

Abstract
The S. cerevisiae RNase III enzyme Rnt1p preferentially binds to dsRNA hairpin substrates with a conserved (A/u)GNN tetraloop fold, via shape-specific interactions by its dsRBD helix α1 to the tetraloop minor groove. To investigate whether conformational flexibility in the dsRBD regulates the binding specificity, we determined the backbone dynamics of the Rnt1p dsRBD in the free and AGAA hairpin-bound states using NMR spin relaxation experiments. The intrinsic μs-ms timescale dynamics of the dsRBD suggests that helix α1 undergoes conformational sampling in the free state, with large dynamics at some residues in the α1-β1 loop (α1-β1 hinge). To correlate free dsRBD dynamics with structural changes upon binding, we determined the solution structure of the free dsRBD used in the previously determined RNA-bound structures. The Rnt1p dsRBD has an extended hydrophobic core comprising helix α1, the α1-β1 loop, and helix α3. Analysis of the backbone dynamics and structures of the free and bound dsRBD reveals that slow-timescale dynamics in the α1-β1 hinge are associated with concerted structural changes in the extended hydrophobic core that govern binding of helix α1 to AGAA tetraloops. The dynamic behavior of the dsRBD bound to a longer AGAA hairpin reveals that dynamics within the hydrophobic core differentiate between specific and non-specific sites. Mutations of residues in the α1-β1 hinge result in changes to the dsRBD stability and RNA-binding affinity, and cause defects in snoRNA processing in vivo. These results reveal that dynamics in the extended hydrophobic core are important for binding site selection by the Rnt1p dsRBD.
Keywords: NMR, Rnt1, RNA binding domain, protein-RNA, spin relaxation
INTRODUCTION
RNase III enzymes process double-stranded RNA (dsRNA) substrates for many non-coding RNA precursors, including pre-rRNAs, -snoRNAs, and -snRNAs, as well as miRNA and siRNA.1–3 RNase III family members typically have one or two double-stranded RNA-binding domains (dsRBDs) and one or two endonuclease domains (endoNDs), which cleave dsRNA substrates as a dimer. Each endoND cleaves the backbone of one RNA strand via a two-Mg2+ catalytic mechanism, leaving a two-nucleotide 3′ overhang on processed RNAs, a defining feature of RNase III cleavage.4,5 In S. cerevisiae, Rnt1p is the only characterized RNase III enzyme, and is involved in the processing of the pre-rRNA precursor,6,7 and of the precursors of many snoRNAs8–10 and snRNAs.11–14 For most of these non-coding RNAs, Rnt1p cleavage provides a site for subsequent processing by the Rat1p or Xrn1p exonucleases or the exosome.14–17 Rnt1p activity is also important for the quality control of mRNA, processing unspliced mRNAs.18,19 Rnt1p cleavage can influence transcription termination by cleaving stem-loop structures that are found downstream from normal polyadenylation signals.20,21 Finally, Rnt1p cleavage limits the expression of a number of mRNAs.15,22–24 Thus, Rnt1p activity controls the production of a large number of cellular transcripts. Rnt1p has a characteristic substrate specificity, cleaving the dsRNA stem of (A/u)GNN tetraloop hairpins 14 and 16 bp from the conserved tetraloop on its RNA targets.25,26 Selective binding by the dsRBD to (A/u)GNN tetraloop hairpins, a unique feature of Rnt1p, determines target site selection.26
Although S. cerevisiae does not have RNAi machinery, other budding yeasts carry out RNAi with a Dicer, called Dcr1, which is evolutionarily related to Rnt1.27 Dcr1 resembles Rnt1 in having a single endoND that dimerizes intermolecularly, unlike other eukaryotic Dicers, which have two tandem endoNDs that dimerize intramolecularly. The Dcr1 endoND is followed by a dsRBD, but has an additional dsRBD separated by a long linker sequence. How these dsRBDs contribute to substrate recognition and processing is unknown, although the endoND-adjacent dsRBD in Dcr1 is required for siRNA processing. Intriguingly, Candida albicans Dcr1 has been found to carry out both RNAi and Rnt1 functions.28
Canonical dsRBDs have an αβββ α secondary structure motif and interact with a broad range of dsRNA substrates. Residues in helix α1, the β1-β2 loop and helix α2 mediate interactions with successive RNA minor, major, and minor grooves on one face of the duplex, respectively.29 The dsRBDs generally recognize dsRNA without any additional substrate specificity, a binding mode typified by the crystal structure of the Xlrbpa dsRBD in complex with A-form dsRNA.30 In contrast, the structure of human ADAR dsRBD in complex with dsRNA revealed that this and other dsRBDs, notably A. aeolicus RNase III dsRBD, can have some sequence specificity for their dsRNA substrates though hydrophobic contacts between dsRBD side chains and nucleotide bases.31 Additionally, some dsRBDs have a canonical dsRBD fold but do not independently bind to dsRNA with high affinity, such as the human Drosha dsRBD.32
The Rnt1p dsRBD is unique among dsRBDs studied to date in recognizing RNA hairpins capped by a tetraloop with the consensus sequence (A/u)GNN,25 through structure-specific recognition of the tetraloop fold by helix α1, with no base-specific contacts.33 Binding of the Rnt1p dsRBD to the conserved tetraloop fold is required for correct substrate cleavage,25 although cleavage independently from the presence of the tetraloop can be observed in vitro in specific conditions.24,26 The structure of the Rnt1p dsRBD differs from canonical dsRBDs in having an additional C-terminal helix α3 that has been proposed to contribute to specific recognition of Rnt1p substrates by indirectly reshaping the RNA binding surface.33,34 Our recent structure of the dsRBD bound to an AAGU tetraloop hairpin,35 a specific but non-canonical substrate,8,36 showed that the dsRBD employs a single binding mode for AGAA and AAGU tetraloop hairpins, with the AAGU tetraloop adopting the same shape as the AGAA tetraloop upon binding by the dsRBD. The identification of a single binding mode for two substrates with dissimilar sequences and conformations in the free state provided further evidence for the structure-specific, rather than sequence-specific, nature of the interaction between the Rnt1p dsRBD and target RNAs. This study further showed that conformational changes in the tetraloop-binding helix α1 are important for allowing the dsRBD to adopt the bound conformation.35
The dynamic properties of biomolecules often contribute to their biological functions by enabling conformational changes necessary for binding and catalysis. Moreover, conformational flexibility can allow proteins to sample functionally important alternative conformations.37,38 Here, we have investigated the intrinsic backbone dynamics of the Rnt1p dsRBD using NMR 15N spin relaxation measurements. Further, we have examined the relationship between dsRBD dynamics and structural changes that occur upon binding to AGAA tetraloop hairpins. Slow-timescale dynamics of the dsRBD indicate that helix α1, which interacts with the tetraloop in the complex, undergoes conformational sampling in the free state, with particularly large dynamics at a hinge within the α1-β1 loop. Upon binding to RNA, dynamics at the α1-β1 hinge are partially quenched. We have determined the solution structure of the free dsRBD for the same construct previously used for the structures of Rnt1p dsRBD/RNA complexes, enabling precise comparison between free and bound states. Changes in the structure and dynamics of the dsRBD upon binding to an AGAA hairpin substrate for regions distal to the binding face reveal a network of hydrophobic residues within α1, the α1-β1 loop, and α3 with specific dynamic properties that facilitate binding to specific tetraloops. Mutation of individual residues in the α1-β1 hinge causes changes in dsRBD conformation and stability and results in defects in snoRNA processing in vivo. These results show that the intrinsic dynamics of the dsRBD contributes to the selection of specific tetraloop-hairpin substrates by Rnt1p, and that helix α1, the α1-β1 loop, and helix α3 cooperatively contribute to regulation of the dynamics of the RNA-binding region of the dsRBD through interactions within an extended hydrophobic core.
MATERIALS AND METHODS
NMR sample preparation
The Rnt1p dsRBD (residues 366–453 of Rnt1p) and single residue mutants were expressed as a glutathione S-transferase (GST) fusion proteins containing a thrombin cleavage site using the pGEX-2T vector (GE Healthcare) in BL21 (DE3) Gold cells (Stratagene).33 The 15N-labeled and 13C,15N-labeled GST-dsRBD fusion proteins were expressed at 30 °C and 37°C, respectively, for 16 h in M9 minimal media containing 1 g/L 15N ammonium chloride and 1g/L 13C glucose. GST-dsRBD was purified using a GSTrap 4B glutathione sepharose column (GE Healthcare), followed by a HiLoad 26/60 Superdex 75 pg (S75) gel-filtration column (GE Healthcare). GST-dsRBD was cleaved with 10 units of thrombin (GE Healthcare) per mg of fusion protein for 24 h at a concentration of about 1 mg/mL in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 2.5 mM CaCl2. Cleaved dsRBD was purified on an S75 gel-filtration column in NMR buffer (20 mM sodium phosphate, pH 6.5, 150 mM NaCl, and 1 mM DTT) and concentrated to about 1 mM.35
RNA samples were prepared by in vitro transcription from a synthetic dsDNA template using mutant T7 RNA polymerase (P266L)39 and purified on a 15% denaturing polyacrylamide gel containing 8 M urea as previously described.40 RNA samples were electroeluted, further purified on a HiTrap Q anion exchange column (GE Healthcare), exchanged into NMR buffer using an Amicon Ultra centrifugal filter, and concentrated to about 1 mM. RNAs were then refolded by heating to 95 °C and slow cooling to 4 °C. dsRBD/RNA complexes for NMR spectroscopy were prepared by adding RNA to protein at a 1.1:1 ratio of RNA:protein and concentrated to 0.8–1 mM.
NMR spectroscopy for structure calculations
NMR spectra for structure determination were recorded on Bruker DRX 500 and 600 MHz spectrometers at 25 °C. The assignments for the Rnt1p dsRBD were derived from analysis of 3D CBCANH, 3D CBCA(CO)NH, 3D HCCH-TOCSY, 3D HCCH-COSY, 3D 13C-NOESY-HSQC and 3D 15N-NOESY-HSQC experiments41–43 acquired on 13C,15N-labeled dsRBD. One-bond 1H-15N RDCs were measured from HSQC-IPAP experiments44 in the presence and absence of the RDC alignment medium C12E5/Hexanol45 on the 600 MHz spectrometer. A total of 84 RDCs were obtained for the free dsRBD. For structure determination of the free dsRBD, a total of 2068 experimental distance restraints were obtained from NOE intensities and classified as strong (1.8–3.0 Å), medium (1.8–4.5 Å) and weak (1.8–6.0 Å). 138 dihedral angles were derived from TALOS.46 Structures were calculated using the NIH-Xplor package47 following standard protocols. Briefly, the calculation started from the extended protein in random orientations. The protein was then folded during 40,000 steps of high temperature dynamics with a time step of 0.002 fs. The structures were cooled down using 75 ps of slow cooling from 2000 K to 100 K. The final structures were obtained after refinement with 52 RDCs (only RDCs from secondary structure elements were used for structure calculations) during a second slow cooling from 1200 K to 100 K. The axial (−30 Hz) and rhombic (0.52) components of the alignment tensor were derived from a grid-search procedure.48 The force constant for the RDCs was gradually increased from 0.001 to 0.2 kcal • mol−1 ↑ Hz−2. The twenty lowest energy structures were selected, and the structures were analyzed using MOLMOL49 and PyMOL.50
NMR spectroscopy for spin relaxation experiments
R1, R2, and 1H-15N nuclear Overhauser enhancement (NOE) values were measured for the free dsRBD and the dsRBD/AGAA and dsRBD/AGAA22 complexes at 20 °C on a Bruker DRX 600 MHz spectrometer. R1 experiments used the following time delays: for the free dsRBD, 41, 161, 299, 299, 437, and 644 ms; for the dsRBD/RNA complexes, 46, 207, 207, 575, 575, and 989 ms. R2 rates were determined with Carr-Purcell-Meiboom-Gill experiments, with the following time delays: for the free dsRBD, 11.2, 22.4, 22.4, 44.8, 67.2, 67.2, and 89.6 ms; for the dsRBD/RNA complexes, 11.2, 22.4, 22.4, 33.5, 33.5, and 44.8 ms. Spectra were processed using NMRPipe/NMRDraw, and peak intensities were obtained using NMRView. Relaxation rates were determined by fitting the expression for relaxation decay, I(R) = I0e−Rt, to the peak intensities using in-house software.
Model-free analysis of relaxation data
Initial estimates of the rotational correlation time and the diffusion tensor for the free dsRBD366–453 (2LUQ; reported here) and RDC-refined dsRBD-AGAA complex (PDB ID 2LUP) were obtained using the program HYDRONMR51,52 and were subsequently optimized using the program ModelFree 4.2153 prior to model selection. Relaxation parameters were interpreted using the Lipari-Szabo model-free formalism to obtain values for motional parameters describing the dynamic behavior of backbone amide bond vectors.54,55 ModelFree53 was used to fit relaxation data for each residue to one of five increasingly complex models using optimized initial estimates of the diffusion tensor and correlation time, where model 1 includes the parameter S2s; model 2, S2s and tm; model 3, S2s and Rex; model 4, S2s, tm, and Rex; and model 5, S2s, S2f, and tm. Following model selection for all residues, global and internal parameters were optimized with a grid-search algorithm using an axially symmetric diffusion tensor (Supplementary Tables 1 and 2). For model-free analysis, bond lengths of 1.02 Å and CSA values of −160 p.p.m. were used. Using a bond length of 1.04 Å results is small changes in the values of model-free parameters (< 5%) but does not change the outcome of our analysis. To confirm that the Rex terms that we observe reflect backbone chemical exchange rather than diffusion anisotropy, we checked that the most significant N-H bond vectors for the free dsRBD are not aligned with the long axis of the diffusion tensor by calculating the angle between the N-H bond and the diffusion tensor z-axis, as defined by the fitted diffusion tensor obtained after model-free analysis.
In vivo analysis of RNT1 hinge mutants
All strains were derived from the BMA64 background. The rnt1::TRP deletion mutant and RNT1 K371A dsRBD mutant were described previously9,56. The dsRBD hinge mutants (I378A, G379A, G379P) and the catalytic mutant (E320K) were constructed using the delitto perfetto method.57 A strain carrying the CORE KanR-URA3 cassette at position S376 was transformed with double-stranded DNA oligonucleotides to excise the CORE sequence and introduce the appropriate mutation in the hinge (I378A, G379A, or G379P), while the E320K mutant was produced from a strain with the CORE KanR-URA3 insertion at position E320. Genomic DNA sequences were confirmed by sequencing. Strains were grown in YPD and RNA was harvested and analyzed by Northern blotting as described9 with the following modifications: 10 ug of RNA was denatured with glyoxal, run on 1X BPTE 2% agarose gels as described58 and transferred to Hybond-N+ membranes (GE Healthcare).
Accession numbers
Coordinates for the 20 lowest energy structures of the Rnt1p dsRBD366–453 have been deposited in the Protein Data Bank under accession code 2LUQ, and chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank under accession code 18535.
RESULTS
Solution structure of the Rnt1 dsRBD366–453
Three structures of the free Rnt1p dsRBD have been reported: a solution structure (PDB ID 1T4N; residues 364–450) and two crystal structures from one asymmetric unit (PDB ID 1T4O; construct includes residues 364–471; crystal structure chain A, residues 362–443; and crystal structure chain B, residues 361–448).34 Helix α3 has a different length and orientation in each of these structures, and this heterogeneity was inferred to reflect dynamics for this helix in solution. Helix α3, unique to the Rnt1p dsRBD, was proposed to contribute to specific RNA binding by reshaping the RNA-binding surface of the dsRBD through steric effects on helix α1 and the α1-β1 loop. However, in the crystal structures, helix α3 of chain A terminates at residue 443 due to disorder in the crystal, and the position of helix α3 of chain B is affected by crystal packing. In the solution structure, there are three non-native residues beyond 447 in helix α3. We previously acquired residual dipolar couplings (RDCs) for the free dsRBD to determine which of the reported structures most closely reflects the conformation of the dsRBD in solution. We showed that the measured RDCs for the free dsRBD correlate best to the back-calculated RDCs for chain A of the crystal structure, although its helix α3 is shorter than in the other reported structures of the free and bound dsRBD.35 However, the large difference between the measured and back-calculated RDCs for helix α3 in the free dsRBD suggested that none of the structures of the free protein accurately describe the orientation of helix α3 in solution (Supplementary Fig. 1).
In order to be able to completely describe the structural changes in the Rnt1p dsRBD upon RNA substrate binding, we determined the solution structure of the free dsRBD (residues 366–453), including an extensive set of RDCs (Fig. 1A, B). This is the same construct used for the solution structures of the dsRBD/RNA hairpin complexes.33,35 The structures of the dsRBD are well converged, with a backbone RMSD to the mean of 0.56 +/−0.11 (Fig. 1A and Table 1). Comparison of our solution structure with the previously determined structures of Rnt1p dsRBD shows that the positions of the three β strands and helix α2, which comprise a hydrophobic core common to all dsRBDs, are nearly identical (RMSD < 1.3; RMSD for the α2 and the β sheets between crystal structures is ~0.8 Å, between the NMR structure is ~1.2 Å) (Fig. 1C). The β1-β2 loop shows evidence of flexibility in all of the reported structures, based on high B factors in the crystal structures and a larger range of conformations in the solution ensembles, especially for the previously determined solution structure.34 This is consistent with our characterization of the dynamics discussed below. However, there are significant differences in the orientations of helix α1, helix α3, and the α1-β1 loop (Fig. 1D, E), particularly between the two solution structures. In our solution structure of the dsRBD366–453, residue I448 is part of helix α3, which is the non-native Ala in the previously determined solution structure. Interestingly, as shown below, I448 has one of the largest chemical shift changes upon binding to RNA substrate (see Fig. 6). The overall fold of the α1-β1 loop is the same for our solution structure and the two crystal structures, although the position of the loop is different (Fig. 1E). Detailed analysis of our solution structure of Rnt1p dsRBD reveals interactions among residues from helices α1 and α3 and the α1-β1 loop that constitute an extended hydrophobic core not present in other dsRBDs. In canonical dsRBDs, residues in helix α1 and the α1-β1 loop are typically solvent exposed. While contiguous with the hydrophobic core common to all dsRBDs, formed by contacts among helix α2 and the β-sheets, this extended hydrophobic core constitutes a distinct internal network of hydrophobic interactions, indicating a potential functional role in tetraloop-specific recognition by the Rnt1p dsRBD.
Fig. 1. Solution structure of the Rnt1p dsRBD366–453.

(A) Superposition of the 20 lowest energy structures of the free dsRBD. (B) Stereoview of the lowest energy structure of the free dsRBD. (C) Comparison of the dsRBD determined here (blue) with previously determined solution (1T4N; residues 364–447) (green) and crystal (IT4O; residues 362–471) chain A (red) and chain B (orange) structures. (D and E) Comparison of helix α1, the α1-β1 loop, and helix α3 in (D) the solution structures of the dsRBD366–453 and dsRBD364–450 (IT4N) and (E) the solution structure of the dsRBD366–453 and the crystal structures chain A and chain B. In all structures, helix α1 begins at residue 369. Helix α3 has three non-native residues beyond residue 447 in the solution structure IT4N. Helix α3 ends at 443 in the crystal structure (IT4O) chain A, at 448 in the crystal structure (IT40) chain B, and at 448 in our solution structure. Structures in C–E are aligned on α2, β1, β2, and β3.
Table 1.
Structural statistics of Rnt1p dsRBD
| Distance and dihedral restraints | |
| Total NOE restraints | 2068 |
| Intraresidue | 581 |
| Sequential | 472 |
| Medium (i+2 to i+4) | 489 |
| Long range (> i+4) | 526 |
| Hydrogen bond restraints | 62 |
| RDC restraints | 55 |
| Dihedral angle restraints | 138 |
| Structure statistics (20 lowest energy structures) | |
| No. of NOE violations > 0.2 Å | 0 |
| No. of Dihedral violations > 5° | 0 |
| No. of RDC violations > 2 Hz | 0 |
| RMSD of RDC (Hz) | 0.20 ± 0.02 |
| RMSD from ideal covalent geometry | |
| Bond lengths (Å) | 0.001 ± 0.0001 |
| Bond angles (°) | 0.313 ± 0.005 |
| Impropers (°) | 0.255 ± 0.008 |
| RMSD from the mean structure (Å) | |
| Backbone (residues 366–448) | 0.56 ± 0.11 |
| Heavy atoms (residues 366–448) | 1.05 ± 0.09 |
| Ramachandran statistics | |
| Most favored regions | 77.3 % |
| Additional allowed regions | 19.9 % |
| Generously allowed regions | 2.4 % |
| Disallowed regions | 0.4 % |
Fig. 6. The effect of substrate length and salt concentration on chemical shift.

Chemical shift mapping of the dsRBD at 150 mM (orange) and 300 mM (purple) NaCl bound to (A) a tetraloop hairpin RNA with a 14 bp stem (AGAA) short and (B) a tetraloop hairpin RNA with a 22 bp stem (AGAA22).
Structural comparison of free and RNA-bound Rnt1p dsRBD reveals concerted changes in the extended hydrophobic core
Comparison of the solution structure of the free dsRBD366–453 with the dsRBD in complex with AGAA (Fig. 2) and AAGU tetraloop hairpins confirms the previously described conformational changes in the dsRBD at the RNA-binding interface that were based on comparison to crystal structure chain A33,35 and provides additional details. Upon binding to the tetraloop minor groove, helix α1 is extended three residues at its N-terminus, rotates 18°, bends between residues L374 and S376, and translates toward the RNA. This reorientation of helix α1 is required for shape-specific binding to the tetraloop minor groove, which is different from the minor groove of A-form RNA. The β1-β2 loop, which interacts with the stem minor groove one helical turn away, moves toward the RNA by about 6 Å compared to its position in the free dsRBD. In the intervening major groove, helix α2 and the β3-α2 loop shift positions for side chain interactions with the phosphodiester backbone. The side chains of the interacting residues all change positions.
Fig. 2. Comparison of the free RDC-refined dsRBD (this work) with the RDC-refined dsRBD/AGAA complex (PDB 2LUP).
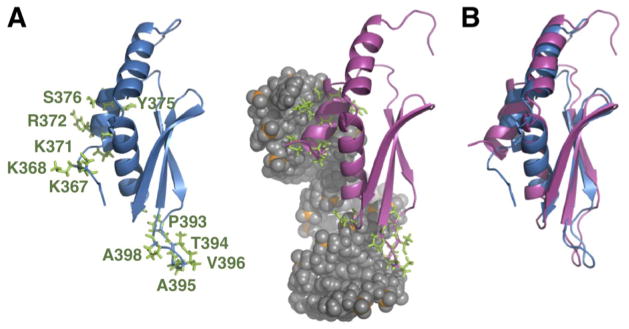
(A) Comparison of free and RNA bound dsRBD, with side chains of residues that interact with the RNA shown. (B) Overlay of free and RNA-bound dsRBD structures, aligned on α2, β1, β2, and β3.
In addition to these conformational changes for residues at the RNA-binding interface, the solution structure of Rnt1p dsRBD366–453 reveals specific changes in the positions of some residues at the interface between helix α1 and helix α3, which are distal from the protein/RNA interface. Superposition of the free and RNA-bound dsRBD on the core α2, β1, β2, and β3 elements reveals that helix α1, helix α3, and the α1-β2 loop all change positions significantly between the free and bound states (Fig. 3A). However, when the free and bound dsRBD are aligned on α1 and α3 it becomes clear that these changes are concerted, i.e. the backbones of all three of these elements are nearly superimposed indicating that they all translate in space together (Fig. 3B). The concerted movement of helix α1 and helix α3 upon RNA binding includes some reorientation of side chains in the hydrophobic core (Fig. 3C–E). The I378 side chain is in the trans rotamer conformation in the free dsRBD, but the gauche- conformation in the RNA-bound state (Fig. 3C, D). This side chain rotation may be necessary to maintain close hydrophobic contacts between α1 and α3 in the complex. The Y380 ring rotates to a position perpendicular to its position in the free state (Fig. 3E). The backbone of the α1-β1 loop moves from its position in the free dsRBD by about 4 Å to accommodate the changes in position of α1 and the α1-β1 side-chains in the bound dsRBD. These changes in side-chain conformation allow the extended hydrophobic core to maintain most of the hyrophobic contacts in the bound state.
Fig. 3. The α1-α3 extended hydrophobic interface.

(A) The hydrophobic interface between helix α1 and α3 in the free dsRBD (blue) and dsRBD/AGAA complex (magenta). Side chains are shown for hydrophobic residues contributing to the interface. The structures are aligned on α2, β1, β2, and β3 (not shown). Space-filling model of the hydrophobic core in the (B) free dsRBD and (C) dsRBD/AGAA complex. (D) The dsRBD and dsRBD/AGAA complex aligned on α1 and α3, illustrating the similar relative orientation of the helices in free and bound states. (E) The dynamic hinge in the α1-β1 loop, comprising residues 378–380, for free (top) and bound (bottom) dsRBDs.
Backbone dynamics of the free dsRBD
To investigate whether the concerted conformational changes in the extended hydrophobic core contribute to tetraloop-specific recognition and, more generally, how conformational flexibility within the free dsRBD affects substrate specificity and binding, we investigated the backbone dynamics of free and RNA bound dsRBDs using NMR spin-relaxation experiments (Fig. 4). The measured 15N relaxation data, R1, R2, and 1H-15N heteronuclear nuclear Overhauser enhancement (NOE) were analyzed using the Lipari-Szabo model-free formalism to obtain a quantitative description of the backbone dynamics, where the order parameter (S2) and the internal correlation time (τe) describe the amplitude and timescale of backbone dynamics, respectively. In addition, to fully describe internal motions, model-free analysis also includes a term to account for chemical exchange at the μs-ms timescale (Rex). Overall, the S2 values obtained from model-free analysis indicate that the dsRBD is relatively rigid for all structured residues at the ps-ns timescale, with an average S2 value of 0.86 (Fig. 5A). The single exception is residue N399, which has an S2 value of 0.6, and is adjacent to a proline (P398) in the β1-β2 loop. We were unable to determine relaxation parameters for residue D397 on the other side of the proline due to spectral overlap. The conformational flexibility in the β1-β2 loop, as evidenced by the low S2 value of N399, is consistent with the multiple conformations for the β1-β2 loop in the NMR structure ensembles and high B factors in the crystal structures of the free dsRBD.34
Fig. 4. NMR spin relaxation parameters.

R1, R2, and heteronuclear NOE values for the free dsRBD.
Fig. 5. Fast- and slow-timescale dynamics of the free dsRBD and the dsRBD/AGAA complex at 150 mM NaCl.

(A) S2 and Rex model-free parameters for the free dsRBD. (B) Structure of free dsRBD with residues that show Rex highlighted. (C) S2 and Rex model-free parameters for the dsRBD /AGAA complex. (D) Structure of dsRBD in the dsRBD/AGAA complex with residues that show Rex highlighted.
Slow-timescale motions, as reflected by the inclusion of an Rex term during model-free analysis, are present in some residues in helix α1, the α1-β1 loop, strand β1, the β1-β2 loop, the β2-β3 loop, and residues Y441 and R445 in helix α3 (Fig. 5A, B). The Rex values in the β1-β2 loop and the end of β1 are consistent with the observed flexibility in this region and proposed P393 cis-trans isomerization34. Within the extended hydrophobic core, a cluster of residues in helix α1 and the α1-β1 loop exhibit notable Rex. One of these residues, I378, has an unusually large value for μs-ms timescale exchange, with an Rex value of 15 s−1. I378 is the C-terminal residue in helix α1 and its hydrophobic side chain is part of the extended hydrophobic core. This large Rex could be due to I378 undergoing jumps between the trans and gauche- rotamers.59 Residue R384, which is in the α1-β1 loop, also has a high Rex value (~8 s−1-) (Fig. 5A, B). Based on the conformational changes in residues within the α1-β1 loop (Fig. 3E) and the large slow-timescale dynamics for residues 378–380 (Fig. 5A), we identify these residues as a dynamic hinge that we propose allows conformational sampling by helix α1. Since I378 is in the trans and gauche- conformations in the free dsRBD and RNA-bound dsRBD, respectively, this would suggest that the hinge samples the bound conformation. The β2-β3 loop has low B-factors in the crystal structure and is well defined in the solution structures, but there is Rex for some residues. Examination of the structure of the free dsRBD reveals that the residues in the β2-β3 loop that exhibit Rex interact with residues in helix α3 and the α1-β1 loop and would be sensitive to conformational changes in the extended hydrophobic core. The presence of a dynamic hinge and chemical exchange in the extended hydrophobic core imply that the tetraloop-binding helix α1 samples the bound state in the free dsRBD.
Dynamics of the dsRBD in the dsRBD/AGAA complex
To determine whether specific RNA substrate binding changes the μs-ms dynamics observed in the free dsRBD, we collected R1, R2 and heteronuclear NOE values for the dsRBD/AGAA hairpin complex whose structure was previously reported33 (Supplementary Fig. 2). The AGAA hairpin, consisting of a 14-bp dsRNA stem capped by an AGAA tetraloop, is a model substrate derived from the Rnt1p recognition motif in the Rnt1p pre-snoRNA substrate snR47. This RNA provides a minimal binding site for the dsRBD, with only 2–3 bp extending below the interaction of the β1-β2 loop in the minor groove. The average R1 and R2 values on the dsRBD/AGAA hairpin complex are lower and higher, respectively, than those of the free protein, as would be expected for the increased molecular weight of the complex. Heteronuclear NOE values indicate that the dsRBD in the complex is rigid overall, except for the N- and C-termini and the β1-β2 loop. Several residues in the β1-β2 loop have heteronuclear NOE values between 0.4 and 0.6, indicating that this loop remains flexible in the dsRBD/AGAA hairpin complex.
Binding of the dsRBD to the AGAA hairpin results in an overall increase in the S2 values of most of the protein residues (average increase of 0.15) (Fig. 5A, C). Exceptions are small decreases (< 0.1) for helix α1 residues K371 and S376, which interact with the minor groove of the AGAA tetraloop, Y380 and L383 in the extended hydrophobic core in the α1-β1 loop, and the single residue R433 between α2 and α3 (Fig. 5C). Rex values increase for most of the residues in helix α1, with particularly large increases for K371 and S376, which contact the RNA backbone. Helix α3, which has only two residues with Rex in the free dsRBD, also shows Rex for most residues. In contrast, the dynamic hinge residues I378 and Y380, both of which exhibit slow-timescale motions in the free protein, have lower Rex values in the complex. Dynamics in helix α1 likely reflect flexibility at the protein-RNA interface. For helix α3, the uniform increase in Rex could originate from propagation of the dynamics in helix α1 via the extended hydrophobic core, and/or from an increase in entropy of the dsRBD in the bound state, an effect that has been observed in other RNA-binding proteins.60 A395 and V396, near P398 in the β1-β2 loop, which contact the minor groove of the dsRNA stem, also have lower Rex values in the complex (Fig. 5C, D). The decrease in slow-timescale motions for residues in the dynamic hinge and the β1-β2 loop indicates that some slow-timescale dynamics present in the free dsRBD are quenched upon binding to RNA.
Helix α1 residue S376 has no Rex term in the free dsRBD but has the largest Rex value in the dsRBD/AGAA complex. Helix α1 bends at S376 to insert into the minor groove, and the S376 side-chain contacts the RNA backbone on the 3′ side of the AGAA tetraloop. This correlation between changes in structure and dynamics suggests that the dynamic properties of S376 might have a functional role in allowing the dsRBD to adopt the bound conformation. Alternatively, chemical exchange at S376 might be caused by exchange between the specifically and nonspecifically bound states, reflecting the role of this residue in recognizing the backbone of the tetraloop.
To verify that the observed Rex is attributable only to the intrinsic dynamics of the dsRBD and not to nonspecific protein-protein interactions or to exchange between the free and bound state,61 we measured R2 values at concentrations of 1 mM and 0.5 mM for the dsRBD/AGAA complex and the free dsRBD (Supplementary Fig. 3). In the absence of these possible additional contributions to chemical exchange, R2 values and NMR linewidths would be expected to be the same at both concentrations. In both cases, R2 values and NMR linewidths for two protein concentrations are nearly identical, indicating that the dynamics determined by model-free analysis arise only from the intrinsic dynamics of the dsRBD and not from other possible contributions to chemical exchange.
In summary, two distinct changes in dsRBD dynamics in the extended hydrophobic core are observed upon substrate binding. First, there is a general increase in slow-timescale dynamics for residues in helix α1 and α3 that are associated with concerted changes in the extended hydrophobic core. Second, there is a decrease in slow-timescale dynamics for residues in the α1-β1 hinge and the β2-β3 loop, due to “locking in” of helix α1 by shape specific binding to the tetraloop minor groove.
Ionic strength dependence of dynamics for the dsRBD in the dsRBD/AGAA complex
Previous NMR titration and isothermal titration calorimetry (ITC) experiments revealed that the dsRBD can bind to the AGAA hairpin both specifically and non-specifically at 150 mM NaCl, with saturation of the RNA at a protein:RNA ratio of 2:1.35 The relaxation data discussed above for the dsRBD/AGAA complex were measured at a protein:RNA ratio of 1:1:1.1 and were expected to primarily reflect values for the dsRBD bound to the specific site. To further confirm this, we investigated the binding and dynamics of the dsRBD/AGAA hairpin complex at 300 mM NaCl (Supplementary Fig. 4). At this salt concentration, non-specific binding should be minimal. Chemical shift mapping for the dsRBD upon RNA binding at 300 mM NaCl revealed chemical shift changes similar in pattern to those for the complex at 150 mM NaCl, but with a much smaller magnitude (Fig. 6). This is consistent with the lower binding affinity of the dsRBD for RNA at a higher salt concentration, as measured by NMR titration and ITC.35 At 300 mM NaCl, the dsRBD exhibits Rex values similar overall to those observed at 150 mM NaCl (compare Fig. 7A, B with Fig. 5C, D). This observation is consistent with a single, specific binding site on the AGAA hairpin. Hence, the dsRBD is fully bound to the AGAA hairpin at the specific binding site under the conditions used for spin relaxation experiments at a high concentration of the complex and 150 mM or 300 mM NaCl.
Fig. 7. Fast- and slow-timescale dynamics of dsRBD/AGAA and dsRBD/AGAA22 complexes at 300 mM NaCl.

(A, B) S2 and Rex model-free parameters for the (A) dsRBD/AGAA complex and (B) dsRBD/AGAA26 complex. (C, D) Rex values mapped onto the structure of the (C) dsRBD/AGAA and (D) dsRBD/AGAA26 complex.
Dynamics of the dsRBD in the presence of both specific and non-specific binding sites
As discussed above, the dsRNA construct was designed such that it has a minimal binding site for the dsRBD. To investigate whether there is a difference in dsRBD dynamics when both non-specific and specific binding sites are present we collected NMR spin relaxation data for Rnt1p dsRBD in complex with an AGAA tetraloop hairpin with a 22-bp stem (AGAA22) at 300 mM NaCl (Supplementary Fig. 5). AGAA22 has a stem that is eight base pairs longer than the AGAA hairpin (14 bp), allowing for non-specific binding to the longer dsRNA stem in addition to specific binding site at the AGAA tetraloop. Because of the longer stem, Rnt1p dsRBD can potentially exchange between the specific site and non-specific sites on AGAA22. AGAA22 more closely reflects native conditions for substrate binding by Rnt1p dsRBD, as the stem length is the same as the stem in the pre-snR47 snoRNA (excluding a single bulge). In general, the values for chemical exchange, as described by Rex, are significantly larger for dsRBD/AGAA22 than for dsRBD/AGAA. Slow-timescale dynamics for dsRBD/AGAA22 are present in residues in helices α1 and α3 in or near the extended hydrophobic core, residues throughout the RNA-binding interface, including helix α2 and the β1-β2 loop, and in β2 and β3. In dsRBD/AGAA, there was no Rex for any residues in the β1-β2 loop, while in dsRBD/AGAA22 most of the β1-β2 loop residues show Rex (Fig. 7C, D). The Rex in the β1-β2 loop is both larger and present in more residues than in the free dsRBD (Fig. 5A). Under the experimental conditions of 300 mM NaCl and excess RNA, the dsRBD is relatively selective for specific binding, so the additional contribution to Rex arising from the presence of additional non-specific binding sites can be attributed to exchange between tetraloop (specific) and stem (non-specific) binding sites. It is notable that all of the elements of the dsRBD that interact with the minor and major groove of the dsRNA stem show more conformational exchange than when only a specific binding site is available. The additional protein dynamics for the dsRBD/AGAA22 complex, compared to the dsRBD/AGAA complex, also reveal the significance of conformational changes in the extended hydrophobic core, in addition to residues at the RNA binding interface, in binding site selection. We conclude that the difference in Rex for the complex with AGAA22 vs AGAA reflects some non-specific binding to the dsRNA on the longer substrate. Furthermore, once helix α1 locks in to the tetraloop, the rest of the dsRBD locks into place, resulting in a decrease of Rex in the β1-β2 loop for AGAA vs AGAA22.
Hydrophobic interactions with the α1-β1 loop maintain dsRBD stability
To extend insights from our characterization of dsRBD structure and dynamics, we further investigated the importance of residues in the α1-β1 loop for RNA binding by generating four dsRBD mutants with single mutations in the α1-β1 loop: I378A, G379P, G379A, and Y380A. These three residues are part of the α1-β1 hinge in the extended hydrophobic core. The side chains of I378 and Y380 change position between the free and RNA-bound states and maintain hydrophobic contacts with residues in helix α3. The ϕ and ψ angles for G379 also change between free and bound states, due to conformational changes in the hydrophobic core (Fig. 3C–E). The 1H-15N HSQC spectrum of I378A was poorly dispersed (Supplementary Fig. 6), and the CD spectrum showed no evidence for secondary structure (Supplementary Fig. 7), indicating that the majority of the protein is unfolded at 25°C. However, in freshly prepared protein samples, there appears to be about 10% folded protein based on the 1H-15N HSQC. Addition of RNA to I378A results in some chemical shift changes indicative of binding for the peaks from the folded protein, but fewer than for the wild-type dsRBD, and the protein unfolds over time. Thus, we conclude that the mutation I378A destabilizes the protein and may also lower RNA binding affinity. Since I378 interacts with residue Y441, which is in helix α3 and part of the extended hydrophobic core, we tested the importance of this interaction by making a Y441A mutation. Y441A is also unstable in solution and precipitates after about 20 min at 25°C, and 1H-15N HSQC spectra indicate that it is unfolded prior to precipitation. We note that the mutation R445A, in helix α3, was previously shown to destabilize the extended hydrophobic core of the dsRBD.33,34 This residue is close to the α1-β1 loop residue S382, and shows Rex in both the free and bound dsRBD.
For the G379P mutant, CD spectra indicate that the Tm decreases by ~6 °C and that melting is less cooperative. The 1H-15N HSQC (Supplementary Fig. 7) has chemical shift changes throughout helix α1 and α3, and none of the resonances for the α1-β1 loop are observed (Supplementary Fig. 6 and 8). Analysis of the backbone chemical shifts for dsRBD G379P indicates the C-terminal end of helix α1 and all of helix α3 are altered relative to the WT dsRBD (Supplementary Fig. 8). Addition of the AGAA hairpin to G379P up to a 2:1 excess of RNA resulted in almost no changes in the 1H-15N HSQC spectrum, indicating that the G379P substitution essentially abrogates binding of the dsRBD to RNA. G379A had a substantially altered 1H-15N HSQC spectrum (Supplementary Fig. 6) and is unstable, as the protein precipitated after 20 min at 25°C. However, in the presence of the AGAA hairpin, the G379A mutant gives 1H-15N HSQC spectra indicating that G379A forms a stable complex (Supplementary Fig. 6). Lastly, we found that Y380A degrades during expression, implying that the mutation of this residue also significantly destabilizes the protein.
In summary, all of the mutations in the α1-β1 loop and α3 destabilize the extended hydrophobic core to some extent and have variable effects on RNA binding. For the G379P mutation, changes to the extended hydrophobic core completely disrupt RNA binding, although this mutation has the smallest effect on dsRBD stability. For G379A, binding to RNA helps stabilize the folded state of the dsRBD. Because all of the residue substitutions in the extended hydrophobic core affect protein stability, we were not able to assess their effects on dynamics independently. Nevertheless, these results support a central structural role for the α1-β1 loop and extended hydrophobic core in maintaining dsRBD stability.
Effect of α1-β1 loop mutations on snoRNA processing in vivo
To determine whether the mutations in the α1-β1 hinge have an effect on cleavage of Rnt1p substrates in vivo, we introduced the single mutations I378A, G379A, and G379P into the RNT1 gene and examined by northern blot the effect of these mutations on the processing of snR36 and snR47 snoRNAs in vivo. For comparison, we included in the analysis strains harboring a previously studied mutation in helix α1 that affect processing (K371A),33 a catalytically inactive mutant (E320K), and a RNT1 deletion (rnt1Δ). Unlike the previously studied K371A mutant, all three α1-β1 loop mutants exhibit temperature-sensitive growth defects (Fig. 8A, B). The growth defects for strains bearing the I378A and G379A mutations are comparable and relatively modest, while G379P strain had a growth defect comparable to the rnt1 strain. This is consistent with the in vitro results that showed that while stable, dsRBD G379P does not bind RNA.
Fig. 8. In vivo analysis of dsRBD α1-β1 loop mutants.

(A) Growth of wild-type dsRBD and dsRBD hinge mutants, with serial dilutions at 16, 30, and 37 °C. (B) Growth curve for the wild-type dsRBD and dsRBD hinge mutants at 30 °C. (C) Northern blot analysis of snR47 and snR46 snoRNA processing for wild-type dsRBD and dsRBD hinge mutants, showing unprocessed precursor (P) and mature snoRNAs (M).
The strain bearing the I378A mutation shows an inhibition of snoRNA processing comparable to the K371A mutation, with a slight processing defect for snR47 and a more pronounced defect for snR36 (Fig. 8C). snR36 was previously observed to be more sensitive than snR47 to mutations in the Rnt1p dsRBD, because of the presence of a large bulge after the fourth base pair below the tetraloop.56 The G379A strain exhibited only minor effects on snoRNA processing in vivo. Although it has a growth defect comparable to I378A, the dsRBD is stabilized by binding to RNA, which may explain the difference in effect on snoRNA processing. The G379P strain, in contrast, showed severe processing defects for both substrates. The processing defects in strains bearing the I378A, G379P, and G379A mutations are consistent with NMR and CD results that indicate that these mutations introduce changes in stability and RNA-binding affinities of the dsRBDs. We conclude that mutations in the α1-β1 hinge, all of which destabilize the extended hydrophobic core and affect RNA binding to different extents, compromise the function of Rnt1p in vivo.
DISCUSSION
dsRBDs recognize dsRNA primarily by interactions with the phosphodiester backbone of successive minor, major, and minor grooves via the β1-β2 loop, the N-terminal end of helix α2, and helix α1, respectively. The dsRBD of Rnt1p is unusual in that helix α1 recognizes a tetraloop through shape specific recognition of its minor groove.33 The Rnt1p dsRBD also has an additional helix α3, which packs against the α1-β1 loop to form a distinctive extended hydrophobic core. All dsRBDs have a conserved hydrophobic core, with residues contributed by the C-terminal residues of helix α1, helix α2, and strand β3. Hydrophobic interactions among these residues in the core of the protein stabilize the folded conformation of the dsRBD.29,62. The unique hydrophobic interface between helices α1 and α3 in the Rnt1p dsRBD is contiguous with the conserved hydrophobic core. Through a detailed analysis of the structures and dynamics of the free and bound dsRBDs, we have shown that this extended hydrophobic core plays an essential role in enabling defined conformational changes associated with RNA substrate recognition. Residues in the α1-β1 loop that interact with helix α3 to form the extended hydrophobic core constitute a dynamic hinge that allows a concerted change in the positions of helix α1 and helix α3 between the free and bound states, a key feature of substrate recognition by the Rnt1p dsRBD (Fig. 3). The importance for RNA binding of residues in the extended hydrophobic core, which includes the α1-β1 hinge, is further supported by the results of mutagenesis of individual residues on snoRNA processing in vivo (Fig. 8).
Helix α3 has been previously proposed to contribute to specific RNA binding indirectly by affecting the length and orientation of helix α1 in the free protein.34 However, the orientation of helix α1 in the free dsRBD is the same as other dsRBDs that bind to dsRNA non-specifically. Our results show that the helix α3 contributes to substrate specific binding by participating in the reorientation of helix α1 in the bound state through concerted structural changes in the extended hydrophobic core.
The free dsRBD samples multiple conformations
Backbone dynamics of the free dsRBD obtained via model-free analysis of NMR spin relaxation data reveal extensive slow-timescale dynamics primarily localized in the RNA binding interface and extended hydrophobic core, including helix α1, α1-β1 loop, and β1-β2 loop, as well as limited dynamics in helix α3. Our comparison of free and bound structures shows that upon binding of the RNA substrate, the α1-β1 loop changes conformation to allow helix α1 and α3 to undergo concerted changes in orientation and side chain position in order for helix α1 to be able to bind to the minor grove of the substrate tetraloop (Fig. 2 and 3). The β1-β2 loop also translocates about 6 Å to bind to the minor grove of dsRNA one helical turn away from the tetraloop (Fig. 2). Thus, the dynamic behavior of the free dsRBD on both fast and slow timescales is associated with conformational changes within the extended hydrophobic core that accompany substrate recognition. This localized flexibility supports the notion that conformational adaptation upon substrate binding is enabled by dynamics of the free dsRBD. Mutations of residues in the α1-β1 loop affect the stability and RNA-binding properties of the dsRBD, revealing that the interactions of the α1-β1 loop with helix α3 are essential for dsRBD stability and function.
The observed dynamics of the α1-β1 loop in the free dsRBD and the concerted movement of helices α1 and α3 led us to propose that residues 378–380 in the α1-β1 loop serve as a dynamic hinge enabling conformational exchange between the free and bound states. One possible model for the contribution of the α1-β1 hinge is that hinge dynamics on the μs-ms timescale backbone facilitate conformational sampling by helix α1. Moreover, hinge dynamics would facilitate concerted movement of helices α1 and α3 upon binding because hinge residues I378 and Y380 are also part of the extended hydrophobic core. These dynamics, along with those of the β1-β2 loop, are partially quenched upon binding to the specific site on target substrates (Fig. 9). We cannot exclude, however, that the dsRBD experiences a combination of conformational selection and induced fit to achieve its final bound conformation.
Fig. 9. dsRBD dynamics are associated with concerted structural changes necessary for binding.

Change in Rex for free vs RNA bound dsRBD for (A) dsRBD/AGAA complex and (B) dsRBD/AGAA22 complex. Model of the dsRBD/AGAA22 complex with dsRBD bound specifically at the tetraloop is based on dsRBD/AGAA complex. ΔRex (Rex free dsRBD minus Rex dsRBD in complex with RNA) is shown in red (increase in Rex) and blue (decrease in Rex). Relative magnitude of the change in Rex is represented by the tube width.
Previous studies of the contribution of protein dynamics to RNA recognition have shown that high-affinity binding to RNA is generally associated with the presence of extensive Rex throughout an RNA-binding domain. NMR relaxation studies of the two dsRBDs of protein kinase R indicated that residues that directly interact with the RNA and throughout helix α1, sheet β1, and helix α2 have slow-timescale motions for PKR dsRBD1, which binds to dsRNA with high affinity. In contrast, there are but few such motions for PKR dsRBD2, which has weaker binding affinity for dsRNA.63 Dynamics within the PKR dsRBD1 were proposed to allow for adaptation to non-uniform RNA substrates. The observed Rex within PKR dsRBD1 and Rnt1p dsRBD are different in both the distribution and extent of slow-timescale dynamics. Thus, dsRBDs with different dsRNA substrates can have different dynamic modes despite having similar structures in the free state. Here, we have shown the first example where dynamic properties of a dsRBD are associated with defined structural changes in the protein that take place upon binding to RNA.
Substrate binding induces changes in backbone dynamics
In complex with RNA, dynamics in Rnt1p dsRBD are present throughout the extended hydrophobic core and RNA-binding interface, corresponding to binding-induced conformational changes. Changes in Rex between free and AGAA tetraloop hairpin-bound dsRBDs are shown in Fig. 9. Residues within the α1-β1 hinge that are dynamic in the free dsRBD are partly or completely quenched, while residues within helix α1 and α3 have even larger slow-timescale dynamics in the complex. Residues at the N-terminus of helix α1 in the complex fold only upon binding to RNA,35 and these residues also have a decrease in slow timescale dynamic upon binding. Two residues within the β1-β2 loop also have lower Rex in the complex than in the free dsRBD, which is consistent with the stable interaction of this loop with the stem minor groove.
An increase of the prevalence of chemical exchange upon substrate binding appears to be a common feature of nucleic acid binding proteins, as well as proteins involved in protein-protein interactions.31,64 Typically, this increase in Rex is observed to occur at sites distant from the interface with the partner molecular and implies an indirect role for conformational flexibility in binding. The increase in Rex is often distributed throughout the protein and is not well correlated with specific structural changes. In contrast, for Rnt1p dsRBD the observed increase in Rex for regions of the protein distal from the RNA binding surface, particularly in the C-terminus of helix α3, is correlated with conformational changes associated with RNA binding. The counterintuitive increase in dynamics for regions of the dsRBD distal to the RNA-binding face reveals that a broad network of residues within the dsRBD contributes to conformational adaptation to the specific tetraloop binding site. The results of these experiments highlight the importance of protein conformational flexibility, particularly within the extended hydrophobic core, in binding of the Rnt1p dsRBD to RNA tetraloop hairpin substrates.
Dynamics reflect binding site exchange on a long substrate
Because the dsRBD binds to the minimal substrate (AGAA) at 300 mM NaCl only at the specific tetraloop site, slow-timescale dynamics are likely to be limited to intrinsic dynamics of the bound state, reflecting intrinsic conformational entropy of the dsRBD in complex with RNA. However, exchange between specific and nonspecific sites becomes significant in the dsRBD/AGA22 complex, as the longer stem allows for non-specific binding away from the tetraloop. In the dsRBD/AGAA22 complex, chemical exchange values are quantitatively higher and are present throughout the dsRBD. Hence, dynamics within the dsRBD in the dsRBD/AGAA22 complex suggest that the dsRBD distinguishes between specific and nonspecific complexes after binding to the dsRNA substrate through conformational exchange. Increased dynamics throughout the extended hydrophobic core and RNA-binding interface, compared to the dsRBD/AGAA complex (Fig. 9B), suggests that flexibility within the RNA-bound dsRBD remains important for selection of the specific binding site even after the dsRBD is bound to RNA. Moreover, elevated Rex values of helix α1 and β1-β2 loop would be expected to be present at the RNA-binding interface for dsRBD/AGAA22 complex if the dsRBD searches between the specific tetraloop site and non-specific stem region, since contacts to the RNA stem in the nonspecific complex would not be identical to the specific complex.
Comparison to budding yeast Dicer
Budding yeast Dicers have two dsRBDs: the first (dsRBD1) is located immediately adjacent to the endoND, as in Rnt1p, and the second (dsRBD2), is at the C-terminus of the protein and separated from dsRBD1 by a long intervening sequence with no known structural motifs. The S. castellii Dcr1 dsRBD1, but not dsRBD2, was shown to be necessary for efficient processing of long dsRNA substrates to 23 nt fragments and for specificity of the enzyme for dsRNA over ssRNA.65 Dcr1, however, does not have tetraloop specificity. A superimposition of the crystal structure of the free K. polysporus Dcr1 dsRBD1 and the solution structure of the free Rnt1p dsRBD (Supplementary Fig. 9) indicates that the two dsRBDs have the same overall conformation, including a short helix α3 in Dcr1, but differ in regions that are important for specific RNA binding by Rnt1p dsRBD. Notably, in Dcr1 there are no interactions between the α1-β1 loop and helix α3 because helix α3 is shorter in the Dcr1 dsRBD1, and the α1-β1 loop adopts a different conformation compared to the Rnt1p dsRBD. Although Dcr1 dsRBD1 has some conserved hydrophobic residues in the α1-β1 loop, e.g. L278 and I280 in K. polysporus Dcr1correspond to Rnt1p dsRBD hinge residues I378 and Y380, it does not appear to have an extended hydrophobic core. L278 cannot undergo the rotameric change that we see for I378, and the I280 sidechain is oriented toward the outside of the protein. These residues do not interact with helix α3 and do not appear to constitute an analogous hinge. Interestingly, K. polysporus Dcr1 L275 has hydrophobic interactions with the conserved residue Y341, which is in the loop extending past helix α3 in the dsRBD1. We speculate that the absence in Dcr1 dsRBD1 of an extended hydrophobic core involving the α1-β1 loop results in the loss of tetraloop specificity for K. polysporus and S. castellii Dcr1. Nevertheless, structural features within this region that are unique to the Dcr1 dsRBD may affect its binding affinity to Dcr1 substrates. Interestingly, C. albicans Dcr1 is able to carry out both Rnt1 and Dcr1 functions,28 suggesting that its dsRBD1 may retain the structural features necessary for tetraloop recognition, including the extended hydrophobic core.
Supplementary Material
The S. cerevisiae dsRBD binds shape specifically to (A/u)GNN tetraloop hairpin RNAs
Helix α1 undergoes conformational sampling on the μs-ms timescale in the free dsRBD
A unique hydrophobic core governs concerted changes in helices α1 and α3 upon binding
dsRBD structural changes correlate with the dynamics of free and bound dsRBDs
Acknowledgments
This work was supported by NIH grant GM37254 to J.F. Qi Zhang was a Baltimore Family Fund Postdoctoral Fellow of the Life Sciences Research Fund. We thank Anni Zhao for help with preparation of the hydrophobic core mutants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–9. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knight SW, Bass BL. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001;293:2269–71. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 4.Ji X. Structural basis for non-catalytic and catalytic activities of ribonuclease III. Acta Crystallogr D Biol Crystallogr. 2006;62:933–40. doi: 10.1107/S090744490601153X. [DOI] [PubMed] [Google Scholar]
- 5.MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol. 2007;17:138–45. doi: 10.1016/j.sbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Kufel J, Dichtl B, Tollervey D. Yeast Rnt1p is required for cleavage of the pre-ribosomal RNA in the 3′ ETS but not the 5′ ETS. RNA. 1999;5:909–17. doi: 10.1017/s135583829999026x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elela SA, Igel H, Ares M., Jr RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell. 1996;85:115–24. doi: 10.1016/s0092-8674(00)81087-9. [DOI] [PubMed] [Google Scholar]
- 8.Ghazal G, Ge D, Gervais-Bird J, Gagnon J, Abou Elela S. Genome-wide prediction and analysis of yeast RNase III-dependent snoRNA processing signals. Mol Cell Biol. 2005;25:2981–94. doi: 10.1128/MCB.25.8.2981-2994.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chanfreau G, Legrain P, Jacquier A. Yeast RNase III as a key processing enzyme in small nucleolar RNAs metabolism. J Mol Biol. 1998;284:975–88. doi: 10.1006/jmbi.1998.2237. [DOI] [PubMed] [Google Scholar]
- 10.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–9. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 11.Chanfreau G, Elela SA, Ares M, Jr, Guthrie C. Alternative 3′-end processing of U5 snRNA by RNase III. Genes Dev. 1997;11:2741–51. doi: 10.1101/gad.11.20.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abou Elela S, Ares M., Jr Depletion of yeast RNase III blocks correct U2 3′ end formation and results in polyadenylated but functional U2 snRNA. EMBO J. 1998;17:3738–46. doi: 10.1093/emboj/17.13.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seipelt RL, Peterson ML. Alternative processing of IgA pre-mRNA responds like IgM to alterations in the efficiency of the competing splice and cleavage-polyadenylation reactions. Mol Immunol. 1995;32:277–85. doi: 10.1016/0161-5890(94)00141-m. [DOI] [PubMed] [Google Scholar]
- 14.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999;18:5399–410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CY, Lee A, Chanfreau G. The roles of endonucleolytic cleavage and exonucleolytic digestion in the 5′-end processing of S. cerevisiae box C/D snoRNAs. RNA. 2003;9:1362–70. doi: 10.1261/rna.5126203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petfalski E, Dandekar T, Henry Y, Tollervey D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol Cell Biol. 1998;18:1181–9. doi: 10.1128/mcb.18.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu LH, Henras A, Lu YJ, Zhou H, Zhou WX, Zhu YQ, Zhao J, Henry Y, Caizergues-Ferrer M, Bachellerie JP. Seven novel methylation guide small nucleolar RNAs are processed from a common polycistronic transcript by Rat1p and RNase III in yeast. Mol Cell Biol. 1999;19:1144–58. doi: 10.1128/mcb.19.2.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danin-Kreiselman M, Lee CY, Chanfreau G. RNAse III-mediated degradation of unspliced pre-mRNAs and lariat introns. Mol Cell. 2003;11:1279–89. doi: 10.1016/s1097-2765(03)00137-0. [DOI] [PubMed] [Google Scholar]
- 19.Egecioglu DE, Kawashima TR, Chanfreau GF. Quality control of MATa1 splicing and exon skipping by nuclear RNA degradation. Nucleic Acids Res. 2012;40:1787–96. doi: 10.1093/nar/gkr864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghazal G, Gagnon J, Jacques PE, Landry JR, Robert F, Elela SA. Yeast RNase III triggers polyadenylation-independent transcription termination. Mol Cell. 2009;36:99–109. doi: 10.1016/j.molcel.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Rondon AG, Mischo HE, Kawauchi J, Proudfoot NJ. Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol Cell. 2009;36:88–98. doi: 10.1016/j.molcel.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavoie M, Ge D, Abou Elela S. Regulation of conditional gene expression by coupled transcription repression and RNA degradation. Nucleic Acids Res. 2012;40:871–83. doi: 10.1093/nar/gkr759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge D, Lamontagne B, Elela SA. RNase III-mediated silencing of a glucose-dependent repressor in yeast. Curr Biol. 2005;15:140–5. doi: 10.1016/j.cub.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Zer C, Chanfreau G. Regulation and surveillance of normal and 3′-extended forms of the yeast aci-reductone dioxygenase mRNA by RNase III cleavage and exonucleolytic degradation. J Biol Chem. 2005;280:28997–9003. doi: 10.1074/jbc.M505913200. [DOI] [PubMed] [Google Scholar]
- 25.Chanfreau G, Buckle M, Jacquier A. Recognition of a conserved class of RNA tetraloops by Saccharomyces cerevisiae RNase III. Proc Natl Acad Sci U S A. 2000;97:3142–7. doi: 10.1073/pnas.070043997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagel R, Ares M., Jr Substrate recognition by a eukaryotic RNase III: the double-stranded RNA-binding domain of Rnt1p selectively binds RNA containing a 5′-AGNN-3′ tetraloop. RNA. 2000;6:1142–56. doi: 10.1017/s1355838200000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, Bartel DP. RNAi in budding yeast. Science. 2009;326:544–50. doi: 10.1126/science.1176945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernstein DA, Vyas VK, Weinberg DE, Drinnenberg IA, Bartel DP, Fink GR. Candida albicans Dicer (CaDcr1) is required for efficient ribosomal and spliceosomal RNA maturation. Proc Natl Acad Sci U S A. 2012;109:523–8. doi: 10.1073/pnas.1118859109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–65. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–13. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, Allain FH. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–37. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller GA, Miller MT, Derose EF, Ghosh M, London RE, Hall TM. Solution structure of the Drosha double-stranded RNA-binding domain. Silence. 2010;1:2. doi: 10.1186/1758-907X-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, Henras A, Chanfreau G, Feigon J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc Natl Acad Sci U S A. 2004;101:8307–12. doi: 10.1073/pnas.0402627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leulliot N, Quevillon-Cheruel S, Graille M, van Tilbeurgh H, Leeper TC, Godin KS, Edwards TE, Sigurdsson ST, Rozenkrants N, Nagel RJ, Ares M, Varani G. A new alpha-helical extension promotes RNA binding by the dsRBD of Rnt1p RNAse III. EMBO J. 2004;23:2468–77. doi: 10.1038/sj.emboj.7600260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Hartman E, Roy K, Chanfreau G, Feigon J. Structure of a Yeast RNase III dsRBD Complex with a Noncanonical RNA Substrate Provides New Insights into Binding Specificity of dsRBDs. Structure. 2011;19:999–1010. doi: 10.1016/j.str.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghazal G, Elela SA. Characterization of the reactivity determinants of a novel hairpin substrate of yeast RNase III. J Mol Biol. 2006;363:332–44. doi: 10.1016/j.jmb.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–72. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 38.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–96. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillerez J, Lopez PJ, Proux F, Launay H, Dreyfus M. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc Natl Acad Sci U S A. 2005;102:5958–63. doi: 10.1073/pnas.0407141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu H, Finger LD, Feigon J. Structure determination of protein/RNA complexes by NMR. Methods Enzymol. 2005;394:525–45. doi: 10.1016/S0076-6879(05)94022-6. [DOI] [PubMed] [Google Scholar]
- 41.Kay LE, Xu GY, Yamazaki T. Enhanced-Sensitivity Triple-Resonance Spectroscopy with Minimal H2O Saturation. J Magn Reson. 1994;109:129–133. [Google Scholar]
- 42.Grzesiek S, Bax A. The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J Amer Chem Soc. 1993;115:12593–12594. [Google Scholar]
- 43.Schleucher J, Schwendinger M, Sattler M, Schmidt P, Schedletzky O, Glaser SJ, Sorensen OW, Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J Biomol NMR. 1994;4:301–6. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 44.Ottiger M, Delaglio F, Bax A. Measurement ofJand Dipolar Couplings from Simplified Two-Dimensional NMR Spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 45.Ruckert M, Otting G. Alignment of Biological Macromolecules in Novel Nonionic Liquid Crystalline Media for NMR Experiments. J Amer Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 46.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 47.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 48.Clore GM, Gronenborn AM, Tjandra N. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J Magn Reson. 1998;131:159–62. doi: 10.1006/jmre.1997.1345. [DOI] [PubMed] [Google Scholar]
- 49.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 50.DeLano WL. The PyMOL Molecular Graphics System. 2002. [Google Scholar]
- 51.Garcia de la Torre J, Huertas ML, Carrasco B. HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J Magn Reson. 2000;147:138–46. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 52.Bernado P, Garcia de la Torre J, Pons M. Interpretation of 15N NMR relaxation data of globular proteins using hydrodynamic calculations with HYDRONMR. J Biomol NMR. 2002;23:139–50. doi: 10.1023/a:1016359412284. [DOI] [PubMed] [Google Scholar]
- 53.Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–63. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 54.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–70. [Google Scholar]
- 55.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–59. [Google Scholar]
- 56.Henras AK, Sam M, Hiley SL, Wu H, Hughes TR, Feigon J, Chanfreau GF. Biochemical and genomic analysis of substrate recognition by the double-stranded RNA binding domain of yeast RNase III. RNA. 2005;11:1225–37. doi: 10.1261/rna.2760705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol. 2001;19:773–6. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook J, Russell DW. Molecular Cloning: A laboratory manual. 3. Cold Spring Harbor Laboratory Press; New York: 2001. [Google Scholar]
- 59.Massi F, Grey MJ, Palmer AG., 3rd Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R1rho relaxation experiments. Protein Sci. 2005;14:735–42. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ravindranathan S, Oberstrass FC, Allain FH. Increase in backbone mobility of the VTS1p-SAM domain on binding to SRE-RNA. J Mol Biol. 2010;396:732–46. doi: 10.1016/j.jmb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 61.Maynard CM, Hall KB. Interactions between PTB RRMs induce slow motions and increase RNA binding affinity. J Mol Biol. 2010;397:260–77. doi: 10.1016/j.jmb.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kharrat A, Macias MJ, Gibson TJ, Nilges M, Pastore A. Structure of the dsRNA binding domain of E. coli RNase III. EMBO J. 1995;14:3572–84. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19:5567–74. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arumugam S, Gao G, Patton BL, Semenchenko V, Brew K, Van Doren SR. Increased backbone mobility in beta-barrel enhances entropy gain driving binding of N-TIMP-1 to MMP-3. J Mol Biol. 2003;327:719–34. doi: 10.1016/s0022-2836(03)00180-3. [DOI] [PubMed] [Google Scholar]
- 65.Weinberg DE, Nakanishi K, Patel DJ, Bartel DP. The inside-out mechanism of Dicers from budding yeasts. Cell. 2011;146:262–76. doi: 10.1016/j.cell.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.