

Abstract
Cryptosporidium parvum (Cp) is a potential biowarfare agent and major cause of diarrhea and malnutrition. This protozoan parasite relies on inosine 5′-monophosphate dehydrogenase (IMPDH) for the production of guanine nucleotides. A CpIMPDH-selective N-aryl-3,4-dihydro-3-methyl-4-oxo-1-phthalazineacetamide inhibitor was previously identified in a high throughput screening campaign. Herein we report a structure-activity relationship study for the phthalazinone-based series that resulted in the discovery of benzofuranamide analogs that exhibit low nanomolar inhibition of CpIMPDH. In addition, the antiparasitic activity of select analogs in a Toxoplasma gondii model of C. parvum infection is also presented.
Cryptosporidium species cause self-limiting gastrointestinal disease that can be chronic and fatal in immunosuppressed patients 1. Oocysts from these organisms are resistant to common methods of water treatment, and therefore pose a potential biowarfare threat. The drugs currently approved for the treatment of cryptosporidiosis are largely ineffective, elevating the need for new chemotherapeutic agents to counter outbreaks. Genomic analysis of the most common human pathogens, C. parvum and C. hominis, revealed that these parasites have a streamlined purine salvage pathway that relies on IMP dehydrogenase (IMPDH) for the production of guanine nucleotides 2–4. This enzyme catalyzes the oxidation of IMP to XMP with the production of NADH as outlined in Scheme 15. Curiously, the Cryptosporidium IMPDH gene appears to have been obtained from an ε-proteobacterium via horizontal gene transfer 6, and is structurally distinct from the host ortholog (the C. parvum and C. hominis enzymes are identical and will be denoted as CpIMPDH). Consequently, CpIMPDH displays unique enzymatic properties compared with the host counterparts 7.
Scheme 1.
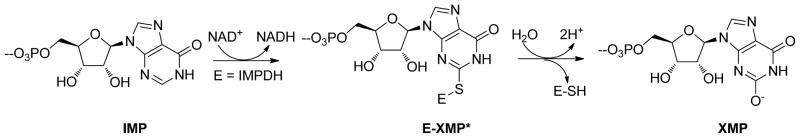
IMPDH catalyzed conversion of IMP to XMP via intermediate E-XMP*.
A previously reported high throughput screening (HTS) campaign identified several structurally distinct classes of selective CpIMPDH inhibitors that bind to the NAD site 8, 9. Structure-activity relationship studies of triazole 10, benzimidazole 9, 11 and urea 12 CpIMPDH inhibitors, exemplified by A110, C64, C97 and P96, respectively, have also now been reported (Figure 1). In addition, a co-crystal structure of the benzimidazole inhibitor C64 with CpIMPDH demonstrated a unique binding mode compared to inhibitors bound to human IMPDH 9. The phthalazinone-based inhibitor 1 was also identified by HTS, with moderate potency (IC50 = 5.4 μM) and good selectivity (IC50 > 50 μM versus both human IMPDH1 and IMPDH2) 8. Compound 1 (50 μM) did not display antiparasitic activity 8. Herein we report a structure-activity relationship study of 1, and the antiparasitic activity of select analogs in a T. gondii model of C. parvum infection.
Figure 1.
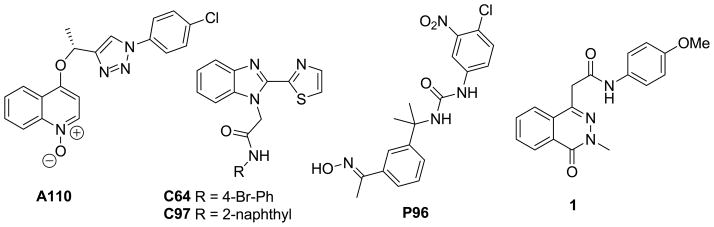
Optimized CpIMPDH inhibitors A110, C64 and C97 and HST identified CpIMPDH inhibitor 1.
Compound 1 and its analogs were prepared following a four-step sequence as depicted in Scheme 2 beginning with the Wittig olefination of phthalic anhydride, 2, in the presence of carbethoxymethylidenetriphenylphosphorane in chloroform to produce (E)-ethyl-2-(3-oxoisobenzofuran-1(3H)-ylidene)acetate, 3. Addition of hydrazines in refluxing ethanol generated the phthalazinone acetoesters 4. Hydrolysis of the esters produced phthalazinone acetic acids 5 13, 14. Finally, coupling of the acetic acid moiety with various aromatic amines in DMSO afforded 6.
Scheme 2.

General procedure for the synthesis of phthalazinone derivatives of 1. a) Ph3PCHCO2Et, CHCl3, reflux, 16 h. b) NH2NH2 or NH2NHMe, EtOH, reflux, 3 h. c) 3 M NaOH/THF, reflux, 2 h followed by acidification. d) R1-NH2, EDC, HOBt, DIPEA, DMSO, 3–5 h.
The synthesized derivatives were evaluated for CpIMPDH inhibition using an assay that monitored the production of NADH in the presence of varying inhibitor concentrations 10. In order to evaluate the effects of non-specific binding, inhibition was also determined in the presence of 0.05% fatty acid free bovine serum albumin (BSA). None of the compounds inhibited human IMPDH2 (IC50 > 5 μM).
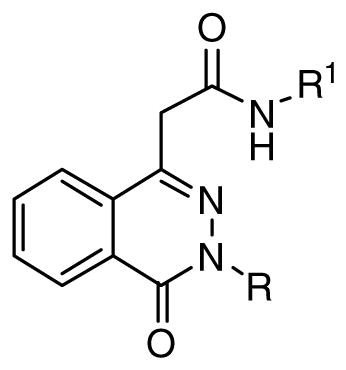
Inhibitor 1 exhibited an IC50 of 1.0 μM upon resynthesis. Gratifyingly, the activity was retained in the presence of 0.05% BSA (IC50 = 970 nM) as shown in Table 1. However, moving the electron donating group to the 2- or 3-position resulted in loss of activity (7 and 10). Replacing the 4-OMe with a variety of other electron donating substituents (11 – 14) was also detrimental. Likewise, replacing the phenyl with an amine (8) or alkyl group (9) or inserting a methylene between the benzene and amide nitrogen atom (15) was not tolerated. Replacement of the electron donating group at the 4-position with electron withdrawing groups improved potency. For example, the 4-chloro and 4-bromo analogs (16 and 17) demonstrated IC50 values of 230 and 130 nM, respectively. However, other electron withdrawing groups (18 – 21) generally were not as well tolerated. Also, transposition of the chlorine from the 4-position to the 3-position (23) resulted in loss of activity. Di-substitution at the 3,4-positions with chlorine and bromine atoms (24 and 25) resulted in increased potency. Furthermore, other 3,4-di-substituted analogs with either an electron-donating or an electron-withdrawing group at the 3-position were also active. Interestingly, in a number of cases substituents at the 3-position that alone were not tolerated when combined with electron withdrawing groups (especially Cl or Br) at the 4-position resulted in potent CpIMPDH inhibitors (e.g. 26, 27 and 37). However, transposition of the substituents from the 3-position to the 2-position (39 – 42) resulted in poor inhibitors. In addition, several other di- and tri-substitutions (43 – 46) were also detrimental. Encouraged by the results of 3,4-disubstitution, the 2-naphthyl analog 47 was prepared and demonstrated an IC50 of 63 nM. Similar increases in CpIMPDH activity by introduction of 3,4-disubstitution or incorporation of 2-naphthylene had also been observed in the structure-activity relationship studies resulting in A110 and C97 10, 11. Finally, analogs that lack methylation of the phthalazinone nitrogen atom (48 and 49) were found to be active indicating that this substituent is not necessary for CpIMPDH inhibitory activity.
Table 1. SAR of anilide and nathylide phthalazinone inhibitors.
Assays as described in 10.
| ||||
|---|---|---|---|---|
| Compound | R | R1 | (−)-BSA (nM) | (+)-BSA (nM) |
| 1 | Me | 4-OMePh | 1000 ± 200 | 930 ± 180 |
| 7 | Me | 2-OMePh | >5000 | ND |
| 8 | Me | NH2 | >5000 | ND |
| 9 | Me | t-Bu | >5000 | ND |
| 10 | Me | 3-OMePh | >5000 | ND |
| 11 | Me | 4-OEtPh | >5000 | ND |
| 12 | Me | 4-OCF3Ph | >5000 | ND |
| 13 | Me | 4-MePh | >5000 | ND |
| 14 | Me | 4-i-PrPh | >5000 | ND |
| 15 | Me | CH2(4-OMePh) | >5000 | ND |
| 16 | Me | 4-ClPh | 230 ± 50 | 280 ± 50 |
| 17 | Me | 4-BrPh | 130 ± 30 | 130 ± 20 |
| 18 | Me | 4-FPh | >5000 | ND |
| 19 | Me | 4-CNPh | 950 ± 90 | 940 ± 90 |
| 20 | Me | 4-CF3Ph | >5000 | ND |
| 21 | Me | 4-SO2MePh | >5000 | ND |
| 22 | Me | 3-CF3Ph | >5000 | ND |
| 23 | Me | 3-ClPh | >5000 | ND |
| 24 | Me | 3,4-di-ClPh | 55 ± 6 | 95 ± 2 |
| 25 | Me | 3-Cl-4-BrPh | 30 ± 1 | 75 ± 2 |
| 26 | Me | 3-CF3-4-BrPh | 24 ± 2 | 48 ± 4 |
| 27 | Me | 3-CF3-4-ClPh | 13 ± 1 | 25 ± 5 |
| 28 | Me | 3-CF3-4-CNPh | 54 ± 5 | 52 ± 6 |
| 29 | Me | 3-Cl-4-CNPh | 290 ± 40 | 470 ± 120 |
| 30 | Me | 3-CF3-4-FPh | 150 ± 20 | 150 ± 30 |
| 31 | Me | 3-CF3-4-OMePh | >5000 | ND |
| 32 | Me | 3-CF3-4-NH2Ph | >5000 | ND |
| 33 | Me | 3-F-4-ClPh | 200 ± 30 | 240 ± 30 |
| 34 | Me | 3-Me-4-ClPh | 79 ± 9 | 96 ± 19 |
| 35 | Me | 3-Me-4-BrPh | 70 ± 10 | 140 ± 10 |
| 36 | Me | 3-Me-4-CNPh | 400 ± 100 | 460 ± 90 |
| 37 | Me | 3-OMe-4-ClPh | 33 ± 1 | 40 ± 7 |
| 38 | Me | 3,4-di-CNPh | >2500 | >2500 |
| 39 | Me | 2-CF3-4-ClPh | >5000 | ND |
| 40 | Me | 2-CF3-4-BrPh | >5000 | ND |
| 41 | Me | 2-F-4-BrPh | 1100 ± 200 | 1700 ± 100 |
| 42 | Me | 2,4-BrPh | >5000 | ND |
| 43 | Me | 2,4-di-ClPh | >5000 | ND |
| 44 | Me | 3,5-di-ClPh | >5000 | ND |
| 45 | Me | 3,4,5-tri-ClPh | >5000 | ND |
| 46 | Me | 3,4,5-tri-FPh | >5000 | ND |
| 47 | Me | 2-naphthyl | 63 ± 3 | 140 ± 20 |
| 48 | H | 3-CF3-4-ClPh | 40 ± 1 | 61 ± 1 |
| 49 | H | 3-CF3-4-BrPh | 32 ± 6 | 130 ± 8 |
All values are average of three independent determinations unless otherwise noted (* two determinations). ND, not determined.
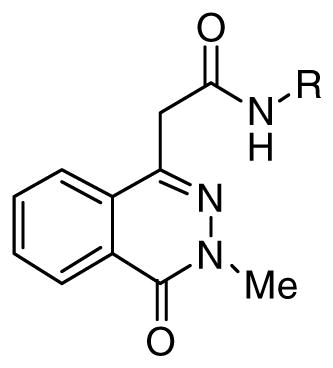
Inspired by the results with 47, a variety of fused heterocycles were explored as replacements of the 2-naphthylene (Table 2). Although the 5-benzofuranyl analog 50 had reduced activity and the 5-benzoxazolyl analog 51 was inactive, the 2-methyl-5-benzofuranyl derivative 52 retained activity with an IC50 of 64 nM. Furthermore, incorporation of an additional ring (53) resulted in enhanced potency. Unsaturation of the ring further increased activity with 54 demonstrating an IC50 of 4 nM. However, the regioisomer 55 and two carbazoles 56 and 57 were not active.
Table 2. SAR of heterotricyclic anilide phthalazinone inhibitors.
Assays as described in 10.
| |||
|---|---|---|---|
| Compound | R | (−)-BSA (nM) | (+)-BSA (nM) |
| 50 |
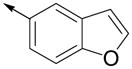
|
93 ± 15 | 85 ± 12 |
| 51 |
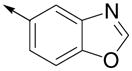
|
>5000 | ND |
| 52 |
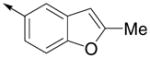
|
64 ± 11 | 72 ± 15 |
| 53 |
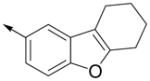
|
20 ± 5 | 150 ± 20 |
| 54 |
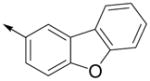
|
4 ± 1 | 30 ± 10 |
| 55 |
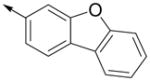
|
>5000 | ND |
| 56 |
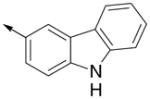
|
>5000 | ND |
| 57 |
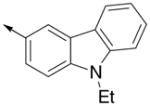
|
>5000 | ND |
All values are average of three independent determinations unless otherwise stated. ND, not determined.
Compounds with IC50 values less than 30 nM were candidates for evaluation of antiparasitic activity in a Toxoplasma gondii model of C. parvum infection 15. In this model, the endogenous T. gondii IMPDH and hypoxanthine-guanine-xanthine phosphoribosyltransferase genes have been knocked out and the CpIMPDH gene inserted to create T. gondii/CpIMPDH, a model parasite that relies on CpIMPDH for the production of guanine nucleotides. Both wild-type and T. gondii/CpIMPDH were cultured in human foreskin fibroblasts immortalized with hTERT, so this assay also reports on host cell toxicity. Compounds 26, 27, 53 and 54 all displayed sub-micromolar activity against T. gondii/CpIMPDH (Table 3). However, only 27 displayed selectivity • 30 versus the wild-type strain, strongly indicating that antiparasitic activity results from the inhibition of CpIMPDH.
Table 3. Antiparasitic activity.
Assays as described in 15.
| Compound | EC50a (μM) | Selectivity b | |
|---|---|---|---|
| Toxo/WT | Toxo/CpIMPDH | ||
| 26 | 4 ± 3 * | 0.3 ± 0.2 | 14 |
| 27 | 23 ± 4 | 0.4 ± 0.3 | 60 |
| 53 | 1 ± 1 * | 0.5 ± 0.2 | 2 |
| 54 | 3 ± 1 | 0.4 ± 0.1 | 7 |
Values are the average and standard deviations of three independent determinations unless otherwise stated
denotes average and range of two determinations).
T. gondii RH (Toxo/WT) should be resistant to CpIMPDH inhibitors while T. gondii/CpIMPDH (Toxo/CpIMPDH) should be sensitive.
Selectivity = EC50(Toxo/WT)/EC50(Toxo/CpIMPDH).
The inhibition of CpIMPDH by compound 27 was characterized further. Whereas compound 1 is a mixed inhibitor of CpIMPDH with respect to NAD+ (Kis = 1.8 μM, Kii = 7μM) 8, 27 is a pure noncompetitive inhibitor (Kis = Kii = 3.4 ± 0.2 nM; Figure 2).
Figure 2.
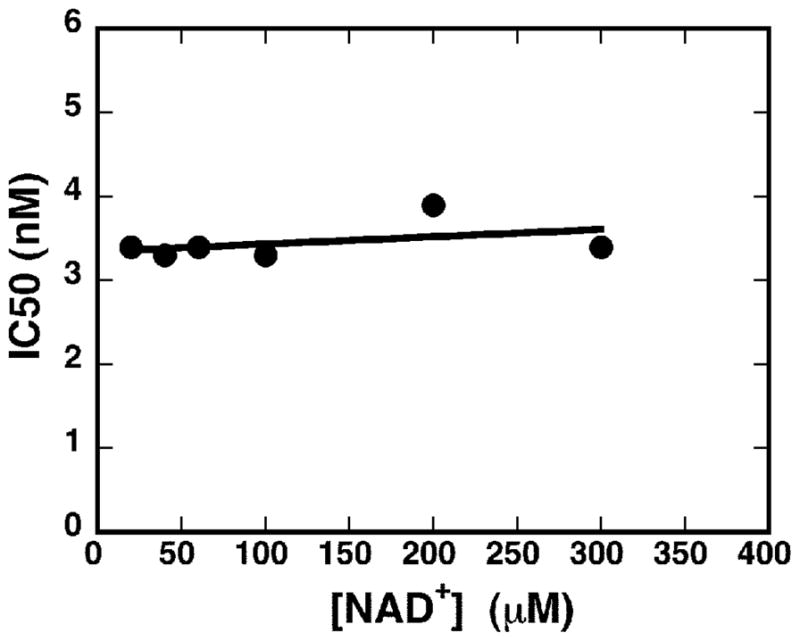
Inhibition of CpIMPDH by 27.
Compound 27 displayed good stability in mouse liver microsomes (T1/2= 79 min). This compound was advanced into the IL12 knockout mouse model of C. parvum infection 16–19. Disappointingly, no antiparasitic activity was observed with once per day oral dosing of 27 (250 mg/kg) for seven days. It may be that alternative dosing regimes or modifications in formulations could improve efficacy in vivo. Additional optimization of pharmacokinetic properties may also be necessary for this compound series in order to achieve in vivo efficacy.
In conclusion, a SAR study of phthalazinone-based CpIMPDH inhibitors revealed that expansion of the aniline ring could increase inhibitory activity, while maintaining selectivity relative to the human orthologs. The phthalazinone-based CpIMPDH inhibitors described herein could serve as lead compounds for the development of effective treatments of cryptosporidiosis as well as useful molecular probes for studying Cryptosporidium parasites.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (U01 AI075466 and U01 AI075466S1) to LH. GDC thanks the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE/BEID) and Harvard NeuroDiscovery Center for financial support. IC50 data for these entire compounds were maintained using ChemAxon, http://www.chemaxon.com/. JRM would like to thank Ms. Nina McNair for technical support. CRJ would like to thank Drs. Sivapriya Kirubakaran and Jihan Khan for insightful and helpful suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Huang DB, White AC. Gastroenterol Clin North Am. 2006;35:291. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Proc Natl Acad Sci U S A. 2004;101:3154. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Science. 2004;304:441. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 4.Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. Nature. 2004;431:1107. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 5.Hedstrom L. Chem Rev. 2009;109:2903. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr, Liu C, Abrahamsen MS. Proc Natl Acad Sci U S A. 2002;99:6304. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. J Biol Chem. 2004;279:40320. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 8.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Chem Biol. 2008;15:70. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D’Aquino JA, Zhang M, Cuny GD, Hedstrom L. J Am Chem Soc. 2010;132:1230. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. J Med Chem. 2009;52:4623. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirubakaran S, Gorla SK, Sharling L, Zhang M, Liu X, Ray SS, Macpherson IS, Striepen B, Hedstrom L, Cuny GD. Bioorg Med Chem Lett. 2012;22:1985. doi: 10.1016/j.bmcl.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorla SK, Kavitha M, Zhang M, Liu X, Sharling L, Gollapalli DR, Striepen B, Hedstrom L, Cuny GD. J Med Chem. 2012;55:7759. doi: 10.1021/jm3007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mylari BL, Larson ER, Beyer TA, Zembrowski WJ, Aldinger CE, Dee MF, Siegel TW, Singleton DH. J Med Chem. 1991;34:108. doi: 10.1021/jm00105a018. [DOI] [PubMed] [Google Scholar]
- 14.Niebel C, Lokshin V, Sigalov M, Krief P, Khodorkovsky V. Eur J Org Chem. 2008:3689. [Google Scholar]
- 15.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. PLoS Negl Trop Dis. 2010;4:e794. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell LD, Stewart JN, Mead JR. J Parasitol. 2002;88:1014. doi: 10.1645/0022-3395(2002)088[1014:STCPII]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 17.Ehigiator HN, Romagnoli P, Borgelt K, Fernandez M, McNair N, Secor WE, Mead JR. Parasite Immunol. 2005;27:17. doi: 10.1111/j.1365-3024.2005.00736.x. [DOI] [PubMed] [Google Scholar]
- 18.Sun XE, Sharling L, Muthalagi M, Mudeppa DG, Pankiewicz KW, Felczak K, Rathod PK, Mead J, Striepen B, Hedstrom L. J Biol Chem. 2010;285:15916. doi: 10.1074/jbc.M110.101543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The anticryptosporidial activity of 27 was assessed in the IL-12 knockout mouse model, which resembles the acute human disease. Mice were inoculated with 1,000 oocysts and treated by gavage with vehicle (10%DMSO/corn oil), vehicle plus 250 mg/kg 27, or 2000 mg/kg paromomycin starting 4 hrs post infection. Treatment was given once daily for 7 days and mice sacrificed on day 8 (peak infection). Parasite load was quantified by FACS assays by the presence of the oocysts in the feces at days 0, 4 and 7. Fecal pellets from the mice were collected and homogenized in adjusted volumes of 2.5% potassium dichromate. Aliquots (200 μl) of vortexed samples were processed over micro-scale sucrose gradients. The oocyst-containing fraction was collected and washed. Purified oocysts were incubated with a fluorescein-labeled oocyst-specific monoclonal antibody (OW5O-FITC) for 20 min. Samples were adjusted to 600 μl and assayed assayed with a 102-s sampling interval (100 μl)l) using logical gating of forward/side scatter and OW5O-FITC fluorescence signal on a Becton Dickinson FACScan flow cytometer. Flow cytometry data were evaluated by analysis of variance (Microsoft Excel; Microsoft Corporation, Redmond, WA).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.