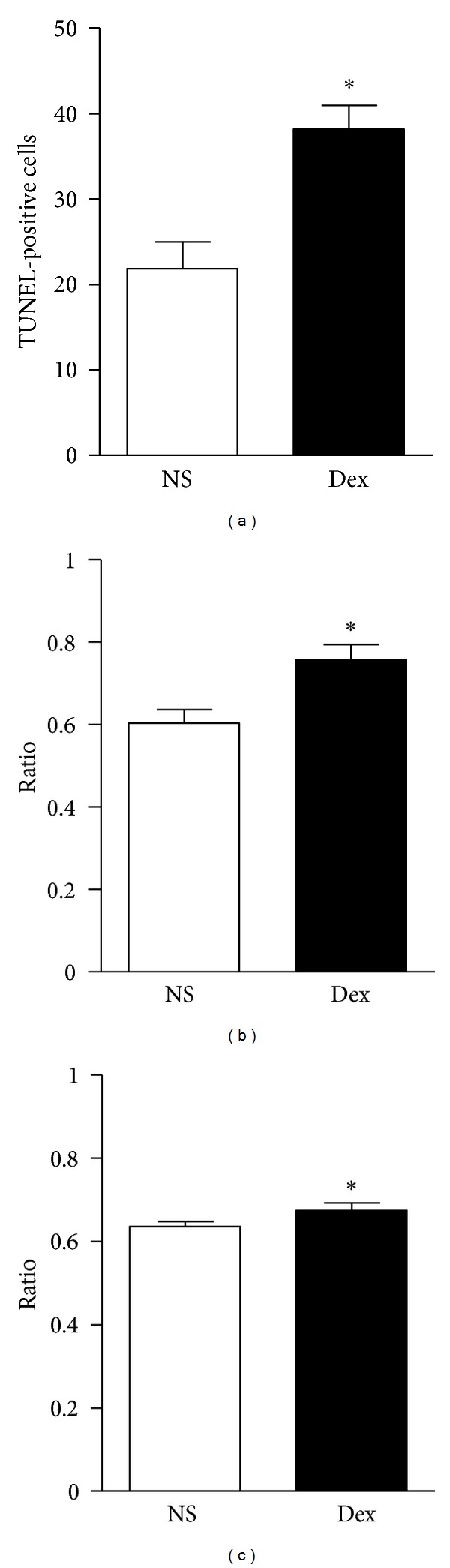
TUNEL-positive cells were more numerous in the hippocampus of Dex-treated D1D2 pups than in control (n = 6, *P < 0.05) (a) and the ratio of TUNEL-positive cells which coexpressed nestin to total TUNEL-positive cells was higher in the Dex treated than in the control groups (n = 6, *P < 0.05) (b). The ratio of TUNEL-positive cells coexpressing NeuN to total TUNEL-positive cells was higher than that of control (n = 6, *P < 0.05) (c). Dex-treated D1D2: P1 pups receiving dexamethasone on postnatal day 1 and sacrificed on day 2.
3.4. Glucocorticoid Receptors (GCRs) and Mifepristone (RU486) in Dex-Induced Apoptosis
Western blot analysis showed that the nuclear fractions of GCRs in the hippocampus were upregulated in the Dex-treated D1D2 group (Figure 4). The Dex-retarded developmental gain in body weight was blocked by RU486 while neither Dex nor RU486 affected the brain weight in this group (Table 2). Furthermore, Dex-induced apoptosis in the P1 D1D2 group was reduced by the preadministration of RU486 (Figure 5). Dex treatment alone increased the apoptotic cell count (41.3 ± 0.60) compared to the control (23.1 ± 0.18); P < 0.05); the number of apoptotic cells in pups treated with DMSO (22.4 ± 0.3) or DMSO plus Dex (41.9 ± 0.56) was similar to that in pups treated with NS or Dex alone (§ P < 0.05). RU486 treatment had no additional effect on apoptosis when compared with the NS or DMSO group. Pretreatment with RU486 followed by Dex reduced the apoptotic cell count (29.2 ± 0.45) (P < 0.05) (Figure 5).
Figure 4.
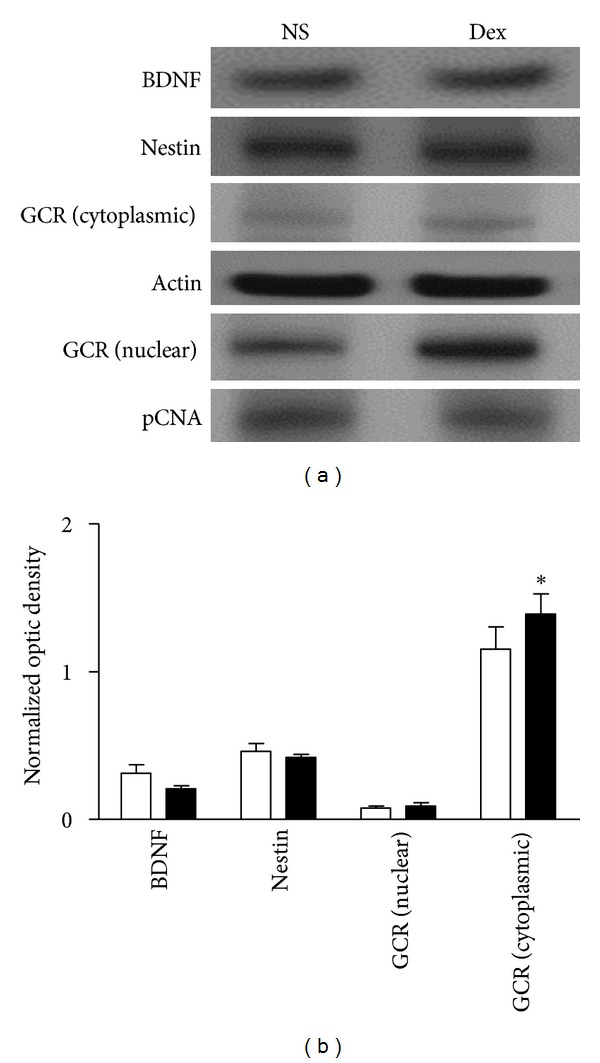
Dexamethasone (Dex) treatment increased levels of nuclear glucocorticoid receptors (GCRs), but not cytosolic GCRs, total brain-derived neurotrophic factor (BDNF) or total nestin in the hippocampus of P1 pups. (a) Representative western blots of hippocampal protein extracts obtained from P1 D1D2 pups, normal saline (NS, control) (left lane), and Dex (right lane). (b) Signals from BDNF, nestin, and cytosolic GCRs were normalized to actin and signals from nuclear GCRs were normalized to pCNA. Quantitative results showed no difference in BDNF, nestin, and cytosolic GCR expression between Dex-treated and control pups. The nuclear fraction of GCR expression in Dex-treated pups differed from control (n = 6, *P < 0.05). P1 D1D2: P1 pups receiving treatment on postnatal day 1 and sacrificed on day 2; white and black bars represent control and Dex-treated pups, respectively.
Table 2.
Effects of dexamethasone and RU486 on D1D2 rat pup body weight and brain weight.
| Treatment | Body weight, Mean ± SE, g (N = 6) | Brain weight, Mean ± SE, g, (N = 6) |
|---|---|---|
| NS | 7.75 ± 0.04 | 0.314 ± 0.004 |
| Dex | 6.75 ± 0.08* | 0.293 ± 0.004 |
| DMSO | 7.50 ± 0.09 | 0.309 ± 0.004 |
| RU486 | 7.16 ± 0.08 | 0.312 ± 0.004 |
| DMSO/Dex | 6.83 ± 0.04 | 0.301 ± 0.004 |
| RU486/Dex | 7.08 ± 0.03 | 0.319 ± 0.004 |
N: number of pups.
Compared pups injected with dexamethasone (Dex) and normal saline (NS) by using Wilcoxon rank sum test, *P < 0.05.
D1D2: administration on postnatal day 1 and sacrificed on day 2; RU486: mifepristone;
DMSO: dimethyl sulfoxide; DMSO/Dex: dimethyl sulfoxide plus dexamethasone;
RU486/Dex: mifepristone plus dexamethasone.
Figure 5.
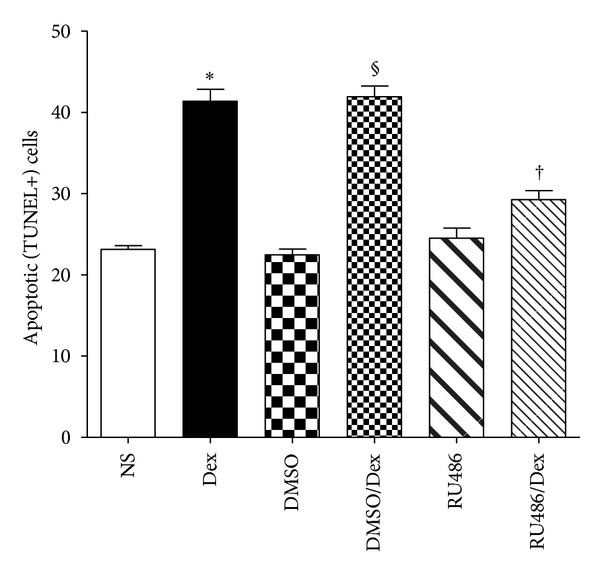
Dexamethasone-(Dex-) induced apoptosis in P1 D1D2 pups was reduced by preadministration of RU486. Dex treatment increased apoptotic cell counts in the hippocampus compared to control (n = 6, *P < 0.05); P1 D1D2 pups treated with DMSO or DMSO plus Dex showed results similar to those from pups treated with either normal saline (NS) or Dex alone (n = 6, § P < 0.05). RU486 treatment showed apoptotic cell counts similar to those from pups treated with NS or DMSO. Pups treated with RU486 plus Dex had lower apoptotic cell counts than those with Dex or DMSO plus Dex (n = 6, † P < 0.05). P1 D1D2: P1 pups receiving treatment on postnatal day 1 and sacrificed on day 2.
3.5. Identification of Inflammatory Cells
Eosin/hematoxylin or IHC staining did not reveal any inflammatory cells in the NS and Dex-treated D1D2. Positive controls after implantation of rat brain tumor cells revealed OX-6 positive cells (stained brown) which helped identification of microglia in the brain (Figure 6).
Figure 6.
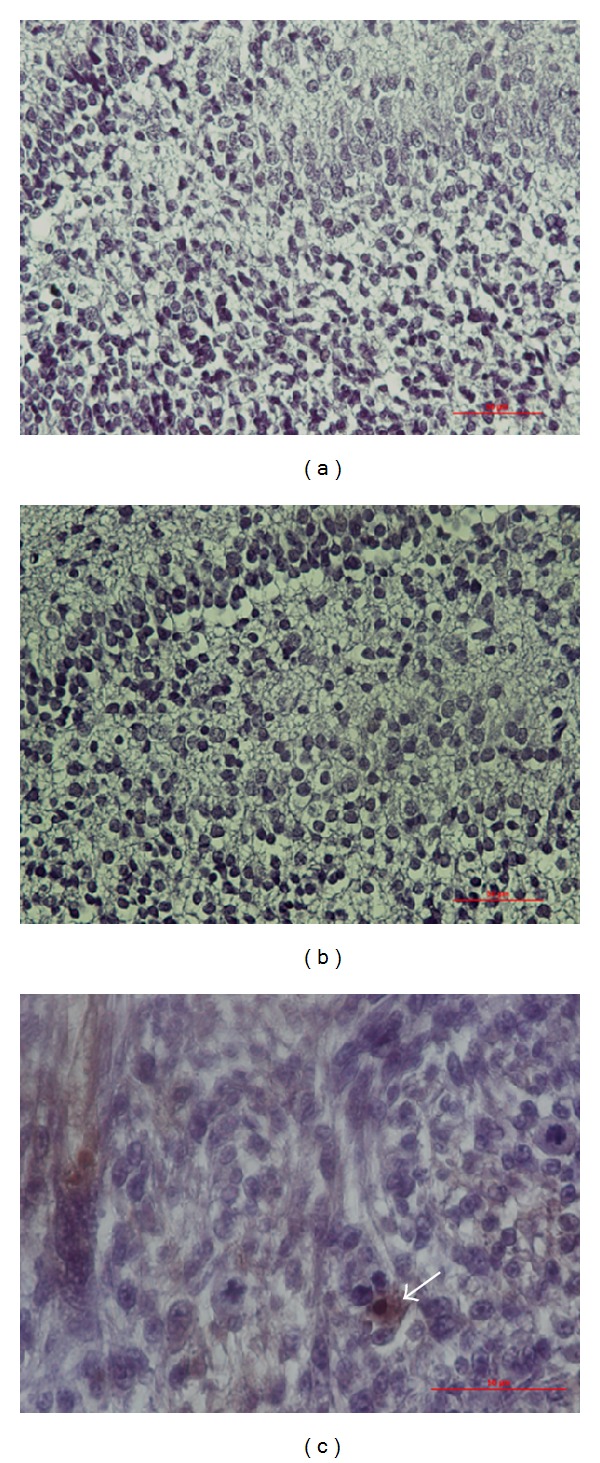
OX-6 IHC staining in the DG. No OX-6 positive cells were identified in P1 D1D2 pups treated with either NS (a) or Dex (b). Brain tumor implant staining as a positive control showed OX-6 positive cells stained brown (c), arrow). Magnification, 400x; scale bar, 50 μm.
4. Discussion
Steroids have long been used in the treatment of respiratory problems in preterm infants [21]. The complications of adverse neurological effects (such as increased risk of cerebral palsy and neurodevelopment impairment [4, 22, 23] demand the reevaluation of steroid-based therapeutic strategies in postnatal practice [23–26]. The safe timing and dosage of Dex remain undecided for preterm patients [4]. Our results from the P1 rat pups, equivalent to 24-week preterm infants, showed that Dex retarded the developmental gain in body weight, consistent with earlier reports [27–29]. Furthermore, neonatal Dex exposure leads to delayed neurodevelopment and physical maturation, suggesting that Dex permanently alters neuronal functions during this period, particularly those associated with the hypothalamic-pituitary-adrenal axis [27–29]. Other studies showed that neonatal Dex exposure reduces brain weight [10, 28–32]. On the contrary, our data showed no such change. We attribute this discrepancy to the differences in timing, dosage regimes, and/or the preparation of Dex [28, 31, 32]. Among clinicians, the general consensus is that a lower dose of Dex (0.1–0.2 mg/kg/day) facilitates tracheal extubation and reduces the risk of chronic-lung disease. This study revealed no deleterious effects on the brain with single low doses of Dex (0.1–0.2 mg/kg), although it became harmful at a higher dose (0.5 mg/kg). The subgranular zone of the DG contains a reserve of neuroglial progenitor cells [33]. Apoptosis is crucial during neuronal development by eliminating excess cells and ensuring proper synaptic connectivity [34, 35]. GCRs are known to be involved in Dex-induced apoptosis [36] and are present at high levels in the hippocampus where progenitor cells capable of dividing reside in the DG. Our results showed increased apoptosis throughout the DG, suggesting that perinatal development in the hippocampus is vulnerable even to a single dose of Dex when given at a critical time. How this effect is related to the GCR density, receptor types, and the proliferation of progenitor cells remains to be studied.
It has been reported that administration of 3.0 mg/kg Dex to P7 mice increases the apoptosis of cerebellar progenitor cells and reduces the number of cerebellar neurons [37, 38]. It is likely that the dosage and timing of Dex treatment and vulnerability of neural progenitor cells to glucocorticoids together determine the effects on neonatal brain development.
Exposure to Dex in the neonatal period results in marked apoptosis among the nestin-expressing cells in the DG [39]. The importance of Dex-induced apoptosis in the hippocampus and the type of cells affected remain unknown. Our results from double staining in the Dex-treated pups showed higher ratios of cells coexpressing TUNEL and nestin to TUNEL-positive cells, indicating that Dex-induced apoptosis affects the neuroprogenitor cells. This result is consistent with previous reports that neuroprogenitor cells are sensitive to Dex during the early neonatal period [39, 40]. Since the ratio of TUNEL and NeuN coexpressing cells to TUNEL-positive cells also increased, Dex treatment might also cause apoptosis in mature neurons and be associated with a transient and acute slowing of cell proliferation during hippocampal development [41, 42]. Whether these neurons were derived from the proliferating progenitors cells or represent existing mature neurons in the DG remains to be determined.
Mifepristone (or RU-486) is a synthetic steroid with both antiprogesterone and antiglucocorticoid properties. In a recent study, mifepristone was the only GCR antagonist found to increase both mineralocorticoid receptor (MCR) and GCR binding in the rat frontal cortex [43]. The effects of mifepristone on corticosteroid receptor expression could explain the neurocognitive improvement it is reported to induce [43, 44]. RU486 has no effect on Dex regarding thymocyte composition and maturation [45]. MCR-mediated responses to glucocorticoids, rather than GCRs, are important in steroid-responsive hearing disorders [46]. GCR activity levels are high in the hippocampus during the first postnatal week [40]. The anti-inflammatory effects of glucocorticoids require the presence of GCRs [47]. Interestingly, results of these studies and those of our own (60% reduction in Dex-induced apoptosis by RU486) are consistent, suggesting that the type of receptor and the timing of Dex treatment determine the effects of glucocorticoids on the hippocampus. Examining pup brain tissues stained with eosin/hematoxylin or IHC revealed no inflammatory cells in the NS and Dex-treated D1D2 groups. These results suggest that while GCRs are likely to be key players in Dex-induced neuroprogenitor cell death, inflammatory cells are not.
In summary, brain development is a dynamic process in which the growth spurt, differentiation, and cell responses to endogenous (and exogenous) steroids occur at critical times. Species differences add further complexity to the process. These species-specific developmental schedules allowed the design of animal models to study the effects of drug treatment in preterm babies, as in this study. Here, we have demonstrated that timing is a major factor in determining Dex-induced apoptosis in the hippocampus, the vulnerable cells are neuronal precursors, and the process is partly regulated by GCRs. We also provided cytological evidence that the administration of a single dose of Dex can result in deleterious effects in the brain.
Acknowledgments
The authors thank Dr. K. S. Hsu for help with experimental design, Ms. Pei-Yu Lee for assistance in image acquisition and analysis using FACS-like Tissue Cytometry at the Center of Clinical Medicine, NCKU Hospital and support from an NCKU Academic Summit Program, Individual Research Project/Integrated Research Project (D100-35001), and the National Science Council (NSC-92-2314-B-006-073).
List of Abbreviations
- Dex:
Dexamethasone
- GCR:
Glucocorticoid receptor
- DG:
Dentate gyrus
- CA:
Cornu ammonias
- RU486:
Mifepristone
- BDNF:
Brain-derived neurotrophic factor
- NS:
Normal saline
- P1/P0:
Day1/day 0 newborn pups
- D1D2:
Pups receiving dexamethasone injection on day 1 and sacrificed on day 2
- IHC:
Immunohistochemical
- IF:
Immunofluorescence
- TUNEL:
Terminal deoxynucleotidyl transferase dUTP nick end labeling.
References
- 1.Halliday HL, Ehrenkranz RA, Doyle LW. Early (<8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews. 2009;(1) doi: 10.1002/14651858.CD001146.pub2.CD001146 [DOI] [PubMed] [Google Scholar]
- 2.Doyle LW, Ehrenkranz RA, Halliday HL. Dexamethasone treatment after the first week of life for bronchopulmonary dysplasia in preterm infants: a systematic review. Neonatology. 2010;98(4):289–296. doi: 10.1159/000286212. [DOI] [PubMed] [Google Scholar]
- 3.Halliday HL, Ehrenkranz RA, Doyle LW. Late (>7 days) postnatal corticosteroids for chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews. 2009;(1):p. CD001145. doi: 10.1002/14651858.CD001145.pub2. [DOI] [PubMed] [Google Scholar]
- 4.Shinwell ES, Eventov-Friedman S. Impact of perinatal corticosteroids on neuromotor development and outcome: review of the literature and new meta-analysis. Seminars in Fetal and Neonatal Medicine. 2009;14(3):164–170. doi: 10.1016/j.siny.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Yeh TF, Lin YJ, Huang CC, et al. Early dexamethasone therapy in preterm infants: a follow-up study. Pediatrics. 1998;101(5, article E7) doi: 10.1542/peds.101.5.e7. [DOI] [PubMed] [Google Scholar]
- 6.Murphy BP, Inder TE, Huppi PS, et al. Impaired cerebral cortical gray matter growth after treatment with dexamethasone for neonatal chronic lung disease. Pediatrics. 2001;107(2):217–221. doi: 10.1542/peds.107.2.217. [DOI] [PubMed] [Google Scholar]
- 7.Ajayi-Obe M, Saeed N, Cowan FM, Rutherford MA, Edwards AD. Reduced development of cerebral cortex in extremely proterm infants. The Lancet. 2000;356(9236):1162–1163. doi: 10.1016/s0140-6736(00)02761-6. [DOI] [PubMed] [Google Scholar]
- 8.Modi N, Lewis H, Al-Naqeeb N, Ajayi-Obe M, Dore CJ, Rutherford M. The effects of repeated antenatal glucocorticoid therapy on the developing brain. Pediatric Research. 2001;50(5):581–585. doi: 10.1203/00006450-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Benesova O, Pavlik A. Perinatal treatment with glucocorticoids and the risk of maldevelopment of the brain. Neuropharmacology. 1989;28(1):89–97. doi: 10.1016/0028-3908(89)90073-7. [DOI] [PubMed] [Google Scholar]
- 10.Baud O. Postnatal steroid treatment and brain development. Archives of Disease in Childhood. 2004;89(2):F96–F100. doi: 10.1136/adc.2003.028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiological Reviews. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 12.Eddie Gabry K, Chrousos G, Gold PW. The hypothalamic-pituitary-adrenal (HPA) axis: a major mediator of the adaptive responses to stress. NeuroImmune Biology. 2003;3:379–414. [Google Scholar]
- 13.Hagberg H, Jacobsson B. Brain injury in preterm infants—what can the obstetrician do? Early Human Development. 2005;81(3):231–235. doi: 10.1016/j.earlhumdev.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Lin HJ, Huang CC, Hsu KS. Effects of neonatal dexamethasone treatment on hippocampal synaptic function. Annals of Neurology. 2006;59(6):939–951. doi: 10.1002/ana.20885. [DOI] [PubMed] [Google Scholar]
- 15.Yeh TF, Lin YJ, Lin HC, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. The New England Journal of Medicine. 2004;350(13):1304–1313. doi: 10.1056/NEJMoa032089. [DOI] [PubMed] [Google Scholar]
- 16.Amaral D, Witter M. Hippocampal formation. In: Paxinos G, editor. Rat Nervous System. Waltham, Mass, USA: Academy Press; 1994. pp. 443–494. [Google Scholar]
- 17.Hagberg H, Bona E, Gilland E, Puka-Sundvall M. Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatrica. 1997;86(422):85–88. doi: 10.1111/j.1651-2227.1997.tb18353.x. [DOI] [PubMed] [Google Scholar]
- 18.Altman J, Bayer SA. Atlas of Prenatal Rat Brain Development. Boca Raton, Fla, USA: CRC Press; 1995. [Google Scholar]
- 19.Lin WY, Chang YC, Lee HT, Huang CC. CREB activation in the rapid, intermediate, and delayed ischemic preconditioning against hypoxic-ischemia in neonatal rat. Journal of Neurochemistry. 2009;108(4):847–859. doi: 10.1111/j.1471-4159.2008.05828.x. [DOI] [PubMed] [Google Scholar]
- 20.Pugh CR, Fleshner M, Rudy JW. Type II glucocorticoid receptor antagonists impair contextual but not auditory-cue fear conditioning in juvenile rats. Neurobiology of Learning and Memory. 1997;67(1):75–79. doi: 10.1006/nlme.1996.3741. [DOI] [PubMed] [Google Scholar]
- 21.Haddad HM, Hsia DY, Gellis SS. Studies on respiratory rate in the newborn, its use in the evaluation of respiratory distress in infants of diabetic mothers. Pediatrics. 1956;17(2):204–213. [PubMed] [Google Scholar]
- 22.Merritt TA, Stuard ID, Puccia J, et al. Newborn tracheal aspirate cytology: classification during respiratory distress syndrome and bronchopulmonary dysplasia. Journal of Pediatrics. 1981;98(6):949–956. doi: 10.1016/s0022-3476(81)80603-8. [DOI] [PubMed] [Google Scholar]
- 23.Shinwell ES, Karplus M, Reich D. Early postnatal dexamethasone treatment and increased incidence of cerebral palsy. Archives of Disease in Childhood. 2000;83(3):F177–F181. doi: 10.1136/fn.83.3.F177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinwell ES, Karplus M, Bader D, et al. Neonatologists are using much less dexamethasone. Archives of Disease in Childhood. 2003;88(5):F432–F433. doi: 10.1136/fn.88.5.F432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh MC, Yao Q, Horbar JD, Carpenter JH, Lee SK, Ohlsson A. Changes in the use of postnatal steroids for bronchopulmonary dysplasia in 3 large neonatal networks. Pediatrics. 2006;118(5):e1328–e1335. doi: 10.1542/peds.2006-0359. [DOI] [PubMed] [Google Scholar]
- 26.Chrysis D, Ritzen EM, Sävendahl L. Growth retardation induced by dexamethasone is associated with increased apoptosis of the growth plate chondrocytes. Journal of Endocrinology. 2003;176(3):331–337. doi: 10.1677/joe.0.1760331. [DOI] [PubMed] [Google Scholar]
- 27.Neal CR, Jr., Weidemann G, Kabbaj M, Vázquez DM. Effect of neonatal dexamethasone exposure on growth and neurological development in the adult rat. American Journal of Physiology. 2004;287(2):R375–R385. doi: 10.1152/ajpregu.00012.2004. [DOI] [PubMed] [Google Scholar]
- 28.Flagel SB, Vázquez DM, Watson SJ, Jr., Neal CR., Jr. Effects of tapering neonatal dexamethasone on rat growth, neurodevelopment, and stress response. American Journal of Physiology. 2002;282(1):R55–R63. doi: 10.1152/ajpregu.2002.282.1.R55. [DOI] [PubMed] [Google Scholar]
- 29.Karemaker R, Kavelaars A, ter Wolbeek M, et al. Neonatal dexamethasone treatment for chronic lung disease of prematurity alters the hypothalamus-pituitary-adrenal axis and immune system activity at school age. Pediatrics. 2008;121(4):e870–e878. doi: 10.1542/peds.2007-2454. [DOI] [PubMed] [Google Scholar]
- 30.Holson RR, Gough B, Sullivan P, Badger T, Sheehan DM. Prenatal dexamethasone or stress but not ACTH or corticosterone alter sexual behavior in male rats. Neurotoxicology and Teratology. 1995;17(4):393–401. doi: 10.1016/0892-0362(94)00074-n. [DOI] [PubMed] [Google Scholar]
- 31.Kanagawa T, Tomimatsu T, Hayashi S, et al. The effects of repeated corticosteroid administration on the neurogenesis in the neonatal rat. American Journal of Obstetrics and Gynecology. 2006;194(1):231–238. doi: 10.1016/j.ajog.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Baud O, Laudenbach V, Evrard P, Gressens P. Neurotoxic effects of fluorinated glucocorticoid preparations on the developing mouse brain: role of preservatives. Pediatric Research. 2001;50(6):706–711. doi: 10.1203/00006450-200112000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. Journal of Neurobiology. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Deshmukh M, Johnson EM. Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Molecular Pharmacology. 1997;51(6):897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 35.Sadoul R. Bcl-2 family members in the development and degenerative pathologies of the nervous system. Cell Death and Differentiation. 1998;5:805–815. doi: 10.1038/sj.cdd.4400438. [DOI] [PubMed] [Google Scholar]
- 36.Amaral JD, Solá S, Steer CJ, Rodriques CM. Role of nuclear steroid receptors in apoptosis. Current Medicinal Chemistry. 2009;16:3886–3902. doi: 10.2174/092986709789178028. [DOI] [PubMed] [Google Scholar]
- 37.Maloney SE, Noguchi KK, Wozniak DF, Fowler SC, Farber NB. Long-term effects of multiple glucocorticoid exposures in neonate mice. Behavioral Sciences. 2011;1:4–30. doi: 10.3390/behavsci1010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death and Differentiation. 2008;15(10):1582–1592. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu S, Patchev AV, Wu Y, et al. Depletion of the neural precursor cell pool by glucocorticoids. Annals of Neurology. 2010;67(1):21–30. doi: 10.1002/ana.21812. [DOI] [PubMed] [Google Scholar]
- 40.Rosenfeld P, van Eekelen JA, Levine S, de Kloet ER. Ontogeny of the type 2 glucocorticoid receptor in discrete rat brain regions: an immunocytochemical study. Brain Research. 1988;470(1):119–127. doi: 10.1016/0165-3806(88)90207-6. [DOI] [PubMed] [Google Scholar]
- 41.Tijsseling D, Wijnberger LDE, Derks DB, et al. Effects of antenatal glucocorticoid therapy on hippocampal histology of preterm infants. Plos ONE. 2012;7(3) doi: 10.1371/journal.pone.0033369.e33369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rayen I, van den Hove DL, Prickaerts J, Steinbusch HW, Pawluski JL. Fluoxetine during development reverses the effects of prenatal stress on depressive-like behavior and hippocampal neurogenesis in adolescence. Plos ONE. 2011;6(9) doi: 10.1371/journal.pone.0024003.e24003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachmann CG, Linthorst AC, Holsboer F, Reul JM. Effect of chronic administration of selective glucocorticoid receptor antagonists on the rat hypothalamic-pituitary-adrenocortical axis. Neuropsychopharmacology. 2003;28(6):1056–1067. doi: 10.1038/sj.npp.1300158. [DOI] [PubMed] [Google Scholar]
- 44.Llorens-Martín M, Trejo JL. Mifepristone prevents stress-induced apoptosis in newborn neurons and increases AMPA receptor expression in the dentate gyrus of C57/BL6 mice. PloS ONE. 2011;6(11) doi: 10.1371/journal.pone.0028376.e28376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berki T, Palinkas L, Boldizsar F, Németh P. Glucocorticoid, (GC) sensitivity and GC receptor expression differ in thymocyte subpopulations. International Immunology. 2002;14:463–469. doi: 10.1093/intimm/14.5.463. [DOI] [PubMed] [Google Scholar]
- 46.Trune DR, Kempton JB. Blocking the glucocorticoid receptor with RU-486 does not prevent glucocorticoid control of autoimmune mouse hearing loss. Audiology and Neurotology. 2009;14(6):423–431. doi: 10.1159/000241899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuckermann JP, Kleiman A, Moriggl R, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. Journal of Clinical Investigation. 2007;117(5):1381–1390. doi: 10.1172/JCI28034. [DOI] [PMC free article] [PubMed] [Google Scholar]