

Abstract
Pituitary adenylate cyclase-activating peptide (PACAP) is a neuroprotective peptide which exerts its effects mainly through the cAMP-protein kinase A (PKA) pathway. Here, we show that in cortical neurons, PACAP-induced PKA signaling exerts a major part of its neuroprotective effects indirectly, by triggering action potential (AP) firing. Treatment of cortical neurons with PACAP induces a rapid and sustained PKA-dependent increase in AP firing and associated intracellular Ca2+ transients, which are essential for the anti-apoptotic actions of PACAP. Transient exposure to PACAP induces long-lasting neuroprotection in the face of apoptotic insults which is reliant on AP firing and the activation of cAMP response element (CRE) binding protein (CREB)-mediated gene expression. Although direct, activity-independent PKA signaling is sufficient to trigger phosphorylation on CREB’s activating serine-133 site, this is insufficient for activation of CREB-mediated gene expression. Full activation is dependent on CREB-regulated transcription co-activator 1 (CRTC1), whose PACAP-induced nuclear import is dependent on firing activity-dependent calcineurin signaling. Over-expression of CRTC1 is sufficient to rescue PACAP-induced CRE-mediated gene expression in the face of activity-blockade, while dominant negative CRTC1 interferes with PACAP-induced, CREB-mediated neuroprotection. Thus, the enhancement of AP firing may play a significant role in the neuroprotective actions of PACAP and other adenylate cyclase-coupled ligands.
Keywords: Ca2+ signaling, CREB, gene regulation, neuroprotective signalling, neurotoxicity, transcription factors
Pituitary adenylate cyclase-activating peptide (PACAP) is a neuropeptide first isolated from the hypothalamus as an activator of cAMP production in pituitary cells (Miyata et al. 1989). It exists in 27 and 38-amino acid forms and binds to three G-protein coupled receptors [PACAP-specific receptor (PAC1) and VIP/PACAP receptor subtypes 1 and 2] which are predominantly coupled to Gαs that promote cAMP production through the activation of adenylate cyclase (AC) (Dickson and Finlayson 2009). PACAP and its receptors are expressed widely in the CNS, where one of their key functions is neuroprotection. PACAP promotes the protection of cerebellar granule neurons against apoptotic and oxidative insults including ceramide, ethanol and H2O2 (Vaudry et al. 2009). PACAP also protects cortical and hippocampal neurons against excitotoxic and apoptotic insults (Shioda et al. 1998; Vaudry et al. 2009). In vivo, administration of PACAP reduces neuronal loss and neurological deficits in models of stroke and traumatic brain injury (Reglodi et al. 2002; Chen et al. 2006; Tamas et al. 2006b; Vaudry et al. 2009), excitotoxic striatal lesions (Tamas et al. 2006a) and Parkinson’s disease (Reglodi et al. 2004, 2006). Given this, PACAP has received considerable attention as a potential therapeutic neuroprotective drug (Somogyvari-Vigh and Reglodi 2004; Shioda et al. 2006; Brenneman 2007; Ohtaki et al. 2008; Vaudry et al. 2009).
PACAP promotes neuroprotection by acting directly on neuronal PACAP receptors (Vaudry et al. 2009). The molecular mechanisms that underlie this neuroprotection centre on activation of the cAMP-dependent protein kinase A (PKA), a major effector of intracellular cAMP (Botia et al. 2007; Vaudry et al. 2009). Activation of de novo gene expression has been implicated in PACAP-mediated neuroprotection, including c-Fos, brain-derived neurotrophic factor, Bcl-2 and PACAP itself (Frechilla et al. 2001; Falluel-Morel et al. 2004; Shintani et al. 2005; Aubert et al. 2006; Dejda et al. 2008). Of note, these genes are all regulated by the cAMP response element (CRE) binding protein (CREB) family of transcription factors, a group of factors that are important for the survival of central and peripheral neurons both pre- and postnatally (Walton et al. 1999; Lonze et al. 2002; Mantamadiotis et al. 2002) and whose activation contribute to the neuroprotective effects of neurotrophins and synaptic activity (Bonni et al. 1999; Riccio et al. 1999; Lee et al. 2005; Papadia et al. 2005). PACAP is known to promote CREB activation under conditions where it is neuroprotective (Racz et al. 2006; Falktoft et al. 2009), however, a causal link has up until now not been tested.
It is generally assumed that PACAP-mediated PKA signaling in neurons triggers neuroprotective gene expression and signal pathways by direct modulation of upstream effectors of these processes. However, we have considered an alternative explanation: that PACAP-induced PKA signaling exerts at least some of its neuroprotective effects indirectly though the enhancement of electrical activity. G-protein coupled receptors that activate cAMP/PKA signals in neurons, such as type I mGluRs and D1-type dopamine receptors, can potentiate synaptic strength and neuronal excitability, and modulate ion channel properties (Nguyen and Woo 2003). PACAP administration has been recently reported to enhance AMPAR currents as well as synaptic NMDAR currents (MacDonald et al. 2007; Costa et al. 2009) and to suppress the Apamin-insensitive slow after-hyperpolarization (IsAHP) current (Hu et al. 2011), which can control neuronal excitability.
Physiological patterns of action potential (AP) bursting are known to be strongly neuroprotective (Bell and Hardingham 2011), activating multiple pathways including CREB-mediated gene expression, antioxidant gene expression and the suppression of apoptotic genes (Hardingham 2006; Hetman and Kharebava 2006; Al-Mubarak et al. 2009; Hardingham and Bading 2010; Soriano et al. 2011; Zhang et al. 2011). An episode of burst activity can confer neuroprotection long after that episode has ceased, via a mechanism involving the activation of nuclear Ca2+- and CREB-dependent gene expression (Papadia et al. 2005; Hardingham 2009; Zhang et al. 2009). Thus, we have studied the effect of PACAP on levels of electrical activity in cortical neurons, and the role this plays in neuroprotection. We find that PACAP-induced PKA signaling triggers sustained increases in AP firing and that this firing activity is essential for PACAP-mediated neuroprotection. Specifically, PACAP-induced AP firing is required in order to trigger nuclear translocation of CREB-regulated transcription co-activator 1 (CRTC1, previously referred to as TORC1: Transducer Of Regulated CREB activity 1) in order to activate CREB-mediated gene expression and subsequent neuroprotection.
Materials and methods
Neuronal cultures and chemicals used
Cortical neurons from E21 Sprague–Dawley rats were cultured as described (Bading and Greenberg 1991; McKenzie et al. 2005) except that growth medium was comprised of Neurobasal A medium with B27 (Invitrogen, Carlsbad, CA, USA), 1% rat serum (Harlan Inc., Indianapolis, IN, USA), 1 mM glutamine. Experiments were performed after a culture period of 9–10 days during which neurons developed a rich network of processes, expressed functional NMDA-type and α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA)/kainate-type glutamate receptors, and formed synaptic contacts (Hardingham et al. 2001, 2002). PKA RIIβ wild-type and knockout mice (Brandon et al. 1998; Watson et al. 2006) were cultured as above from E17 animals. PACAP-27 was purchased from NeoMPS (Strasbourg, France). PACAP-27 and PACAP-38 are both found in the brain, and PACAP-27 was chosen since it represents the core functional region for activating PACAP receptors (Vaudry et al. 2009). Since we were concerned with events downstream of PACAP receptor activation, PACAP-27 was deemed sufficient for this purpose. MK801, KN-62 and forskolin from Tocris Cookson, Ballwin, MO, USA, bicuculline from Sigma, St Louis, MO, USA, PD-98059 from Ascent Scientific (Bristol, UK), H-89 from LC Laboratories (Woburn, MA, USA), staurosporine, tetrodotoxin (TTX) and 4-aminopyridine from Calbiochem, San Diego, CA, USA.
Models of neuronal apoptosis, PACAP-induced protection
Trophic deprivation
Neurons were transferred from growth medium to a trophically-deprived medium containing 10% minimal essential medium (Invitrogen), 90% Salt-Glucose-Glycine (SGG) medium (Papadia et al. 2005) (SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 0.5 mM sodium pyruvate, 0.1% Phenol Red; osmolarity 325 mOsm/L). When placed in this trophically-deprived medium, neurons exhibit significant levels of caspase-dependent apoptosis after 72 h (Papadia et al. 2005; Leveille et al. 2010). To assess neuroprotective signaling by continuous PACAP exposure, PACAP-27 (10 nM, NeoMPS) was administered at the point of trophic deprivation and left throughout the course of the experiment (72 h). The importance of PACAP-induced firing activity for neuroprotection was assessed by administering PACAP in the presence or absence of tetrodotoxin (1 μM). To assess the long-lasting neuroprotective effect of PACAP exposure (and its dependence on firing activity), PACAP ± TTX was administered at the point of trophic deprivation for 24 h, after which the medium was replaced with PACAP-free, TTX-containing medium. Cell death was quantified after a total of 72 h trophic deprivation.
Staurosporine-induced apoptosis
To induce rapid apoptotic cell death, and assess PACAP-induced protection, staurosporine treatment was employed as described (Papadia et al. 2005; Leveille et al. 2010). Briefly, neurons were placed in trophically deprived medium ± PACAP for 23 h, at which point the PACAP-treated neurons were given a second dose of PACAP. After a further 1 h, neurons were treated with 100 nM staurosporine, and death was assessed after a further 24 h. We have previously established that 100 nM staurosporine induced caspase-dependent apoptosis over this time period (Papadia et al. 2005; Leveille et al. 2010).
Assessment of cell death
To quantify cell death, neurons were fixed and subjected to DAPI (Vector Laboratories, Burlingame, CA, USA) staining and cell death quantified by counting (blind) the number of apoptotic nuclei as a percentage of the total. Approximately 1500 cells were counted per treatment, across at least four independent experiments (performed on different cultures). Morphologically, staurosporine-treated and trophically deprived neurons show typical signs of apoptotic-like cell death (shrunken cell body and large round chromatin clumps). Images were taken using a Leica AF6000 LX imaging system (Milton Keynes, UK), with a DFC350 FX digital camera.
Calcium imaging and analysis of imaging data
For pre-conditioning experiments, neurons were treated as indicated for 2 h, then transferred to aCSF (150 mM NaCl, 3 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 1 mM glucose) Ca2+ imaging was performed as described (Hardingham et al. 1997; Soriano et al. 2008b). Briefly, cells were loaded with 11 μM Fluo-3 AM [from a stock solution of 2.2 mM Fluo-3 dissolved in anhydrous dimethylsulfoxide containing 20% (w/v) Pluronic detergent] for 30 min at 37°C. Fluo-3 fluorescence images (excitation 488 nm, emission ≥ 515 nm) were taken at one frame per second. To calibrate images, Fluo-3 was saturated by adding 50 μM ionomycin to the perfusion chamber (to obtain Fmax) and quenched with 10 mM MnCl2 +50 μM ionomycin to levels corresponding to 100 nM Ca2+ (Minta et al. 1989), which was in turn used to calculate Fmin. Free Ca2+ concentrations were calculated from fluorescence signal (F) according to the equation [Ca2+] = Kd(F–Fmin)/(Fmax–F), and expressed as a multiple of the Kd of Fluo-3 (which is approximately 315 nM). In order to quantitate the effect of PACAP on firing activity-induced Ca2+ influx, the mean [Ca2+] 30 s before and 30 s after TTX treatment was calculated in either control neurons or neurons treated with PACAP ± H-89. For each cell, the degree of TTX-sensitive Ca2+ changes was calculated as the difference between mean [Ca2+] before and after TTX treatment. For each treatment, 60 cells were analysed within six independent experiments.
Electrophysiological recording and analysis
Coverslips containing cortical neurons were transferred to a recording chamber and perfused (at a flow rate of approximately 5 mL/min) with an external recording solution composed of 150 mM NaCl, 2.8 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 50 μM glycine, 2 μM strychnine and 10 mM glucose, pH 7.3 (320–330 mOsm). Patch-pipettes were made from thick-walled borosilicate glass (Harvard Apparatus, Kent, UK) and filled with a K-gluconate-based internal solution containing (in mM): 155 K-gluconate, 2 MgCl2, 10 Na-HEPES, 10 Na-PiCreatine, 2 Mg2-ATP and 0.3 Na3-GTP, pH 7.3 (300 mOsm). Electrode tips were fire-polished for a final resistance ranging between 5 and 10 MΩ. Currents were recorded at room temperature (21 ± 2°C) using an Axopatch-1C amplifier (Molecular Device, Union City, CA, USA) and stored on digital audio tape. Data were subsequently digitized and analyzed using WinEDR v6.1 software (John Dempster, University of Strathclyde, UK). Neurons were voltage-clamped at −70 mV, and recordings were rejected if the holding current was greater than −100 pA or if the series resistance drifted by more than 20% of its initial value (< 35 MΩ). Neurons were treated ± PACAP (10 nM) for 1–2 h prior to spontaneous excitatory post-synaptic currents being recorded in voltage-clamp for 5 min. Recordings were studied to determine whether they showed evidence of burst-like activity, defined as long periods of activity (> 1 s), peaking at > 50 pA.
Western blotting
In order to minimize the chance of post-translational modifications during the harvesting process, neurons were lysed immediately in 1.5× sample buffer (1.5 M Tris pH 6.8; Glycerol 15%; sodium dodecyl sulfate 3%; β-mercaptoethanol 7.5%; bromophenol blue 0.0375%) and boiled at 100°C for 5 min. Approximately 30 μg of protein was loaded onto a gel and subjected to gel electrophoresis and western blotting were performed using the Xcell Surelock system (Invitrogen) with precast gradient gels (4–20%) according to the manufacturer’s instructions. The gels were blotted onto polyvinylidene difluoride membranes, which were blocked for 1 h at 21 ± 2°C with 5% (w/v) non-fat dried milk in Tris-buffered saline with 0.1% Tween 20. The membranes were incubated at 4°C overnight with the primary antibodies diluted in blocking solution: Anti-phospho-CREB serine-133 (1 : 500, Upstate Biotechnology, Lake Placid, NY, USA) and CREB (1 : 500, Upstate). For visualisation of western blots, horseradish peroxidase-based secondary antibodies were used followed by chemiluminescent detection on X-Omat film (Kodak, Hemel Hempstead, UK). Western blots were analyzed by digitally scanning the blots, followed by densitometric analysis (ImageJ, National Institutes of Health, Washington DC, USA). All analysis involved normalizing to CREB expression as a loading control.
Transfection and luciferase assays
Neurons were transfected at DIV8 using Lipofectamine 2000 as described (McKenzie et al. 2006) using a total of 0.6–0.7 μg cDNA/well and 2.33 μL/well of Lipofectamine 2000 (1 μg/mL, from Invitrogen). Under these conditions transfection efficiency is approximately 2–5%, with > 99% of transfected cells NeuN-positive, and < 1% glial fibrillary acidic protein-positive (Papadia et al. 2008; Soriano et al. 2008a) confirming their neuronal identity. For CRE-reporter assays, neurons were transfected with 0.5 μg of CRE-Firefly Luciferase + 0.1 μg of pTK-Renilla (Promega, Madison, WI, USA); or 0.2 μg CRE-Luc, 0.1 μg Renilla and 0.4 μg of either β-globin control vector, or vectors encoding inducible cAMP early repressor 1 (ICER1) [a gift from Dr. Paulo Sassone-Corsi (Stehle et al. 1993)], CRTC1 (TORC1) or Dominant negative CRTC1 [CRTC1-DN (TORC1-N44), a gift from Dr Yang Zhou (Zhou et al. 2006)]. Stimulations were performed 24 h post-transfection. Neurons were treated with 10 nM PACAP or 5 μM Forskolin for 4 h, or with 50 μM bicuculline and 250 μM 4-aminopyridine for 8 h, with inhibitors added 1 h before. Assays were performed using the Dual Glo assay kit (Promega) and were performed on a FLUOstar OPTIMA (BMG Labtech, Aylesbury, UK). Firefly luciferase activity was normalized to the Renilla control in all cases and each experiment was performed at least four times.
Following the fate of transfected cells
The overall method to do this is as described (Papadia et al. 2008) with some modifications to the timing. Neurons were transfected with 0.5 μg of vectors expressing β-globin or ICER1 or CRTC1-DN plus 0.1 μg of a plasmid encoding enhanced green fluorescent protein (GFP) as a transfection marker. At 24 h post-transfection, neurons were placed in trophically-deprived medium and treated ± 10 nM PACAP. After a further 24 h, images were taken of GFP-expressing neurons using a Leica AF6000 LX imaging system, with a DFC350 FX digital camera, prior to the transfer of cell to PACAP-free, TTX-containing medium to block AP firing. Using mark-and-find software, the fate of the photographed neurons was followed at 24 h and 48 h after TTX treatment. 250–500 cells were analysed per treatment within 3–5 independent experiments.
CRTC1-localisation
For CRTC1-localisation studies, neurons were transfected with 0.6 μg GFP tagged CRTC1 [peGFP-C2/TORC1 a gift from Dr Dong-Yan Jin, Department of Biochemistry, University of Hong Kong, Hong Kong China (Siu et al. 2006)]. At 24 h post-transfection, neurons were treated with 20 ng/mL Leptomycin B (LC Laboratories) for 30 min in order to visualise CRTC1 import more clearly (Kovacs et al. 2007) and then 10 nM PACAP for a further 30 min in the presence of 1 μM TTX, 10 μM H-89 or 10 μM FK-506 (added 1 h before). Neurons were then fixed and stained for anti-GFP (1 : 750, Invitrogen) and visualised using biotinylated secondary antibody/cy3-conjugated streptavidin. Nuclei were counter-stained with DAPI. Subcellular distribution of CRTC1 was scored as being nuclear if levels were higher in the nucleus than in the surrounding perinuclear cytoplasm. 400–800 cells were analysed per treatment across 4–8 independent experiments.
Statistical analysis
Statistical testing involved a 2-tailed paired Student’s T-test. For studies employing multiple testing, we used a one-way ANOVA followed by Fisher’s least significant difference post-hoc test.
Results
PACAP triggers sustained increases in AP firing in cortical neurons
PACAP is known to promote PKA-dependent neuroprotection in a variety of systems in vitro and in vivo (see above). However, PKA activation is also capable of altering neuronal network activity through the control of intrinsic excitability and synaptic strength (Nguyen and Woo 2003). To investigate the effect of PACAP on levels of electrical activity we performed Ca2+ imaging experiments on cortical neurons pre-treated with PACAP. This pre-treatment resulted in enhanced AP firing as evidenced by strong oscillatory intracellular Ca2+ transients that were blocked by the Na+ channel antagonist TTX (Fig. 1a, quantitation in Fig. 1d). In contrast, control neurons exhibited far smaller TTX-sensitive Ca2+ transients (Fig. 1b and d). Pre-treatment of neurons with the PKA inhibitor H-89 prevented any PACAP-induced changes in Ca2+ oscillations (Fig. 1c and d). We also performed whole-cell voltage-clamp recordings of neurons pre-treated with PACAP which corroborated the Ca2+ imaging data: nine out of nine PACAP-treated neurons exhibited incoming excitatory post-synaptic currents consistent with burst-like activity (> 1 s in duration, > 50 pA at peak), compared to zero out of eight control neurons (Fig. 1e). Further Ca2+ imaging experiments revealed that acute administration of PACAP also had a similar effect, indicating that the potentiating effect of PACAP on AP firing is fast-acting as well as long-lasting (Fig. 1f). Thus, PACAP-induced PKA signaling in cortical neurons induces long-lasting increases in AP firing and associated Ca2+ transients.
Fig. 1.
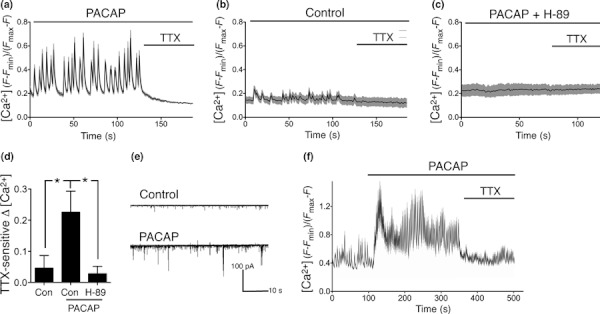
PACAP enhances AP firing in cortical neurons. (a–d) Pre-treatment with PACAP (10 nM PACAP-27 here and throughout the study) causes an increase in AP firing-dependent Ca2+ transients. Neurons were treated where indicated with PACAP ± H-89 (10 μM). After 2 h, the neurons were subjected to Fluo-3 Ca2+ imaging studies (see Methods for details) to monitor the size of Ca2+ transients in the different stimulation conditions. TTX (1 μM) was added where indicated to determine the extent to which the observed Ca2+ transients were because of action potential firing. Example traces are shown: black line indicates the mean Ca2+ concentration within a field of cells, and the grey shaded region indicates ± SEM of the Ca2+ concentration within that field. Free Ca2+ concentrations were calculated from fluorescence signal (F) according to the equation [Ca2+] = Kd(F–Fmin)/(Fmax–F), and expressed as a multiple of the Kd of Fluo-3 (which is approximately 315 nM). (d) shows quantification of data shown in (a–c), that is, quantification of the difference in mean amplitude of [Ca2+] before and after 1 μM TTX treatment. In order to quantitate the effect of PACAP on firing activity-induced [Ca2+] influx, the mean [Ca2+] 30 s before and 30 s after TTX treatment was calculated in either control neurons or neurons treated with PACAP ± H-89. For each cell, the degree of TTX-sensitive Ca2+ changes was calculated as the difference between mean [Ca2+] before and after TTX treatment. For each condition, 60 cells were analysed within six independent experiments (*p < 0.05). (e) Example trace of a whole-cell voltage-clamp recording of a control and PACAP-treated cortical neurons. PACAP causes an increase in burst-like activity, consistent with the Ca2+ imaging data. (f) Ca2+ imaging of acute PACAP treatment, a typical example trace is shown representative of six independent experiments.
Enhanced AP firing is essential for PACAP-induced neuroprotection
It is known that elevated electrical activity can promote neuroprotection in cortical neurons (Mennerick and Zorumski 2000; Bell and Hardingham 2011), raising the possibility that PACAP-induced AP firing contributes to its neuroprotective effect. We studied the capacity of PACAP to protect neurons against two different apoptotic insults, and studied the effect of blocking AP firing by TTX treatment. We first used staurosporine which induces caspase-dependent apoptosis of cortical neurons (Papadia et al. 2005). Pre-treatment of cortical neurons with PACAP before exposure to staurosporine for 24 h reduced levels of apoptosis (Fig. 2a and b). TTX treatment alone enhanced basal levels of neuronal apoptosis, however, staurosporine treatment caused additional neuronal loss. Importantly, we found that PACAP treatment failed to protect neurons against apoptosis in the presence of TTX.
Fig. 2.
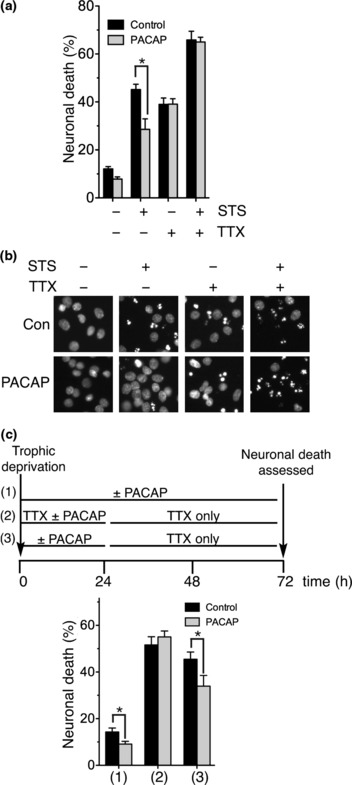
PACAP promotes resistance to apoptotic stimuli which is dependent on AP firing. (a,b) PACAP protects against staurosporine-induced cell death, but not in the presence of TTX. Neurons were treated with PACAP in the presence or absence of TTX 24 h and 1 h before treatment with 100 nM staurosporine. After a further 24 h, the cells were then fixed and DAPI stained and death was measured by counting pyknotic and non-pyknotic nuclei (*p < 0.05, n = 4); (b) shows example pictures. (c) PACAP-induced AP firing protects against trophic deprivation and promotes long lasting neuroprotection. At t = 0, the neurons were placed in trophically-deprived medium and given one of the three treatment regimes outlined in the upper schematic (1–3). At t = 72 h, cells were fixed, DAPI stained and levels of neuronal death analysed (*p < 0.05, n = 3).
We next employed a model of prolonged trophic deprivation (72 h) that also induces progressive caspase-dependent apoptosis (Papadia et al. 2005; Leveille et al. 2010). Once again, PACAP treatment protected neurons against apoptosis in control, although overall levels of apoptosis were not high [Fig. 2c, treatment (1)]. An episode of AP firing can promote neuroprotection that lasts well beyond the point at which that activity ends (Papadia et al. 2005). We hypothesised that PACAP-induced AP firing would similarly be able to exert long-lasting neuroprotection. Neurons subjected to trophic deprivation were treated with or without PACAP for 24 h, after which all neurons were placed in PACAP-free, TTX-containing medium. Levels of apoptosis were then assessed after a further 48 h [Fig. 2c, treatment (3)]. PACAP treatment was found to confer significant neuroprotection [Fig. 2c, treatment (3)] and this was dependent on PACAP-induced AP firing, since no protection was observed if PACAP was administered in the presence of TTX [Fig. 2c, treatment (2)]. Thus, PACAP-induced AP firing confers long-lasting neuroprotection. We conclude from these experiments that PACAP-induced enhancement of AP firing is important for its neuroprotective effects in these models of cortical neuronal apoptosis. This suggests that activation of PKA signaling is insufficient to directly activate certain neuroprotective pathways, and that it activates them indirectly by inducing AP firing which in turn triggers Ca2+-dependent signaling pathways that induce pro-survival events.
Induction of CREB-mediated gene expression contributes to PACAP-mediated neuroprotection
We next investigated the mechanism by which PACAP-induced AP firing leads to long-lasting neuroprotection. The CREB family controls the expression of a number of pro-survival genes containing CRE promoter elements and is a target for activation by both cAMP/PKA signals as well as activity-dependent Ca2+ signaling (Lonze and Ginty 2002). CREB itself is the predominant member in forebrain neurons (Papadia et al. 2005) and so is likely to be responsible for the majority of CRE-mediated gene expression. PACAP treatment resulted in the strong activation of a CRE-reporter (which was blocked by a PACAP receptor antagonist, Fig. 3a, lower), raising the possibility that CREB activation contributes to PACAP-mediated neuroprotection. We studied the effect of blocking CRE-dependent gene expression by transfecting neurons with a vector encoding ICER1, which is an inhibitory isoform of the CREB family (De Cesare and Sassone-Corsi 2000). We confirmed the efficacy of ICER1: expression of ICER1 blocked PACAP-induction of a CRE-reporter gene (Fig. 3a, upper).
Fig. 3.
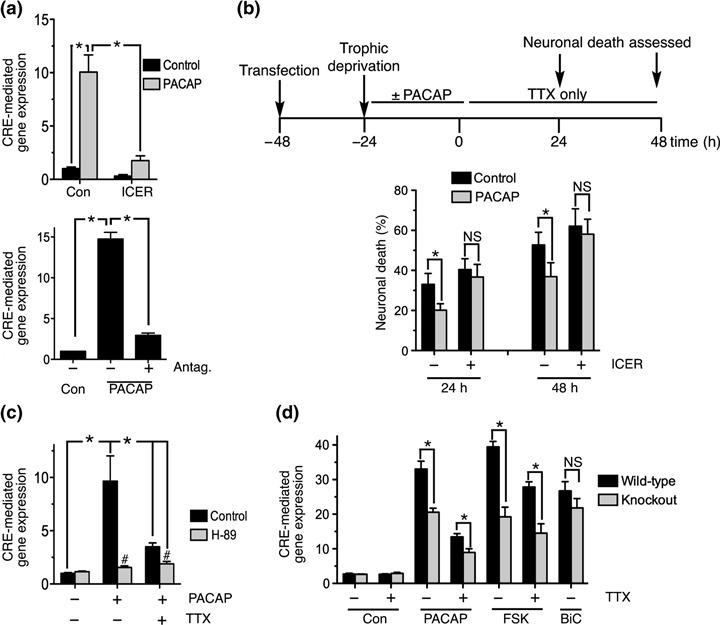
PACAP induces CRE-dependent gene expression, which is neuroprotective, and relies on AP firing. (a) Upper-PACAP induces CRE-mediated gene expression. Neurons were transfected with a CRE-Firefly luciferase vector, pTK renilla transfection control and vectors encoding either ICER1 or control (β-globin). See Methods section for exact quantities used. At 24 h post-transfection, neurons were treated with PACAP and luciferase expression was measured after a further 4 h. CRE-Firefly luciferase activity was normalised to Renilla control (*p < 0.05, n = 3). Lower-Effect of the PACAP antagonist (Antag, PACAP6–38, 1 μM) on PACAP induction of CRE-luciferase (*p < 0.05, n = 3). (b) PACAP mediated long-lasting neuroprotection depends on activation of CRE-mediated gene expression. Upper panel illustrates the experimental protocol. Briefly, neurons expressing GFP plus either ICER1 or β-globin control were treated ± PACAP 24 h post-transfection and then all cells were placed in TTX-containing medium after a further 24 h, at which point images of GFP-expressing neurons were taken (t = 0 in the upper schematic). The fate of these cells was then monitored at 24 and 48 h after this medium change. 250–400 cells were analysed per treatment across six cultures within three independent experiments. (*p < 0.05). (c) PACAP induced CRE-dependent gene expression is dependent on AP firing. Neurons were treated with PACAP where indicated for 4 h; all other drugs were added 1 h beforehand (*p < 0.05, #p < 0.05 comparing H-89 with control for that particular PACAP/TTX condition, n = 7). (d) PACAP and forskolin-induced activation of CRE-mediated gene expression is disrupted in RIIβ-deficient neurons: both AP firing-dependent and independent components. Forskolin was used at 5 μM. For comparison is an illustration of the RIIβ-independence of CRE activation triggered by promoting AP firing by network disinhibition through treatment with the GABAA receptor blocker bicuculline (50 μM) plus 250 μM 4-aminopyridine, which is a PKA-independent way of inducing AP firing (Papadia et al. 2005) (*p < 0.05, n = 6).
To assess the importance of CREB in PACAP-induced long-lasting protection, neurons were transfected with vectors expressing either β-globin (control) or ICER1 plus a peGFP transfection marker. At 24 h post-transfection, neurons were placed in trophically-deprived medium and treated ± PACAP. After a further 24 h, images were taken of GFP-expressing neurons, prior to the transfer of the neurons to PACAP-free, TTX-containing medium to block AP firing. The fate of the transfected neurons was followed at 24 h and 48 h after TTX treatment (see Fig. 3b, schematic). We found that the transfection procedure caused slightly higher rates of neuronal death than in untransfected cells. However, in control-transfected neurons, PACAP treatment promoted significant protection both 24 h and 48 h after the removal of PACAP (Fig. 3b). Importantly, PACAP treatment was not significantly neuroprotective in ICER1-expressing neurons (Fig. 3b), indicating a role for CRE/CREB-dependent gene expression in PACAP-mediated long-lasting neuroprotection.
AP firing underlies PACAP-induced CREB activation
The fact that PACAP-induced neuroprotection is not observed when neurons are co-treated with TTX suggested that activation of CRE-dependent gene expression could be dependent on AP firing. Indeed, we found this to be the case: TTX treatment alone had little effect on basal activity of a CRE- reporter, but inhibited PACAP-mediated activation by around 80% (Fig. 3c). PKA inhibition by H-89 treatment completely blocked the induction of the CRE reporter by PACAP, including the small TTX-insensitive component. Taken together, these data show that direct signaling by PKA is able to support weak activation of CRE-dependent gene expression, but that AP firing is needed for strong CRE-induction and resultant neuroprotection.
To further confirm the role of PKA in both activity-dependent and -independent activation of CREB by PACAP, we studied activation of a CRE reporter in neurons cultured from a mouse deficient in the RIIβ subunit of PKA. In the RIIβ−/− mouse, levels of cAMP-inducible PKA activity within the cortex are lower than wild-type, while basal PKA activity is similar (Brandon et al. 1998). We found that both TTX-sensitive and -insensitive components of PACAP-induced CRE-mediated gene expression were lower in RIIβ-null neurons (Fig. 3d). The level of reduction in PACAP-induced CRE-activation in the RIIβ-null neurons was comparable to that seen in the context of the adenylate cyclase activator forskolin (Fig. 3d), confirming that PKA is central to both AP-dependent and -independent components of CREB activation by PACAP.
Given that activity-dependent Ca2+ influx can activate Ca2+-dependent adenylate cyclases, it was theoretically possible that PKA could play a role in CREB activation downstream of AP firing. However, it has been shown that strong firing activity does not cause global levels of cAMP to rise sufficiently high to support PKA signaling to CREB in the nucleus (Pokorska et al. 2003). Nevertheless, to investigate this directly we studied the activation of CRE-mediated gene expression by AP firing induced via a PKA-independent mechanism: network disinhibition by the GABAA receptor blocker bicuculline plus the K+ channel blocker 4-aminopyridine [to enhance burst frequency (Hardingham et al. 2001)]. Induction of CRE-mediated gene expression by bicuculline/4-AP-induced AP firing was not lower in RIIβ-null neurons (Fig. 3d). This indicates that cAMP/PKA signaling is not a major mediator of CRE-dependent gene expression downstream of AP firing, in agreement with previous studies (Pokorska et al. 2003). Collectively these observations support a model whereby PACAP-induced PKA signaling weakly activates CREB directly, but triggers strong CREB activation by promoting AP firing which in turn activates CREB via PKA-independent pathways.
PACAP-induced CREB phosphorylation does not require AP firing
We next investigated which CRE-activating molecular events triggered by PACAP treatment are actually reliant on activity-dependent Ca2+ signals, and whether any can be triggered in an activity-independent manner by direct PKA signaling. CREB phosphorylation on serine-133 is essential for CREB activation since it triggers the recruitment of the co-activator CREB binding protein (CBP; Chrivia et al. 1993). CREB phosphorylation was induced by PACAP treatment (Fig. 4a). Interestingly, TTX treatment did not interfere with PACAP-induced CREB phosphorylation (Fig. 4b and c), while the PKA inhibitor H-89 blocked CREB phosphorylation (Fig. 4a and c). Serine-133 of CREB is a good substrate for PKA (Gonzalez and Montminy 1989), and these data indicate that PACAP-induced PKA activity is sufficient to result in the direct phosphorylation of CREB, and that AP firing is not needed for this particular activation step. However, while CREB serine-133 phosphorylation is necessary for full activation of CREB, it is not sufficient. A key secondary activation step involves the co-activator CRTC (CREB-regulated transcription co-activator), which is subject to Ca2+-dependent nuclear import, where it binds to CREB and enhances its affinity for both CBP and the basal transcriptional machinery (Conkright et al. 2003; Screaton et al. 2004; Zhou et al. 2006; Kovacs et al. 2007; Li et al. 2009).
Fig. 4.
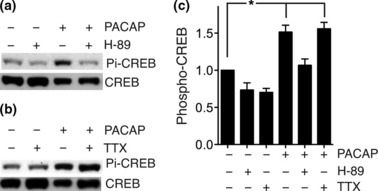
PACAP-mediated induction of serine-133 CREB phosphorylation does not require AP firing. (a–c) PACAP induces phosphorylation of CREB at serine-133 in a TTX-insensitive, PKA-dependent manner. Neurons were pre-treated with TTX or H-89 and then treated for 15 min with PACAP. Protein was harvested and subject to western analysis for phospho-CREB (see Methods, normalized in all cases to total CREB, *p < 0.05, n = 4, example blots are shown). (a) and (b) show example westerns and (c) shows quantitation of phospho-CREB levels (normalized to total CREB).
PACAP-induced AP firing mediates calcineurin-dependent CRTC1 nuclear import
We confirmed the importance of CRTC for CRE activation: expression of a dominant negative mutant of CRTC1 (CRTC1-DN; Zhou et al. 2006) strongly inhibited PACAP-induction of the CRE-mediated gene expression, as well as that induced by bicuculline/4-AP treatment (Fig. 5a). We also investigated the importance of CRTC signaling in PACAP-mediated long-lasting neuroprotection, using an identical protocol to that used in Fig. 3(b), except that the ICER-encoding vector was replaced with that of CRTC1-DN. At the 48 h timepoint (after removal of PACAP from the medium), a very small, but statistically significant, amount of PACAP-dependent neuroprotection was still observed at 48 h in CRTC1-DN-expressing neurons. However, levels of neuronal death in CRTC1-DN-expressing neurons previously exposed to PACAP were significantly higher than control-transfected cells previously exposed to PACAP (Fig. 5b and c). Thus, CRTC1-DN interferes with neuroprotection evoked by transient exposure to PACAP, consistent with the role of CREB in this process, and the importance of CRTCs in CREB-mediated gene expression.
Fig. 5.
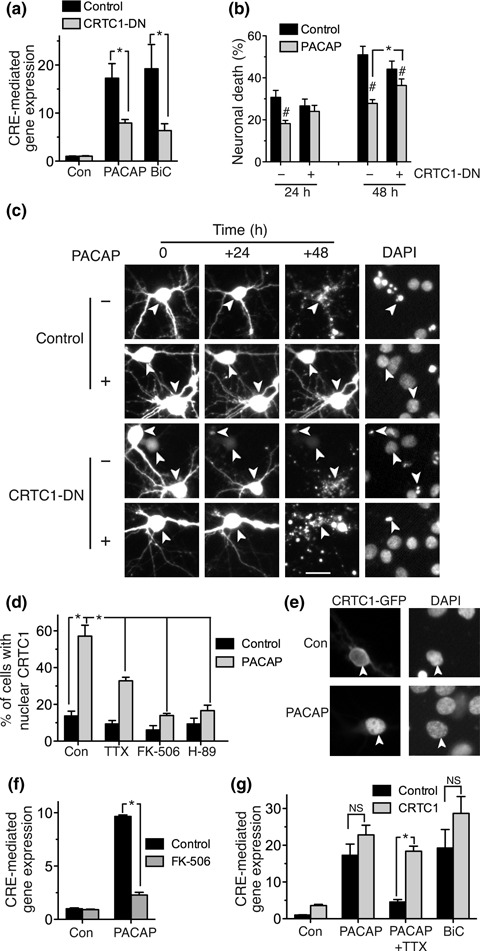
PACAP induces nuclear translocation of CRTC1, necessary for the AP firing-dependent component of CREB activation. (a) CRTC1 dominant negative inhibits PACAP mediated activation of CREB. Neurons were transfected with CRE-luciferase, pTK-Renilla and vectors encoding either a CRTC1 dominant negative mutant or control (β-globin). Neurons were stimulated PACAP or bicuculline plus 4-AP (BiC) (*p < 0.05, n = 4). (b,c) PACAP mediated long-lasting neuroprotection depends on CRTC1. The experimental protocol is the same as that illustrated schematically in Fig. 3(b). Briefly, neurons expressing GFP plus either CRTC1-DN (dominant negative) or β-globin control were treated ± PACAP 24 h post-transfection and then all cells were placed in TTX-containing medium after a further 24 h, at which point images of GFP-expressing neurons were taken. The fate of these cells was then monitored at 24 and 48 h after this medium change (*p < 0.05, paired T-test, n = 3; #p < 0.05, paired T-test comparing control to PACAP within each condition/timepoint). (c) shows example pictures. Scale bar = 20 μm. (d,e) PACAP induces CRTC1 nuclear translocation via activity-dependent calcineurin signaling. Neurons were transfected with a vector encoding GFP-tagged CRTC1. At 24 h post-transfection, neurons were treated with 20 ng/mL leptomycin B for 30 min to block nuclear export [to enable import to be observed more clearly (Kovacs et al. 2007)], plus the indicated inhibitors (1 μM TTX, 10 μM H-89 or 10 μM FK-506) and then PACAP added for 30 min prior to fixing of the cells and analysing localisation of GFP-CRTC1 in 400–800 cells per treatment (*p < 0.05, n = 4–8). (e) shows example pictures. (f) PACAP-induced activation of CRE-mediated gene expression requires the Ca2+-dependent phosphatase calcineurin. Where used, FK-506 was added 1 h prior to PACAP stimulation (*p < 0.05, n = 4). (g) CRTC1 over-expression rescues the inhibition of PACAP-mediated CRE activation by TTX. Neurons were transfected with CRE-luciferase, pTK-Renilla and either vectors encoding CRTC1 or β-globin control. 24 h post-transfection the neurons were stimulated with PACAP ± TTX or bicuculline + 4-AP (BiC) as indicated. Over-expression of CRTC1 does not further enhance CRE activation by BiC or PACAP, suggesting that levels are not limiting, however, it strongly enhances levels induced by PACAP in the presence of TTX (*p < 0.05, n = 4).
Nuclear translocation of CRTC is an important step in the full activation of CREB-dependent gene expression (Screaton et al. 2004). We found that PACAP treatment caused the nuclear translocation of CRTC1 that was inhibited by TTX (Fig. 5d and e). Activity-dependent Ca2+ influx is known to induce CRTC1 translocation through activation of the Ca2+-dependent phosphatase calcineurin. Calcineurin subsequently dephosphorylates CRTC, triggering its nuclear import and co-activation of CREB (Li et al. 2009). This mechanism is employed in the case of PACAP signaling: inhibition of calcineurin activity by treatment with the inhibitor FK-506 inhibited PACAP-mediated CRTC1 translocation (Fig. 5d) and PACAP-mediated induction of CRE-mediated gene expression (Fig. 5f). As a control, we wanted to confirm that FK-506 was not inhibiting the induction of activity-dependent Ca2+ transients. Using the same methodology as for Fig. 1(d), we found that TTX-sensitive Ca2+ elevation in PACAP + FK-506-treated neurons was 100 ± 6.4% of that in neurons treated with PACAP alone (n = 3, 25–30 cells analysed per treatment). Thus, the inhibitory effect of FK-506 on PACAP-mediated CRTC1 translocation is not because of any indirect interference in the induction of activity-dependent Ca2+ transients.
Taken together, these observations suggest that a key reason why TTX inhibits PACAP-activation of CRE-mediated gene expression is that blockade of CRTC nuclear import renders nuclear levels of CRTC too low to efficiently co-activate CREB. We postulated that if CRTC was indeed limiting, then if we over-expressed CRTC1, then this might rescue the inhibitory effect of TTX on PACAP activation of the CRE reporter. Although over-expressed CRTC1 would be mainly cytoplasmic, we reasoned that since a proportion of it is nuclear then this could rescue the deficiency in nuclear levels. We found this to be the case: over-expression of CRTC1 reversed the inhibitory effect of TTX on PACAP-induction of CREB-dependent gene expression (Fig. 5g). CRTC1 over-expression, however, did not further enhance PACAP activation of CREB-mediated gene expression in the absence of TTX (Fig. 5g), indicating that PACAP-induced firing causes sufficient CRTC nuclear import such that levels of nuclear CRTC are not limiting for efficient co-activation of CREB. Thus, while PACAP activation of direct PKA signaling is sufficient to induce CREB phosphorylation, this is insufficient to activate CREB on its own. Enhancement of AP firing is critical in order to induce calcineurin-dependent CRTC nuclear translocation, an important step in CREB activation and consequent neuroprotection.
Discussion
This study shows that certain PACAP-mediated anti-apoptotic signals in cortical neurons are not mediated by direct cAMP/PKA-dependent activation. Instead, the primary role of cAMP/PKA signaling is to enhance neuronal network activity. The resulting AP-dependent Ca2+ transients are the direct activators of neuroprotection, and induce a long-lasting phase of protection dependent on activation of CREB-mediated gene expression. These events are illustrated schematically in Fig. 6.
Fig. 6.
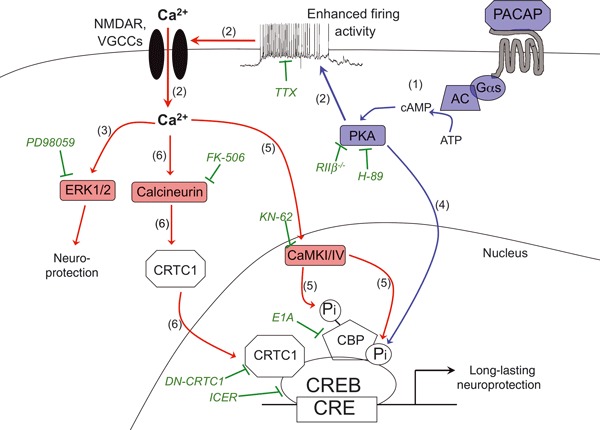
Schematic illustration of the role of activity-dependent Ca2+ signaling in PACAP-mediated neuroprotection. Activation of PACAP receptors leads to activation of PKA via the classical G-protein-adenylate cyclase (AC)-cAMP: pathway (1). PKA activation causes an increase in synaptic strength and/or neuronal excitability leading to a strong increase in levels of action potential firing which in turn triggers intracellular Ca2+ influx, likely through synaptic receptors (e.g. NMDA receptors) or voltage-gated Ca2+ channels (VGCCs): pathway (2). Activation of long-lasting neuroprotection by PACAP requires induction of gene expression mediated by the transcription factor CREB. CREB phosphorylation on serine-133 can be triggered directly by PKA in an AP firing-independent manner: pathway (3). However, this is insufficient to fully activate CREB-mediated gene expression. A key Ca2+/activity-dependent pathway involves CRTC1 nuclear translocation through activation of the Ca2+-dependent phosphatase calcineurin: pathway (4). Blue arrows and molecules indicate AP firing activity-independent events, while red arrows and molecules highlight the events dependent on AP firing. The pharmacological and genetic inhibitors of the various pathways used in this study are shown in green.
Modulation of neuronal electrical activity by PACAP and other AC-coupled ligands
The ability of PACAP to induce AP firing in networks of cortical neurons is consistent with the known influence of intracellular cAMP on neuronal excitability. Neurotransmitters, neuropeptides and pharmacological compounds that activate AC are well-known to modulate neuronal excitability, ion channel conductance, and synaptic transmission and plasticity, predominantly through PKA activation (Nguyen and Woo 2003). At the synapse, pharmacological activators of AC, and agonists of AC-coupled receptors such as the D1/D5 dopamine receptor, or the β-adrenergic receptor, all mimic long term potentiation (LTP) and/or enhance excitatory post-synaptic potentials (Nguyen and Woo 2003). Mice deficient in AC1 and AC8 show deficits in LTP and spatial memory (Nguyen and Woo 2003; Ferguson and Storm 2004). At the molecular level, PKA-mediated GluR1 phosphorylation at serine-845 increases AMPA receptor open probability and stabilizes synaptic location of AMPA receptors trafficked to the synapse during LTP (Banke et al. 2000; Esteban et al. 2003; Lee et al. 2003). PACAP at low doses is known to enhance AMPAR currents via PKA activation (Costa et al. 2009) as well as synaptic NMDAR currents (MacDonald et al. 2007). Moreover, mice deficient in PACAP have defective LTP at the mossy fibre synapse, implicating endogenous PACAP signaling in synaptic potentiation (Otto et al. 2001). In addition to modifying the properties of synaptic glutamate receptors, AC-coupled PKA signaling also can modulate neuronal excitability by controlling the IsAHP. IsAHP is mediated by a Ca2+ activated potassium current which is activated in response to bursts of AP firing. This is a key negative regulator of neuronal excitability, inducing a prolonged state of hyperpolarization, and this is in turn negatively regulated by AC-coupled PKA activity induced either pharmacologically (e.g. forskolin) or by treatment with AC-coupled ligands (e.g. dopamine) (Pedarzani and Storm 1995; Lancaster et al. 2006; Hu et al. 2011). PACAP treatment itself leads to inhibition of IsAHP in cortical pyramidal neurons (Hu et al. 2011), which could contribute to the enhanced AP firing that we observe (Fig. 1).
PACAP-induced AP firing promotes CREB-dependent neuroprotection
The CREB family of transcription factors is known to be an important mediator of activity-dependent gene expression (Lonze and Ginty 2002). The potential of CREB family-regulated gene products to promote neuronal survival was first demonstrated in the context of neurotrophin signaling (Bonni et al. 1999; Riccio et al. 1999) and exogenous over-expression (Walton et al. 1999). In addition, studies of mice where CREB and/or CREB family members have been deleted also point to a pro-survival role for CREB in vivo both pre- and postnatally (Lonze et al. 2002; Mantamadiotis et al. 2002). CREB-dependent gene expression is causally linked to the long-lasting phase of activity-dependent neuroprotection against apoptotic insults (Papadia et al. 2005) and data presented in this study supports the idea that PACAP treatment recruits this activity-dependent neuroprotective pathway.
Activation of CREB-mediated gene expression requires serine-133 phosphorylation which is necessary to recruit CBP, a transcriptional cofactor, to CREB (Chrivia et al. 1993). Several Ca2+-activated kinase cascades can mediate this event, including the Ras-extracellular signal regulated kinase 1/2 pathway and also nuclear Ca2+/calmodulin-dependent protein kinase (CaM kinase) (Soriano and Hardingham 2007). However, PACAP induced CREB phosphorylation does not require these activity-dependent pathways, since even when AP firing is blocked CREB phosphorylation is still observed (Fig. 4). This is consistent with the fact that PKA is also a CREB kinase and indicates that PACAP-induced PKA activity is strong enough to mediate this event directly.
However, direct PKA activity induced by PACAP is not sufficient to induce subsequent activation steps, including nuclear translocation of CRTCs. CRTCs enhance the interaction of CREB with the TAF(II)130 component of TFIID following its recruitment to the promoter (Conkright et al. 2003). Calcineurin promotes nuclear translocation of CRTC2 through calcineurin-mediated dephosphorylation of serine-171 (Screaton et al. 2004). Translocation can be enhanced/synergized by PKA signaling with causes the inhibition of the serine-171-kinase – salt-inducible kinase-2 (Screaton et al. 2004). CRTC1 is the major isoform in the brain and is a key regulator of CREB-dependent gene expression (Kovacs et al. 2007; Li et al. 2009). Ca2+ signals promote the nuclear translocation of CRTC1, dependent on calcineurin signaling which directly dephosphorylates CRTC1 (Bittinger et al. 2004). Analogously with CRTC2, cAMP signals can also trigger the translocation of CRTC1 (Bittinger et al. 2004), most likely through the inhibition of salt-inducible kinase-mediated phosphorylation. In neurons, calcineurin activation is sufficient to trigger CRTC1 translocation (Li et al. 2009). The requirement for AP firing and calcineurin signaling for PACAP treatment to induce CRTC1 translocation strongly indicates that PACAP-induced PKA activity is not strong enough on its own to promote sufficient CRTC1 translocation directly, although may be playing a supporting role.
Another more recently discovered role for CRTC is in assisting the recruitment of CREB’s co-activator CBP to phospho(serine-133) CREB (Ravnskjaer et al. 2007). We and others have shown previously that CBP itself is activated by Ca2+ influx in neurons (via CaM kinase-dependent phosphorylation) which contributes to activation of CRE-mediated gene expression (Chawla et al. 1998; Hardingham et al. 1999; Impey et al. 2002). Thus, CBP activation may also contribute to the activation of CRE-mediated gene expression by PACAP-induced firing activity. Indeed, we observe strong activity-dependent activation of CBP’s transactivating potential by PACAP, which is both dependent on firing activity and CaM kinase activity (Baxter and Hardingham, unpublished observations).
PACAP prevents neuronal loss and dysfunction in vivo: potential role of enhanced AP firing
PACAP has been reported to protect neurons against a variety of insults including ceramide, glutamate and hydrogen peroxide-induced death (Vaudry et al. 2009), insults that synaptic activity also protects against (Lee et al. 2005; Papadia et al. 2005, 2008). Importantly, activation of CREB-mediated gene expression is implicated in activity-dependent protection against both apoptotic and excitotoxic insults (Lee et al. 2005; Papadia et al. 2005). Based on this study, it may be that indirect activity-dependent signaling to CREB contributes to the neuroprotective effects of PACAP in vitro and also begs the question as to whether any of its in vivo effects are similarly because of enhancing neuronal activity.
In vivo administration of PACAP reduces neuronal loss in the substantia nigra in acute models of Parkinson’s Disease: 6-Hydroxydopamine and MPTP treatment (Reglodi et al. 2004, 2006; Wang et al. 2008). However, most neuroprotective studies on PACAP have centred on excitotoxic trauma: principally stroke, traumatic brain injury (TBI) and retinal injury. PACAP crosses the blood brain barrier and can be administered intravenously to decrease damage in several models of ischemia and is effective even when administered several hours after the ischemic episode (Uchida et al. 1996; Reglodi et al. 2000; Chen et al. 2006; Ohtaki et al. 2008). Enhanced neuronal AP firing is known to protect neurons against excitotoxic cell death including ischemic conditions (Lee et al. 2005; Tauskela et al. 2008) and so the notion that PACAP can reduce neuronal damage in part by promoting AP firing is plausible. PACAP is also highly effective in ameliorating damage to the retina in a variety of trauma models, including excitotoxic and ischemic injury (Atlasz et al. 2010). In addition, post-insult PACAP treatment also reduces the extent of axonal damage following TBI (Farkas et al. 2004; Tamas et al. 2006b). TBI is characterised by brief acute hyperactivity of ionotropic glutamate receptors, including NMDA receptors, which mediate acute excitotoxic damage, followed by sustained loss of function (Biegon et al. 2004; Yaka et al. 2007). As such the NMDA receptor has been proposed to rapidly switch between ‘destructive’ and ‘recovery’ roles (Biegon et al. 2004; Yaka et al. 2007). In the immature brain, treatment with NMDAR antagonists reduces primary excitotoxic death but exacerbates secondary apoptosis, resulting in increased overall death (Pohl et al. 1999). By promoting AP firing, PACAP may boost the recovery phase post-injury by mechanisms related to those described in this study, as well as others more specific to the activity-dependent protection of axons.
Of course, enhanced neuronal activity is very unlikely to mediate all the effects of PACAP in the CNS: direct activity-independent effects are likely to be exerted in neurons as well. Moreover, there are well-documented neuroprotective effects of PACAP acting indirectly via non-neuronal cells. For example, PACAP stimulates the astrocytic release of neuroprotective IL-6 (Ohtaki et al. 2008) and also suppresses microglial activation, thus reducing the release of potentially harmful cytokines that can form part of the post-ischemic response (Gonzalez-Rey et al. 2007; Vaudry et al. 2009). Nevertheless, the impact of PACAP on neuronal activity should be taken into account when assessing the mechanism and extent of any therapeutic effect.
Acknowledgments
We thank Paulo Sassone-Corsi, Yang Zhou and Dong-Yan Jin for plasmids. This work is funded by the Wellcome Trust, the MRC and the BBSRC.
Glossary
Abbreviations used
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionate
- AP
action potential
- CaM kinase
Ca2+/calmodulin-dependent protein kinase
- CBP
CREB binding protein
- CRE
cAMP response element
- CREB
CRE binding protein
- CRTC
CREB-regulated transcription co-activator
- CRTC1-DN
dominant negative CRTC1
- GFP
green fluorescent protein
- ICER
inducible cAMP early repressor
- IsAHP
Apamin-insensitive slow after-hyperpolarization current
- LTP
long term potentiation
- PACAP
pituitary adenylate cyclase-activating peptide
- PKA
protein kinase A
- TBI
traumatic brain injury
- TTX
tetrodotoxin
- TORC1
transducer of regulated CREB activity 1
References
- Al-Mubarak B, Soriano FX, Hardingham GE. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels (Austin, Tex) 2009;3:233–238. doi: 10.4161/chan.3.4.9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlasz T, Szabadfi K, Kiss P, et al. Pituitary adenylate cyclase activating polypeptide in the retina: focus on the retinoprotective effects. Ann. N Y Acad. Sci. 2010;1200:128–139. doi: 10.1111/j.1749-6632.2010.05512.x. [DOI] [PubMed] [Google Scholar]
- Aubert N, Falluel-Morel A, Vaudry D, et al. PACAP and C2-ceramide generate different AP-1 complexes through a MAP-kinase-dependent pathway: involvement of c-Fos in PACAP-induced Bcl-2 expression. J. Neurochem. 2006;99:1237–1250. doi: 10.1111/j.1471-4159.2006.04148.x. [DOI] [PubMed] [Google Scholar]
- Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KF, Hardingham GE. The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 2011;21:299–305. doi: 10.1016/j.conb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc. Natl Acad. Sci. USA. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr. Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Botia B, Basille M, Allais A, et al. Neurotrophic effects of PACAP in the cerebellar cortex. Peptides. 2007;28:1746–1752. doi: 10.1016/j.peptides.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Logue SF, Adams MR, et al. Defective motor behavior and neural gene expression in RIIbeta-protein kinase A mutant mice. J. Neurosci. 1998;18:3639–3649. doi: 10.1523/JNEUROSCI.18-10-03639.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE. Neuroprotection: a comparative view of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Peptides. 2007;28:1720–1726. doi: 10.1016/j.peptides.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Chen Y, Samal B, Hamelink CR, Xiang CC, Chen M, Vaudry D, Brownstein MJ, Hallenbeck JM, Eiden LE. Neuroprotection by endogenous and exogenous PACAP following stroke. Regul. Pept. 2006;137:4–19. doi: 10.1016/j.regpep.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Mol. Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Costa L, Santangelo F, Li Volsi G, Ciranna L. Modulation of AMPA receptor-mediated ion current by pituitary adenylate cyclase-activating polypeptide (PACAP) in CA1 pyramidal neurons from rat hippocampus. Hippocampus. 2009;19:99–109. doi: 10.1002/hipo.20488. [DOI] [PubMed] [Google Scholar]
- De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog. Nucleic Acid Res. Mol. Biol. 2000;64:343–369. doi: 10.1016/s0079-6603(00)64009-6. [DOI] [PubMed] [Google Scholar]
- Dejda A, Jolivel V, Bourgault S, Seaborn T, Fournier A, Vaudry H, Vaudry D. Inhibitory effect of PACAP on caspase activity in neuronal apoptosis: a better understanding towards therapeutic applications in neurodegenerative diseases. J. Mol. Neurosci. 2008;36:26–37. doi: 10.1007/s12031-008-9087-1. [DOI] [PubMed] [Google Scholar]
- Dickson L, Finlayson K. VPAC and PAC receptors: from ligands to function. Pharmacol. Ther. 2009;121:294–316. doi: 10.1016/j.pharmthera.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Falktoft B, Georg B, Fahrenkrug J. Signaling pathways in PACAP regulation of VIP gene expression in human neuroblastoma cells. Neuropeptides. 2009;43:387–396. doi: 10.1016/j.npep.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Falluel-Morel A, Aubert N, Vaudry D, Basille M, Fontaine M, Fournier A, Vaudry H, Gonzalez BJ. Opposite regulation of the mitochondrial apoptotic pathway by C2-ceramide and PACAP through a MAP-kinase-dependent mechanism in cerebellar granule cells. J. Neurochem. 2004;91:1231–1243. doi: 10.1111/j.1471-4159.2004.02810.x. [DOI] [PubMed] [Google Scholar]
- Farkas O, Tamas A, Zsombok A, Reglodi D, Pal J, Buki A, Lengvari I, Povlishock JT, Doczi T. Effects of pituitary adenylate cyclase activating polypeptide in a rat model of traumatic brain injury. Regul. Pept. 2004;123:69–75. doi: 10.1016/j.regpep.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Ferguson GD, Storm DR. Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda) 2004;19:271–276. doi: 10.1152/physiol.00010.2004. [DOI] [PubMed] [Google Scholar]
- Frechilla D, Garcia-Osta A, Palacios S, Cenarruzabeitia E, Del Rio J. BDNF mediates the neuroprotective effect of PACAP-38 on rat cortical neurons. Neuroreport. 2001;12:919–923. doi: 10.1097/00001756-200104170-00011. [DOI] [PubMed] [Google Scholar]
- Gonzalez G, Montminy M. cAMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine-133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Varela N, Chorny A, Delgado M. Therapeutical approaches of vasoactive intestinal peptide as a pleiotropic immunomodulator. Curr. Pharm. Des. 2007;13:1113–1139. doi: 10.2174/138161207780618966. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem. Soc. Trans. 2006;34:936–938. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009;37:1147–1160. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr. Top. Med. Chem. 2006;6:787–799. doi: 10.2174/156802606777057553. [DOI] [PubMed] [Google Scholar]
- Hu E, Demmou L, Cauli B, et al. VIP, CRF, and PACAP act at distinct receptors to elicit different cAMP/PKA dynamics in the neocortex. Cereb. Cortex. 2011;21:708–718. doi: 10.1093/cercor/bhq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Kovacs KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc. Natl Acad. Sci. USA. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Gibb B, Storm JF. Kinetics of ion channel modulation by cAMP in rat hippocampal neurones. J. Physiol. 2006;576:403–417. doi: 10.1113/jphysiol.2006.115295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling stimulus intensity, and temporal regulation of CREB phosphorylation at Serine 133. J. Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveille F, Papadia S, Fricker M, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J. Neurosci. 2010;30:2623–2635. doi: 10.1523/JNEUROSCI.5115-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang C, Takemori H, Zhou Y, Xiong ZQ. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J. Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. G protein-coupled receptors control NMDARs and metaplasticity in the hippocampus. Biochim. Biophys. Acta. 2007;1768:941–951. doi: 10.1016/j.bbamem.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- McKenzie GJ, Stevenson P, Ward G, Papadia S, Bading H, Chawla S, Privalsky M, Hardingham GE. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005;93:171–185. doi: 10.1111/j.1471-4159.2005.03010.x. [DOI] [PubMed] [Google Scholar]
- McKenzie G, Ward G, Stallwood Y, Briend E, Papadia S, Lennard A, Turner M, Champion B, Hardingham GE. Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals. BMC Cell Biol. 2006;7:10. doi: 10.1186/1471-2121-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Neural activity and survival in the developing nervous system. Mol. Neurobiol. 2000;22:41–54. doi: 10.1385/MN:22:1-3:041. [DOI] [PubMed] [Google Scholar]
- Minta A, Kao J, Tsien R. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem. Biophys. Res. Commun. 1989;164:567–574. doi: 10.1016/0006-291x(89)91757-9. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog. Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Ohtaki H, Nakamachi T, Dohi K, Shioda S. Role of PACAP in ischemic neural death. J. Mol. Neurosci. 2008;36:16–25. doi: 10.1007/s12031-008-9077-3. [DOI] [PubMed] [Google Scholar]
- Otto C, Kovalchuk Y, Wolfer DP, et al. Impairment of mossy fiber long-term potentiation and associative learning in pituitary adenylate cyclase activating polypeptide type I receptor-deficient mice. J. Neurosci. 2001;21:5520–5527. doi: 10.1523/JNEUROSCI.21-15-05520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J. Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. Dopamine modulates the slow Ca(2+)-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J. Neurophysiol. 1995;74:2749–2753. doi: 10.1152/jn.1995.74.6.2749. [DOI] [PubMed] [Google Scholar]
- Pohl D, Ishmaru MJ, Bittigau P, Stadhaus D, Hubner C, Olney JW, Turski L, Ikonomidou C. NMDA antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc. Natl Acad. Sci. USA. 1999;96:2508–2513. doi: 10.1073/pnas.96.5.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorska A, Vanhoutte P, Arnold FJ, Silvagno F, Hardingham GE, Bading H. Synaptic activity induces signalling to CREB without increasing global levels of cAMP in hippocampal neurons. J. Neurochem. 2003;84:447–452. doi: 10.1046/j.1471-4159.2003.01504.x. [DOI] [PubMed] [Google Scholar]
- Racz B, Tamas A, Kiss P, et al. Involvement of ERK and CREB signaling pathways in the protective effect of PACAP in monosodium glutamate-induced retinal lesion. Ann. N Y Acad. Sci. 2006;1070:507–511. doi: 10.1196/annals.1317.070. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR, III, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reglodi D, Somogyvari-Vigh A, Vigh S, Kozicz T, Arimura A. Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke; a journal of cerebral circulation. 2000;31:1411–1417. doi: 10.1161/01.str.31.6.1411. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Somogyvari-Vigh A, Szanto Z, Kertes E, Lenard L, Arimura A, Lengvari I. Effects of pretreatment with PACAP on the infarct size and functional outcome in rat permanent focal cerebral ischemia. Peptides. 2002;23:2227–2234. doi: 10.1016/s0196-9781(02)00262-0. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Lubics A, Szalontay L, Lengvari I. Morphological and functional effects of PACAP in 6-hydroxydopamine-induced lesion of the substantia nigra in rats. Regul. Pept. 2004;123:85–94. doi: 10.1016/j.regpep.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Lengvari I, Toth G, Szalontay L, Lubics A. Comparative study of the effects of PACAP in young, aging, and castrated males in a rat model of Parkinson’s disease. Ann. N Y Acad. Sci. 2006;1070:518–524. doi: 10.1196/annals.1317.072. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Shintani N, Suetake S, Hashimoto H, et al. Neuroprotective action of endogenous PACAP in cultured rat cortical neurons. Regul. Pept. 2005;126:123–128. doi: 10.1016/j.regpep.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Shioda S, Ozawa H, Dohi K, et al. PACAP protects hippocampal neurons against apoptosis: involvement of JNK/SAPK signaling pathway. Ann. N Y Acad. Sci. 1998;865:111–117. doi: 10.1111/j.1749-6632.1998.tb11169.x. [DOI] [PubMed] [Google Scholar]
- Shioda S, Ohtaki H, Nakamachi T, et al. Pleiotropic functions of PACAP in the CNS: neuroprotection and neurodevelopment. Ann. N Y Acad. Sci. 2006;1070:550–560. doi: 10.1196/annals.1317.080. [DOI] [PubMed] [Google Scholar]
- Siu YT, Chin KT, Siu KL, Yee Wai Choy E, Jeang KT, Jin DY. TORC1 and TORC2 coactivators are required for tax activation of the human T-cell leukemia virus type 1 long terminal repeats. J. Virol. 2006;80:7052–7059. doi: 10.1128/JVI.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyvari-Vigh A, Reglodi D. Pituitary adenylate cyclase activating polypeptide: a potential neuroprotective peptide. Curr. Pharm. Des. 2004;10:2861–2889. doi: 10.2174/1381612043383548. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J. Physiol. 2007;584:381–387. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Leveille F, Papadia S, Higgins LG, Varley J, Baxter P, Hayes JD, Hardingham GE. Induction of sulfiredoxin expression and reduction of peroxiredoxin hyperoxidation by the neuroprotective Nrf2 activator 3H-1,2-dithiole-3-thione. J. Neurochem. 2008a;107:533–543. doi: 10.1111/j.1471-4159.2008.05648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Martel MA, Papadia S, et al. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J. Neurosci. 2008b;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Leveille F, Papadia S, Bell KF, Puddifoot C, Hardingham GE. Neuronal activity controls the antagonistic balance between PGC-1α and SMRT in regulating antioxidant defences. Antioxid. Redox Signal. 2011;14:1425–1436. doi: 10.1089/ars.2010.3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle JH, Foulkes NS, Molina CA, Simonneaux V, Pevet P, Sassone-Corsi P. Adrenergic signals direct rhythmic expression of transcriptional repressor CREM in the pineal gland. Nature. 1993;365:314–320. doi: 10.1038/365314a0. [DOI] [PubMed] [Google Scholar]
- Tamas A, Lubics A, Lengvari I, Reglodi D. Protective effects of PACAP in excitotoxic striatal lesion. Ann. N Y Acad. Sci. 2006a;1070:570–574. doi: 10.1196/annals.1317.083. [DOI] [PubMed] [Google Scholar]
- Tamas A, Zsombok A, Farkas O, Reglodi D, Pal J, Buki A, Lengvari I, Povlishock JT, Doczi T. Postinjury administration of pituitary adenylate cyclase activating polypeptide (PACAP) attenuates traumatically induced axonal injury in rats. J. Neurotrauma. 2006b;23:686–695. doi: 10.1089/neu.2006.23.686. [DOI] [PubMed] [Google Scholar]
- Tauskela JS, Fang H, Hewitt M, Brunette E, Ahuja T, Thivierge JP, Comas T, Mealing GA. Elevated synaptic activity preconditions neurons against an in vitro model of ischemia. J. Biol. Chem. 2008;283:34667–34676. doi: 10.1074/jbc.M805624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida D, Arimura A, Somogyvari-Vigh A, Shioda S, Banks WA. Prevention of ischemia-induced death of hippocampal neurons by pituitary adenylate cyclase activating polypeptide. Brain Res. 1996;736:280–286. doi: 10.1016/0006-8993(96)00716-0. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J. Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- Wang G, Pan J, Tan YY, Sun XK, Zhang YF, Zhou HY, Ren RJ, Wang XJ, Chen SD. Neuroprotective effects of PACAP27 in mice model of Parkinson’s disease involved in the modulation of K(ATP) subunits and D2 receptors in the striatum. Neuropeptides. 2008;42:267–276. doi: 10.1016/j.npep.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Watson RF, Abdel-Majid RM, Barnett MW, Willis BS, Katsnelson A, Gillingwater TH, McKnight GS, Kind PC, Neumann PE. Involvement of protein kinase A in patterning of the mouse somatosensory cortex. J. Neurosci. 2006;26:5393–5401. doi: 10.1523/JNEUROSCI.0750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Biegon A, Grigoriadis N, et al. D-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J. 2007;21:2033–2041. doi: 10.1096/fj.06-7856com. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genetics. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Buchthal B, Lau D, et al. A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J. Neurosci. 2011;31:4978–4990. doi: 10.1523/JNEUROSCI.2672-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS ONE. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]