

Abstract
The anti-arrhythmic efficacy of the late sodium channel current (late INa) inhibition has been convincingly demonstrated in the ventricles, particularly under conditions of prolonged ventricular repolarization. The value of late INa block in the setting of atrial fibrillation (AF) remains poorly investigated. All sodium channel blockers inhibit both peak and late INa and are generally more potent in inhibiting late vs. early INa. Selective late INa block does not prolong the effective refractory period (ERP), a feature common to practically all anti-AF agents. Although the late INa blocker ranolazine has been shown to be effective in suppression of AF, it is noteworthy that at concentrations at which it blocks late INa in the ventricles, it also potently blocks peak INa in the atria, thus causing rate-dependent prolongation of ERP due to development of post-repolarization refractoriness. Late INa inhibition in atria is thought to suppress intracellular calcium (Cai)-mediated triggered activity, secondary to a reduction in intracellular sodium (Nai). However, agents that block late INa (ranolazine, amiodarone, vernakalant, etc) are also potent atrial-selective peak INa blockers, so that the reduction of Nai loading in atrial cells by these agents can be in large part due to the block of peak INa. The impact of late INa inhibition is reduced by the abbreviation of the action potential that occurs in AF patients secondary to electrical remodeling. It stands to reason that selective late INa block may contribute more to inhibition of Cai-mediated triggered activity responsible for initiation of AF in clinical pathologies associated with a prolonged atrial APD (such as long QT syndrome). Additional studies are clearly needed to test this hypothesis.
Keywords: Atrial fibrillation, Late sodium channel current, Ranolazine, Action potential, Pharmacology
Introduction
Specific inhibition of late sodium channel current (late INa) can effectively suppress ventricular arrhythmias, particularly under conditions of prolonged ventricular repolarization (such as long QT syndrome and heart failure) [1–5]. These anti-arrhythmic actions of selective late INa inhibition are largely due to a reduction of intracellular calcium (Cai) loading, secondary to a decrease of intracellular sodium (Nai). Augmented late INa may significantly contribute to Nai loading, which brings calcium into the cell via reverse mode of sodium-calcium exchange. The electrophysiology and pharmacology of late INa as well as the antiarrhythmic benefit of its inhibition have been studied mostly in ventricles. The value of block of late INa for the suppression of atrial fibrillation (AF) remains poorly investigated. This review examines available data relative to the electrophysiology, pharmacology, and anti-AF ability of late INa inhibition.
Electrophysiology of Late Sodium Current
Cardiac sodium channel current is comprised of two basic components: peak and late INa. Peak INa is responsible for phase 0 of the action potential, whereas late INa contributes to the inward charge maintaining phase 2 and phase 3 (Fig. 1). Several mechanism are thought to contribute to the manifestation of late INa, including 1) slow inactivation of the sodium channel [6], 2) single as well as bursts of late reopening of the sodium channel [7], and 3) a steady state current occurring within a window of voltage representing the overlap of steady state activation and inactivation of the sodium channel (window current [8]).
Fig. 1.
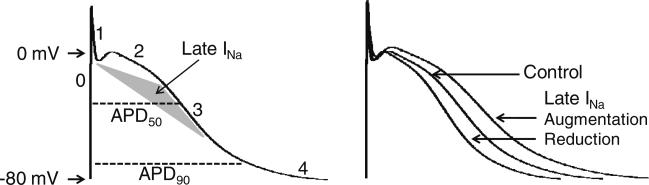
Role of late INa in the action potential (AP) of normal cardiomyocyte. The cardiac AP is comprised of 5 phases (denoted by the numbers). Phase 0, 1, 2, 3, and 4 represent depolarization, early repolarization, “plateau”, late repolarization, and resting state, respectively. Peak INa flows into the cell during phase 0 (upstroke of the AP; 1–2 ms duration) and late INa during phase 2 and early phase 3 (at potentials positive to –60 mV, peaking at –20 mV; 100–300 ms duration). The density of peak INa is significantly greater than that of late INa. Action potential duration (APD) is measured at the level of 50–90 % of full repolarization (as depicted by the dashed lines). Specific augmentation of late INa (an inward current) prolongs APD and block of this current abbreviates APD
Late INa in Ventricular Cells
The amplitude of late INa in the ventricles ranges from 0.1 to 1.0 % of that of peak INa, depending on species, cell type, pathology, and conditions under which the currents are measured [2, 5]. However, due to a much longer duration during the action potential (100–300 for late INa vs. 1–2 ms for peak INa), the total charge carried by late INa can be considerable. Late INa flows into the cell at potentials positive to –60 mV, peaking at –20 mV [9]. Prolongation of action potential duration (APD) increases and abbreviation of APD reduces the integral or total charge of late INa.
The relative contribution of late INa to Nai loading increases under pathophysiological conditions associated with an increase in late INa density and prolongation of APD (such as heart failure) [2]. Myocardial ischemia in the ventricles is also associated with increased late INa density [10]. In contrast, late INa has been reported to be reduced in remodeled hypertrophied left ventricles (LV) in a canine chronic AV node block model [4]. It is also noteworthy that Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) can increase late INa density [11]. There is significant regional heterogeneity in late and peak INa density in canine ventricles. Left ventricular midmyocardial to subendocardial M cells have larger late and peak INa densities compared to epicardial and endocardial myocytes [9].
Late INa in Atrial Cells
Late INa is less studied in atrial vs. ventricular cells. The general characteristics of peak and late INa appear to be similar in atrial and ventricular cells. However, there are atrioventricular electrophysiological differences that can modulate the relative contribution of late INa to Nai loading. Because APD50–75 is shorter in atrium vs. ventricle (Fig. 2), the atrial late INa integral is likely to be smaller, as is its relative contribution to Nai loading. While the density of late INa is similar in atrial and ventricular cells [12], the density of peak INa is significantly greater in atrial vs. ventricular myocytes [13, 14]. However, a more negative steady-state inactivation voltage in atrial vs. ventricular myocytes (~12 mV) [13, 14] as well as a more depolarized resting membrane potential (RMP) in atrial vs. ventricular myocytes reduce the availability of the sodium channel in atria vs. ventricles at the physiologically relevant RMP range. As a result, the range of available peak INa appears to be similar in the two chambers as reflected by a similar range of maximum rate of rise of the action potential up-stroke (Vmax, an index of peak INa) in the canine heart [15, 16]. The exception is Purkinje fiber action potentials, which display the largest peak INa and Vmax in the heart. The value of Vmax generally positively correlates with conduction velocity, i.e., the larger the Vmax the faster conduction velocity and vice versa. There is a regional Vmax difference in healthy canine right atria (ranging from 356±60 V/s in the endocardial crista terminalis to 234±54 V/s in the epicardial appendage) [15].
Fig. 2.
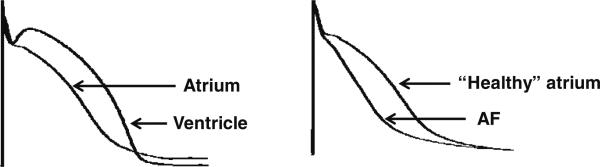
Action potential duration (APD) is shorter in atria than in. ventricles under baseline conditions and the APD in atrium is significantly abbreviated in remodeled atria (in the setting of atrial fibrillation or prone to develop atrial fibrillation). APD abbreviation reduces the period during which late INa flows into the cell, and thus the relative contribution of this current to intracellular sodium (Nai) loading
Because APD is commonly significantly abbreviated in remodeled atria (Fig. 2), the integral of late INa is likely to be smaller in remodeled vs. healthy atria. Properties of atrial late INa in AF are poorly studied. The integral of late INa (total charge) has been shown to be increased in atrial myocytes isolated from right atrial appendage of persistent AF vs. sinus rhythm patients (by 26 %) [17]. In this study, however, the late INa integral in AF and non-AF patients was measured at the same pulse duration, not adjusted to the shorter APD observed in AF. It not known whether late INa density is altered in patients with paroxysmal AF. The density of late INa in left atria (but not in right atria) has been reported to be increased in a rabbit LV hypertrophy model (caused by hypertension) [18]. Available data relative to alterations of peak INa in AF models and patients are controversial. Peak INa density in atrial myocytes has been reported to be reduced in persistent AF vs. SR patients in some studies [17] but not others [19]. A decrease in peak INa activity was reported in remodeled canine [20], but not goat atria [21]. Interestingly, Nai was shown to be reduced in canine atrial cells after 48 h of atrial-tachypacing [22].
The magnitude of late INa is reverse-rate dependent, i.e., the faster the rate the less the magnitude of this current [9, 23–25]. The rate-dependent reduction of late INa contributes to rate-dependent APD abbreviation [9, 25]. The acceleration-induced reduction in late INa is much greater than that of peak INa (Fig. 3). Thus, the relative contribution of late INa in cardiac electrophysiology is thought to be more important when APD is longer and heart rate is slower.
Fig. 3.

Acceleration of heart rate can lead to a reduction in late INa total charge in the cardiac cell because of direct rate-dependent reduction in late INa density as well as abbreviation of APD. The latter is in part due to the former. Acceleration of pacing rate reduces late INa density more than that of peak INa [9, 23–25]
Pharmacology of Late Sodium Current
All sodium channel blockers inhibit both peak and late INa, typically with a higher potency of blocking late INa vs. peak INa [26]. Relatively low concentrations of INa blockers (including TTX, lidocaine, quinidine, ranolazine, etc) can potently inhibit late INa without affecting peak INa [1, 26]. Selective pharmacological augmentation of late INa prolongs APD/ERP and reduction of this current leads to an abbreviation of APD/ERP (Fig. 1). Block of late INa reduces Nai (and thus Cai) and does not directly affect cardiac excitability or conduction properties. Most data dealing with the pharmacology of late INa were obtained using ventricular myocytes.
Pharmacology of Late INa in Ventricles
The role played by late INa is often investigated using ranolazine, currently the most potent late INa blocker in ventricles [5]. Ranolazine has been shown to be 9 to 45 times more potent in blocking late vs. peak INa in isolated ventricular myocytes and expression systems [5, 27–29]. The study by Udrovinas and colleagues [28] reported an exceptional selectivity of ranolazine to inhibit late vs. peak INa in canine ventricular cells (6.5 vs. 294 μM, respectively; i.e., demonstrating an IC50 ratio of 45). Contributing to this high selectivity was the fact that ranolazine's blocking potency was measured at a cycle length (CL) of 10 s, at which the use-dependent effects of the drug are not apparent [14, 30]. Indeed, in canine ventricular myocytes, at slow rates and negative holding potentials (at which all sodium channels are available), 285 μM of ranolazine was needed to cause a 50 % reduction (IC50) of peak INa, whereas at faster rates and more “physiological” holding potentials IC50 was less than 10 % of that value [30]. Late INa IC50 values for ranolazine inhibition also depend on recording conditions and can range between from 5 to 21 μM in canine ventricular myocytes [24]. Thus, the IC50 of INa blockers to inhibit both late and peak INa can vary significantly depending on the conditions at which IC50 is measured, including stimulation rate, diastolic interval, holding and test potentials, and temperature [24, 29, 30]. The differences in functional manifestation of late vs. peak INa inhibition in multicellular preparations can be even more pronounced, as discussed below.
INa blocker-induced inhibition of both peak and late INa is rate-dependent (i.e., the faster the rate the greater the blocking efficacy) [12, 24, 26, 29]. The rate-dependence of peak INa block is particularly steep for INa blockers with rapid unbinding kinetics, such as ranolazine, vernakalant, and amiodarone [14, 26, 30–32]. The rate-dependence of peak vs. late INa inhibition is not well investigated. In HEK293 cells expressing a LQT3 mutation (SCN5A-R1623Q), rate-dependent effect of ranolazine to block late and peak INa is similar [29]. In cardiac cells, the functional manifestations of rate-dependent peak INa block is generally greater than that of late INa block (Fig. 4). INa blockers with rapid kinetics (e.g., ranolazine and lidocaine) can be highly selective late INa blockers at slow heart rates and much less selective or non-selective for late INa block at rapid activation rates. Indeed, ranolazine and lidocaine (10–20 μM) significantly abbreviate APD (due to late INa block) but have little to no affect on Vmax in Purkinje fiber and ventricular M cell preparations at CLs ≥1000 ms [24, 33], thus demonstrating high selectivity for block of late vs. peak INa. At rapid rates, these agents significantly reduce Vmax with little or no change in APD in ventricular muscles and Purkinje fibers, consistent with much less selective block of late INa. Ranolazine (20 μM) causes a 30–40 % reduction of Vmax at a CL of 300 ms in canine and human ventricular slice preparations [34]. In canine and human ventricular preparations, ranolazine (10 μM) reduces Vmax by 8 and 20 % at a CL of 500 and 300 ms, respectively [14, 34].
Fig. 4.

Effect of acceleration of rate on total charge of atrial peak and late INa in the absence and presence of sodium channel blockers with rapid unbinding kinetics. This schema applies only to INa blockers that dissociate rapidly from the sodium channel, such as ranolazine, lido-caine, amiodarone (prominent late INa blockers). Under baseline conditions, acceleration of pacing rates significantly reduces total charge of late INa much more than that of peak INa (due to decrease in late INa density and APD abbreviation; see Fig. 3). At slow pacing rates and long APDs, block of late INa is evidenced by a prominent abbreviation of APD and lack of block of peak INa is evidenced by a lack of effect on Vmax. At rapid activation rates and relatively short APD, drug-induced block of both late and peak INa is augmented. Despite a rate-dependent increase in potency of drug-induced late INa block, there is a reduction in absolute amount of total charge of late INa blocked following acceleration due to a significant reduction of available pre-drug total charge of late INa integral secondary to a reduction of late INa density and abbreviation of APD. Due to a much steeper rate-dependent bock of peak INa in atria vs. ventricles, the degree of rate-dependent augmentation of peak vs. late INa block is likely to be greater in atria vs. ventricles
Pharmacology of Late INa in Atria
There are fundamental differences in the response of atria and ventricles to block of peak INa. INa blockers with relatively rapid unbinding kinetics (like ranolazine amiodarone, vernakalant, AZD1305, Wenxin Keli, etc) are atrial-selective peak INa blockers (Figs. 5 and 6). The rate-dependent potency of these agents to inhibit peak INa and to depress peak INa-mediated parameters (Vmax, conduction velocity, excitability, etc) in atrial cells is much greater than in ventricular cells (Figs. 5 and 6) [14, 30, 32, 34–38]. Ranolazine (10 μM), for example, produces little to no change in Vmax at a CL of ≥1000 ms in the canine right atrium, but reduces Vmax by 25 and 60 % at CLs of 500 and 300 ms, respectively [14, 39]. A major electrophysiological effect of ranolazine, amiodarone, vernakalant, etc (most relevant to their anti-AF action) is the induction atrial-selective post-repolarization refractoriness (PRR), a peak INa-mediated parameter (Fig. 5). Our current understanding of the mechanisms of atrial selectivity of INa blockers have been discussed in detail elsewhere [39–41] and includes a more depolarized RMP, more negative half-inactivation voltage (V0.5), and more gradual phase 3 of the action potential in atrial cells as compared with ventricular cells. It is not known whether atrial-selective peak INa blockers cause atrial-selective inhibition of late INa.
Fig. 5.

Ranolazine selectively induces prolongation of the effective refractory period (ERP) and development of post-repolarization refractoriness in atria (PRR, the difference between ERP and APD75 in atria and between ERP and APD90 in ventricles; ERP corresponds to APD75 in atria and APD90 in ventricles). CL=500 ms. C – control. The arrows in panel A illustrate the position on the action potential corresponding to the end of the ERP in atria and ventricles and the effect of ranolazine to shift the end of the ERP in atria but not ventricles. * p<0.05 vs. control. † = p<0.05 vs. APD75 values in atria and APD90 in ventricles; (n=5–18). From Burashnikov et al. [14], with permission. PRR is a peak INa-mediated parameter
Fig. 6.

Ranolazine produces a much greater rate-dependent inhibition of the maximal action potential upstroke velocity (Vmax) in atria than in ventricles. a Normalized changes in Vmax of atrial and ventricular cardiac preparations paced at a cycle length (CL) of 500 ms. b Ranolazine prolongs late repolarization in atria, but not ventricles and acceleration of rate leads to elimination of the diastolic interval, resulting in a more positive take-off potential in atrium and contributing to atrial selectivity of ranolazine. The diastolic interval remains relatively long in ventricles. * p<0.05 vs. control. † p<0.05 vs. respective values of M cell and Purkinje (n=7–21). From Burashnikov et al. [14], with permission
It is noteworthy that despite the rate-dependence of late INa inhibition, the total charge of blocked late INa may actually decrease with acceleration of pacing rate due to significant rate-dependent reduction of baseline late INa density and abbreviation of APD (Figs. 3 and 4). This suggests that the anti-arrhythmic action of late INa block is likely reduced with acceleration of heart rate. Considering the shorter APD in atria vs. ventricles and atrial selectivity of peak INa block, INa blockers with rapid unbinding kinetics are likely to be less specific late INa blockers in atria vs. ventricles, particularly at rapid activation rates.
Pathological conditions often modulate the potency of antiarrhythmic drugs. The efficacy of INa blockers to inhibit late INa in the setting of AF is poorly defined. The efficacy of ranolazine to inhibit late INa is increased and that of peak INa is reduced in atrial myocytes isolated from patients with persistent AF [17]. The physiological and anti-AF consequences of these altered blocking efficacies are not clear. However, it is known that INa blockers, including ranolazine [42], tend to lose their anti-AF effectiveness in persistent AF patients/models (as discussed in the next section). A reduced ability of ranolazine to block peak INa in persistent AF patients is expected to decrease the effectiveness of the drug to induce PRR, thus limiting its anti-AF potency. Selective block of late INa is expected to cause a greater APD abbreviation in “healthy” vs. remodeled atria (Fig. 7).
Fig. 7.

Selective late INa inhibition produces a greater APD abbreviation in healthy vs. remodeled atrial cells
Anti-Arrhythmic Efficacy of Late Sodium Current Inhibition
The anti-arrhythmic benefit of late INa inhibition and the role of late INa augmentation in arrhythmogenesis is considerably more studied in ventricles than in atria [5, 43, 44]. Understanding the anti-arrhythmic utility of late INa inhibition requires an understanding of the electrophysiological mechanisms underlying the generation of cardiac arrhythmias as well as some atrioventricular differences in arrhythmogeneicity. The occurrence of AF is commonly associated with abbreviation of APD and ERP. In contrast, ventricular tachycardia (VT) and fibrillation (VF) are relatively rarely associated with an abbreviated APD and ERP (i.e., short QT syndrome, acute ischemia, etc.). VT/VF more often occurs under conditions of prolonged APD (heart failure, hypotrophy, long QT syndrome, etc). Sustained electrical remodeling (due to prolonged rapid activation) is associated abbreviation of APD and ERP in atria but prolongation of APD in the ventricles [21, 45].
Tachycardia and fibrillation are normally triggered by a focal source and maintained by a reentrant mechanism [46]. Focal mechanisms include early (EAD) and delayed (DAD) afterdepolarization-induced triggered activity and automaticity [47]. EAD is normally associated with a prolonged APD and bradycardia. An exception is the late phase 3 EAD which requires a significant abbreviation of APD. Phase 3 EAD-induced triggered activity usually develops following an episode of tachycardia followed by a pause [48]. The appearance of DAD is normally associated with tachycardia and relatively short APD. Cai overload is a common source of induction of EAD- and DAD-induced triggered activity as well as accelerated automaticity. Reentrant mechanisms are often associated with conduction disturbances and structural and electrical heterogeneities [46]. Most arrhythmic mechanisms are commonly suppressed by APD/ERP prolongation; an exception is bradycardia-mediated EAD activity, which can be inhibited by APD abbreviation [46].
The electrophysiological consequences of late INa inhibition (described in the previous sections) suggest that the major anti-arrhythmic value of late INa block is in suppression of Cai-mediated triggered activity, particularly those occurring in conjunction with APD prolongation and bradycardia.
Ventricular Arrhythmias
Late INa has attracted a great deal of attention as an anti-arrhythmic target to suppress ventricular arrhythmias, particularly under conditions of prolonged repolarization, such as those associated with the long QT syndrome, heart failure and bradycardia. These anti-arrhythmic actions of inhibition of late INa are due to reduction of repolarization heterogeneity secondary to preferential abbreviation of M cell action potential and suppression of EAD- and DAD-induced triggered activity [5, 24, 49]. EADs associated with a prolongation of repolarization can be suppressed by direct reduction of late INa, leading to APD abbreviation (Fig. 8). Abbreviation of APD with late INa block may also reduce the total charge carried by the L-type Ca channel current as well, which may contribute to reduction of Cai. Because of the presence of a high density of late INa in M cells, late INa can importantly abbreviate APD in these cells, thus reducing dispersion of repolarization, and suppressing EADs in all long QT types [5, 50].
Fig. 8.

Late INa block is effective in suppressing early afterdepolarization-induced triggered activity. The principal mechanisms underlying suppression of early afterdepolarizations by selective late INa inhibition include: 1) reduction of inward current and 2) decrease of Nai and thus Cai
Ranolazine has been shown to suppress VF in the setting of myocardial ischemia in pigs in vivo [51, 52]. In addition to its late INa inhibition, peak INa block by ranolazine may have contributed to this anti-arrhythmic effect of ranolazine, since ERP was significantly prolonged [51]. Ischemia commonly produces depolarization of RMP, which strongly promotes peak INa block. Ranolazine (10 μM) effectively suppresses H202-induced VF in rat [53]. Because H202 promotes late INa, ranolazine's inhibition of late INa is likely to importantly contribute to VF suppression. In this study, Vmax and conduction velocity were significantly decreased by ranolazine [53], indicating block of peak INa.
Atrial Arrhythmias
The therapeutic benefits of peak INa block in the setting of AF have long been recognized. The anti-AF action of peak INa blockers is due largely to rate-dependent reduction of excitability, prolongation of ERP (secondary to the development of post-repolarization refractoriness), and to conduction block in a critical part of reentrant circuit. Reduction of peak INa can also significantly decrease Nai and, thus, Cai, which may suppress Cai-mediated triggered activity. There is little information regarding the role of late INa in the generation of AF and even less information on potential pharmacological outcomes of the inhibition of this current on AF. In a recent comprehensive review of ranolazine's anti-arrhythmic actions in ventricles and atria, it was concluded that anti-AF ability of ranolazine is largely due to inhibition of peak INa and that anti-AF value of late INa inhibition appears to be limited to the suppression of trigger(s) under conditions of prolonged atrial repolarization [5].
Clinical and experimental experience indicates that practically all effective anti-AF agents prolong atrial ERP [41, 54]. Potassium channel blockers prolong ERP due to APD prolongation, whereas sodium channel blockers prolong ERP due to induction of rate-dependent post-repolarization refractoriness (PRR); mixed blockers prolong ERP due to both APD prolongation and PRR induction. Because selective late INa block does not prolong ERP, reduction of late INa alone is unlikely to be significantly effective for the suppression of AF. In fact, inhibition of late INa acts to abbreviate APD (Fig. 7 and 8) which may promote AF.
AF is believed to be largely maintained by a reentrant mechanism. Reentrant AF can be suppressed by peak INa blockers via reduction of excitability and prolongation of ERP and are unlikely to be significantly affected by selective late INa inhibition.
Late INa inhibition may however suppress the trigger(s) that initiate AF, particularly under conditions of prolonged APD and bradycardia. Several experimental and clinical studies point to the development of atrial arrhythmias under “atrial” long QT [55–57]. However, clinical experience indicate that repolarization prolonging agents induce proarrhythmias in ventricles but not in atria [58–60], and that only a minority of congenital long QT patients develop AF (<2 %) [61]. Among these patients only one out of 59 LQT3 patients had AF [61]. In another study, 3.2 % of patients with early-onset AF have a SCN5A mutation or rare variant previously associated with long QT3 [62]. Consistent to these clinical data, experimental conditions mimicking LQT1, 2 and 3 and which produce EADs and Torsade de Pointes in canine ventricles [63, 64], do not induce EAD or any arrhythmias in canine atria [65]. It appears that late INa inhibition can prevent AF initiation in the “atrial long QT” patients, but the number of patients is likely to be limited.
Apart from long QT syndrome, there are a number of pathological conditions prone to AF which may be associated with a prolongation of atrial APD/ERP and AF occurrence, such as the congestive heart failure [66], atrial dilatation [67, 68] and hypertension [18, 69]. It is noteworthy that in most of these studies [67–69], ERP but not APD was measured and that in many pathological conditions (including heart failure) atrial ERP can prolong without APD prolongation (due to PRR development) [70]. Interestingly, aging, a major pro-AF factor, seems also to be associated with a prolongation of APD and ERP [71, 72]. It is tempting to speculate that prolonged atrial APD in these pathologies and aging is due at least in part to an increase in total charge of late INa. Lengthening of APD in atrial myocytes (secondary to an increase in late INa) promotes Nai and then Cai loading, leading to the generation of triggered activity and ranolazine has been shown to suppress these activities [73]. Atrial APD is likely to be short in patients experiencing AF or having frequent episodes of AF with any pathology, due to rapid activation-mediated electrical remodeling [21]. Atrial APD can be prolonged in patients who are prone to develop new-onset AF and patients experiencing rare/short episodes of AF, not causing sustained electrical remodeling in atria. In these cases, abbreviation of APD by block of late INa may prevent AF initiation.
Tachycardia-mediated triggered activity appears to be less responsive to inhibition of late INa compared to bradycardia-mediated triggered activity in atria (Figs. 3 and 4). Ranolazine (10 μM) has been shown to prevent DAD-induced triggered activity (2.0–0.5 Hz) appearing following a period of rapid pacing (5–10 Hz) in superfused canine pulmonary vein (PV) preparations [74]. The primary mechanism of this action of ranolazine appears to be due largely to block of peak INa, because ranolazine (10 μM) causes a significant Vmax reduction and development of PRR in these PVs [74]. Also, under conditions of a very short APD (APD85<100 ms) and rapid pacing rates (CL0 200–100 ms), ranolazine-induced reduction of Nai in PV muscular sleeves is likely to be largely due to block of peak INa rather than late INa. It needs to be recognized that the contribution of late vs. peak INa block in the anti-arrhythmic action of ranolazine (or any other INa blocker) is difficult to determine. In fact, block of peak INa without inhibition of late INa is not possible. It is likely that it is a combination of late and peak INa inhibition that suppresses the appearance of the Cai–mediated triggered activity.
Experimental evidence indicates that ranolazine effectively prevents, terminates, and/or shortens AF only at concentrations that potently inhibit peak INa in atria (>5–10 μM), causing a significant rate-dependent depression of excitability and prolongation of PRR [14, 42, 75–77]. Relatively low concentrations of ranolazine (4–5 μM, which are more selective for block of late INa but still capable of causing measurable inhibition peak INa (Figs. 5 and 6) [14, 75], are less effective against AF [42, 75]. An exception to this rule is AF occurring in the setting of ischemia/reperfusion in atria, where 5 μM ranolazine causes a strong rate-dependent ERP prolongation due to development of PRR and effectively prevents the induction of AF (Fig. 9) [14]. Ranolazine also inhibit late ICa current (25–30 % at 2–6 μM [24]) and cause weak block of β-adrenergic receptors [78], which may contribute to its anti-arrhythmic effect. A combination of ranolazine and either dronedarone or amiodarone causes synergistic atrial-selective block of peak INa, thus effectively suppressing and preventing the induction of AF in a canine experimental model of AF [75, 79].
Fig. 9.

Ranolazine suppresses AF and/or prevents its induction in two experimental models involving isolated canine arterially-perfused right atria. a Persistent ACh (0.5 μM)-mediated AF is suppressed by ranolazine (10 μM). AF initially converts to flutter and then to sinus rhythm. b ERP measured at a CL of 500 ms is 140 ms. Attempts to re-induce AF fail because ranolazine-induced depression of excitability leads to 1:1 activation failure soon after CL is reduced from 500 to 200 ms (right panel). c Rapid-pacing induces non-sustained AF (48 s duration) following ischemia/reperfusion plus isoproterenol (Iso, 0.2 μmol/L) (left panel). Ranolazine (5 μM) prevents pacing-induced AF due to 1:1 activation failure (right panel). In both models, ranolazine causes prominent use-dependent induction of post-repolarization refractoriness. Reproduced with permission from Burashnikov et al. [14]
The clinical anti-AF efficacy of ranolazine has been demonstrated in several studies. In the MERLIN-TIMI 36 trial, ranolazine treatment was associated with a significant reduction of supraventricular tachyarrhythmias (p<0.001) as well as a 30 % reduction in new onset AF (p=0.08) [80]. Subsequently, a number of small exploratory clinical studies showed the effectiveness of ranolazine to terminate paroxysmal AF [81, 82] and prevent post-operative AF [83]. Ranolazine has been shown also to facilitate electrical cardioversion of AF in cardioversion-resistant patients [84] as well as to improve effectiveness of amiodarone to terminate paroxysmal AF [85].
Apart from ranolazine, there is an abundance of data on the effectiveness of INa blockers against AF in both experimental and clinical studies [41, 54, 59], from which the utility of late INa inhibition can be deduced. Highly selective INa blockers (lidocaine, mexiletine etc, at therapeutically relevant concentrations) are usually not effective against AF, suggesting that reduction of peak and late INa (and thus Nai) alone may be insufficient to effectively terminate and/or prevent AF. In experimental studies, lidocaine suppresses AF only at concentrations causing strong suppression of excitability due to peak INa inhibition (clinically toxic concentrations) [14, 86]. Of note, all clinically effective INa blockers (including ranolazine) inhibit other currents, particularly IKr [40]. In fact, IKr block-induced prolongation of atrial repolarization (APD90) greatly synergizes the effect of these agents to depress peak INa, at rapid activation rates (Fig. 6) [14, 35, 87].
The anti-AF efficacy of anti-arrhythmic drugs that block INa (flecainide, propafenone, amiodarone, etc) is relatively high with paroxysmal AF but low or absent with persistent AF [59]. Available evidence indicates that ranolazine loses its anti-AF efficacy in experimental models of persistent lone AF in the goat [42]. In this respect, the anti-AF value of the increased efficacy of ranolazine to inhibit late INa in persistent AF [17] is not clear. A reduced efficacy of ranolazine to block peak INa in persistent AF [17] may account for or contribute to the failure of this agent (and perhaps the other INa blockers) to terminate persistent AF.
The anti-AF efficacy of ranolazine in specific pathological conditions associated with AF remains poorly studied. It has been reported that ranolazine (5 μM) is effective in preventing AF in canine ischemia/reperfusion-induced AF model [14] as well as AF in canine ventricular tachypacing-induced heart failure model [70]. In both studies, anti-AF efficacy of ranolazine has been largely attributed to block of peak INa.
Conclusion
At present there is little specific information regarding the role of late INa in the generation of AF and even less information on potential pharmacological outcomes of the specific inhibition of this current on AF. Agents that potently block late INa in the ventricles, such as ranolazine and amiodarone, are also atrial-selective peak INa blockers and their anti-AF efficacy is due largely to inhibition of peak INa. The anti-arrhythmic effectiveness of selective late INa inhibition is thought to be greater when APD is long and heart rate is slow. The anti-arrhythmic efficacy of late INa block is reduced following acceleration of heart rates and/or abbreviation of APD. Available data suggest that block of late INa contributes to prevention of Cai-mediated triggered activity capable of initiating AF, particularly in clinical pathologies associated with a prolonged atrial APD (such as long QT syndrome)
Acknowledgments
Supported by grants HL 47678 from the National Institutes of Health (CA), Gilead Sciences, and the Masons of New York State and Florida.
References
- 1.Antzelevitch C, Belardinelli L, Wu L, Fraser H, Zygmunt AC, Burashnikov A, et al. Electrophysiologic properties and antiar-rhythmic actions of a novel anti-anginal agent. J Cardiovasc Pharmacol Therapeut. 2004;9(Suppl 1):S65–83. doi: 10.1177/107424840400900106. [DOI] [PubMed] [Google Scholar]
- 2.Undrovinas A, Maltsev VA. Late sodium current is a new therapeutic target to improve contractility and rhythm in failing heart. Cardiovasc Hematol Agents Med Chem. 2008;6:348–59. doi: 10.2174/187152508785909447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- 4.Antoons G, Oros A, Beekman JDM, Engelen MA, Houtman MJC, Belardinelli L, et al. Late Na+ current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J Am Coll Cardiol. 2010;55:801–9. doi: 10.1016/j.jacc.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 5.Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–90. doi: 10.1016/j.hrthm.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gintant GA, Daytner NB, Cohen IS. Slow inactivation of a tetrodotoxin-sensitive current in canine cardiac Purkinje fibers. Biophys J. 1984;45:509–12. doi: 10.1016/S0006-3495(84)84187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patlak JB, Ortiz M. Slow currents through single sodium channels of the adult rat heart. J Gen Physiol. 1985;86:89–104. doi: 10.1085/jgp.86.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attwell D, Cohen IS, Eisner DA, Ohba M, Ojeda C. The steady-state tetrodotoxin-sensitive (“window”) sodium current in cardiac Purkinje fibers. Pflugers Arch. 1979;379:137–42. doi: 10.1007/BF00586939. [DOI] [PubMed] [Google Scholar]
- 9.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–97. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 10.Belardinelli L, Shryock JC, Fraser H. The mechanism of ranolazine action to reduce ischemia-induced diastolic dysfunction. Eur Heart J Suppl. 2006;8:A10–3. [Google Scholar]
- 11.Sossalla S, Maier LS. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther. 2012;133:311–23. doi: 10.1016/j.pharmthera.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Persson F, Andersson B, Duker G, Jacobson I, Carlsson L. Functional effects of the late sodium current inhibition by AZD7009 and lidocaine in rabbit isolated atrial and ventricular tissue and Purkinje fibre. Eur J Pharmacol. 2007;558:133–43. doi: 10.1016/j.ejphar.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 13.Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol. 2002;34:1185–94. doi: 10.1006/jmcc.2002.2053. [DOI] [PubMed] [Google Scholar]
- 14.Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–57. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burashnikov A, Mannava S, Antzelevitch C. Transmembrane action potential heterogeneity in the canine isolated arterially-perfused atrium: effect of IKr and Ito/IKur block. Am J Physiol. 2004;286:H2393–400. doi: 10.1152/ajpheart.01242.2003. [DOI] [PubMed] [Google Scholar]
- 16.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell. Circ Res. 1991;68:1729–41. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 17.Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, et al. Altered Na+ currents in atrial fibrillation: effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–42. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 18.Guo D, Young LH, Wu Y, Belardinelli L, Kowey PR, Yan GX. Increased late sodium current in left atrial myocytes of rabbits with left ventricular hypertrophy: its role in the genesis of atrial arrhythmias. Am J Physiol Heart Circ Physiol. 2010 doi: 10.1152/ajpheart.01145.2009. [DOI] [PubMed] [Google Scholar]
- 19.Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–31. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 20.Gaspo R, Bosch RF, Bou-Abboud E, Nattel S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ Res. 1997;81:1045–52. doi: 10.1161/01.res.81.6.1045. [DOI] [PubMed] [Google Scholar]
- 21.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–68. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 22.Akar JG, Everett TH, Ho R, Craft J, Haines DE, Somlyo AP, et al. Intracellular chloride accumulation and subcellular elemental distribution during atrial fibrillation. Circulation. 2003;107:1810–5. doi: 10.1161/01.CIR.0000058462.23347.93. [DOI] [PubMed] [Google Scholar]
- 23.Clancy CE, Rudy Y. Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation. 2002;105:1208–13. doi: 10.1161/hc1002.105183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, et al. Electrophysiologic effects of ranolazine: a novel anti-anginal agent with antiarrhythmic properties. Circulation. 2004;110:904–10. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo D, Lian J, Liu T, Cox R, Margulies KB, Kowey PR, et al. Contribution of late sodium current (I(Na-L)) to rate adaptation of ventricular repolarization and reverse use-dependence of QT-prolonging agents. Heart Rhythm. 2011;8:762–9. doi: 10.1016/j.hrthm.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 26.Carmeliet E, Mubagwa K. Antiarrhythmic drugs and cardiac ion channels: mechanisms of action. Prog Biophys Mol Biol. 1998;70:1–72. doi: 10.1016/s0079-6107(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 27.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S161–77. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajamani S, El-Bizri N, Shryock JC, Makielski JC, Belardinelli L. Use-dependent block of cardiac late Na+ current by ranolazine. Heart Rhythm. 2009;6:1625–31. doi: 10.1016/j.hrthm.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zygmunt AC, Nesterenko VV, Rajamani S, Hu D, Barajas-Martinez H, Belardinelli L, et al. Mechanisms of atrial-selective block of sodium channel by ranolazine I. Experimental analysis of the use-dependent block. Am J Physiol Heart Circ Physiol. 2011;301:H1606–14. doi: 10.1152/ajpheart.00242.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whalley DW, Wendt DJ, Grant AO. Basic concepts in cellular cardiac electrophysiology: Part II: Block of ion channels by antiarrhythmic drugs. PACE. 1995;18:1686–704. doi: 10.1111/j.1540-8159.1995.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 32.Burashnikov A, Pourrier M, Gibson JK, Lynch JJ, Antzelevitch C. Rate-dependent effects of vernakalant in the isolated non-remodeled canine left atria are primarily due to block of the sodium channel. Comparison with ranolazine and dl-sotaol. Circ Arrhythm Electrophysiol. 2012;5:400–8. doi: 10.1161/CIRCEP.111.968305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wasserstrom JA, Salata JJ. Basis for tetrodotoxin and lidocaine effects on action potentials in dog ventricular myocytes. Am J Physiol. 1988;254:H1157–66. doi: 10.1152/ajpheart.1988.254.6.H1157. [DOI] [PubMed] [Google Scholar]
- 34.Szel T, Koncz I, Jost N, Baczko I, Husti Z, Virag L, et al. Class I/B antiarrhythmic property of ranolazine, a novel antianginal agent, in dog and human cardiac preparations. Eur J Pharmacol. 2011;662:31–9. doi: 10.1016/j.ejphar.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 35.Burashnikov A, Di Diego JM, Sicouri S, Ferreiro M, Carlsson L, Antzelevitch C. Atrial-selective effects of chronic amiodarone in the management of atrial fibrillation. Heart Rhythm. 2008;5:1735–42. doi: 10.1016/j.hrthm.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burashnikov A, Zygmunt AC, Di Diego JM, Linhardt G, Carlsson L, Antzelevitch C. AZD1305 exerts atrial-predominant electrophysiological actions and is effective in suppressing atrial fibrillation and preventing its re-induction in the dog. J Cardiovasc Pharmacol. 2010;56:80–90. doi: 10.1097/FJC.0b013e3181e0bc6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nesterenko VV, Zygmunt AC, Rajamani S, Belardinelli L, Antzelevitch C. Mechanisms of atrial-selective block of Na+ channels by ranolazine: II. Insights from a mathematical model. Am J Physiol Heart Circ Physiol. 2011;301:H1615–24. doi: 10.1152/ajpheart.00243.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burashnikov A, Petroski A, Hu D, Barajas-Martinez H, Antzelevitch C. Atrial-selective inhibition of sodium channel current by Wenxin Keli is effective in suppressing atrial fibrillation. Heart Rhythm. 2012;9:125–31. doi: 10.1016/j.hrthm.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burashnikov A, Antzelevitch C. Atrial-selective sodium channel block for the treatment of atrial fibrillation. Expert Opin Emerg Drugs. 2009;14:233–49. doi: 10.1517/14728210902997939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burashnikov A, Antzelevitch C. New development in atrial antiarrhythmic drug therapy. Nat Rev Cardiol. 2010;7:139–48. doi: 10.1038/nrcardio.2009.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burashnikov A, Antzelevitch C. Novel pharmacological targets for the rhythm control management of atrial fibrillation. Pharmacol Ther. 2011;132:300–13. doi: 10.1016/j.pharmthera.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verheule S, Tuyls E, van Hunnik A, Kuiper M, Wang WQ, Dhalla A, et al. Effects of ranolazine in a goat model of lone atrial fibrillation. Circulation. 2009;120:S675. [Google Scholar]
- 43.Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28:287–94. doi: 10.1111/j.1755-5922.2010.00210.x. [DOI] [PubMed] [Google Scholar]
- 44.Moreno JD, Clancy CE. Pathophysiology of the cardiac late Na current and its potential as a drug target. J Mol Cell Cardiol. 2012;52:608–19. doi: 10.1016/j.yjmcc.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zipes DP. Electrophysiological remodeling of the heart owing to rate. Circulation. 1997;95:1745–8. doi: 10.1161/01.cir.95.7.1745. [DOI] [PubMed] [Google Scholar]
- 46.Antzelevitch C. Mechanisms of cardiac arrhythmias and conduction disturbances. In: Fuster V, RA O'Rourke, Walsh RA, Poole-Wilson P, editors. Hurst's the heart. 12th ed. Mc-Graw-Hill; New York: 2008. pp. 913–45. [Google Scholar]
- 47.Wit AL, Rosen MR. Afterdepolarizations and triggered activity: distinction from automaticity as an arrhythmogenic mechanism. 1992:2113–64. [Google Scholar]
- 48.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–60. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 49.Antzelevitch C. Electrical heterogeneity, cardiac arrhythmias, and the sodium channel. Circ Res. 2000;87:964–5. doi: 10.1161/01.res.87.11.964. [DOI] [PubMed] [Google Scholar]
- 50.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–47. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 51.Kumar K, Nearing BD, Bartoli CR, Kwaku KF, Belardinelli L, Verrier RL. Effect of ranolazine on ventricular vulnerability and defibrillation threshold in the intact porcine heart. J Cardiovasc Electrophysiol. 2008;19:1073–9. doi: 10.1111/j.1540-8167.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- 52.Nieminen T, Nanbu DY, Datti IP, Vaz GR, Tavares CA, Pegler JR, et al. Antifibrillatory effect of ranolazine during severe coronary stenosis in the intact porcine model. Heart Rhythm. 2011;8:608–14. doi: 10.1016/j.hrthm.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 53.Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN, et al. Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2011;52:366–75. doi: 10.1016/j.jacc.2010.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Workman AJ, Smith GL, Rankin AC. Mechanisms of termination and prevention of atrial fibrillation by drug therapy. Pharmacol Ther. 2011;131:221–41. doi: 10.1016/j.pharmthera.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satoh T, Zipes DP. Cesium-induced atrial tachycardia degenerating into atrial fibrillation in dogs: atrial torsades de pointes? J Cardiovasc Electrophysiol. 1998;9:970–5. doi: 10.1111/j.1540-8167.1998.tb00137.x. [DOI] [PubMed] [Google Scholar]
- 56.Kirchhof P, Eckardt L, Franz MR, Monnig G, Loh P, Wedekind H, et al. Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1027–33. doi: 10.1046/j.1540-8167.2003.03165.x. [DOI] [PubMed] [Google Scholar]
- 57.Zellerhoff S, Pistulli R, Monnig G, Hinterseer M, Beckmann BM, Kobe J, et al. Atrial arrhythmias in long-QT syndrome under daily life conditions: a nested case control study. J Cardiovasc Electrophysiol. 2009;20:401–7. [Google Scholar]
- 58.Vincent GM. Atrial arrhythmias in the inherited long QT syndrome: laboratory quirk or clinical arrhythmia? J Cardiovasc Electrophysiol. 2003;14:1034–5. doi: 10.1046/j.1540-8167.2003.03365.x. [DOI] [PubMed] [Google Scholar]
- 59.Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, Ernst S, et al. Guidelines for the management of atrial fibrillation: the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;12:1360–420. doi: 10.1093/eurheartj/ehq278. [DOI] [PubMed] [Google Scholar]
- 60.van der Hooft CS, Heeringa J, van Herpen G, Kors JA, Kingma JH, Stricker BH. Drug-induced atrial fibrillation. J Am Coll Cardiol. 2004;44:2117–24. doi: 10.1016/j.jacc.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 61.Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. 2008;5:704–9. doi: 10.1016/j.hrthm.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–9. doi: 10.1161/CIRCGENETICS.111.962597. [DOI] [PubMed] [Google Scholar]
- 63.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–52. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 64.Burashnikov A, Antzelevitch C. Prominent IKs in epicardium and endocardium contributes to development of transmural dispersion of repolarization but protects against development of early after-depolarizations. J Cardiovasc Electrophysiol. 2002;13:172–7. doi: 10.1046/j.1540-8167.2002.00172.x. [DOI] [PubMed] [Google Scholar]
- 65.Burashnikov A, Antzelevitch C. Absence of early afterdepolarizations in canine atria under long QT syndrome. Heart Rhythm. 2011;8(5S):S328. (Abstract) [Google Scholar]
- 66.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–8. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 67.Verheule S, Wilson E, Everett T, Shanbhag S, Golden C, Olgin J. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation. 2003;107:2615–22. doi: 10.1161/01.CIR.0000066915.15187.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts-Thomson KC, Stevenson I, Kistler PM, Haqqani HM, Spence SJ, Goldblatt JC, et al. The role of chronic atrial stretch and atrial fibrillation on posterior left atrial wall conduction. Heart Rhythm. 2009;6:1109–17. doi: 10.1016/j.hrthm.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 69.Kistler PM, Sanders P, Dodic M, Spence SJ, Samuel CS, Zhao C, et al. Atrial electrical and structural abnormalities in an ovine model of chronic blood pressure elevation after prenatal corticosteroid exposure: implications for development of atrial fibrillation. Eur Heart J. 2006;27:3045–56. doi: 10.1093/eurheartj/ehl360. [DOI] [PubMed] [Google Scholar]
- 70.Burashnikov A, Di Diego JM, Moise NS, Kornreich BG, Belardinelli L, Antzelevitch C. Ranolazine causes atrial-selective electrophysiological effects and suppresses the induction of atrial fibrillation in a canine model of heart failue. Heart Rhythm. 2012;9 (Abstract, in press) [Google Scholar]
- 71.Anyukhovsky EP, Sosunov EA, Chandra P, Rosen TS, Boyden PA, Danilo P, Jr, et al. Age-associated changes in electrophysiologic remodeling: a potential contributor to initiation of atrial fibrillation. Cardiovasc Res. 2005;66:353–63. doi: 10.1016/j.cardiores.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 72.Kistler PM, Sanders P, Fynn SP, Stevenson IH, Spence SJ, Vohra JK, et al. Electrophysiologic and electroanatomic changes in the human atrium associated with age. J Am Coll Cardiol. 2004;44:109–16. doi: 10.1016/j.jacc.2004.03.044. [DOI] [PubMed] [Google Scholar]
- 73.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–9. doi: 10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- 74.Sicouri S, Glass A, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm. 2008;5:1019–26. doi: 10.1016/j.hrthm.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burashnikov A, Sicouri S, Di Diego JM, Belardinelli L, Antzelevitch C. Synergistic effect of the combination of dronedarone and ranolazine to suppress atrial fibrillation. J Am Coll Cardiol. 2010;56:1216–24. doi: 10.1016/j.jacc.2010.08.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar K, Nearing BD, Carvas M, Nascimento BC, Acar M, Belardinelli L, et al. Ranolazine exerts potent effects on atrial electrical properties and abbreviates atrial fibrillation duration in the intact porcine heart. J Cardiovasc Electrophysiol. 2009;20:796–802. doi: 10.1111/j.1540-8167.2009.01437.x. [DOI] [PubMed] [Google Scholar]
- 77.Carvas M, Nascimento BC, Acar M, Nearing BD, Belardinelli L, Verrier RL. Intrapericardial ranolazine prolongs atrial refractory period and markedly reduces atrial fibrillation inducibility in the intact porcine heart. J Cardiovasc Pharmacol. 2010;55:286–91. doi: 10.1097/FJC.0b013e3181d26416. [DOI] [PubMed] [Google Scholar]
- 78.Letienne R, Vie B, Puech A, Vieu S, Le Grand B, John GW. Evidence that ranolazine behaves as a weak beta1- and beta2-adrenoceptor antagonist in the cat cardiovascular system. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:464–71. doi: 10.1007/s002100000378. [DOI] [PubMed] [Google Scholar]
- 79.Sicouri S, Burashnikov A, Belardinelli L, Antzelevitch C. Synergistic electrophysiologic and antiarrhythmic effects of the combination of ranolazine and chronic amiodarone in canine atria. Circ Arrhythm Electrophysiol. 2010;3:88–95. doi: 10.1161/CIRCEP.109.886275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–52. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 81.Murdock DK, Overton N, Kersten M, Kaliebe J, Devecchi F. The effect of ranolazine on maintaining sinus rhythm in patients with resistant atrial fibrillation. Indian Pacing Electrophysiol J. 2008;8:175–81. [PMC free article] [PubMed] [Google Scholar]
- 82.Murdock DK, Kersten M, Kaliebe J, Larrian G. The use of oral ranolazine to convert new or paroxysmal atrial fibrillation: a reveiw of experience with implications for possible “pill in the pocket” approach to atrial fibrillation. Indian Pacing Electrophysiol J. 2009;9:260–7. [PMC free article] [PubMed] [Google Scholar]
- 83.Miles RH, Passman R, Murdock DK. Comparison of effectiveness and safety of ranolazine versus amiodarone for preventing atrial fibrillation after coronary artery bypass grafting. Am J Cardiol. 2011;108:673–6. doi: 10.1016/j.amjcard.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 84.Murdock DK, Kaliebe J, Larrain G. The use of ranolazine to facilitate electrical cardioversion in cardioversion-resistant patients: a case series. Pacing Clin Electrophysiol. 2012;35:302–7. doi: 10.1111/j.1540-8159.2011.03298.x. [DOI] [PubMed] [Google Scholar]
- 85.Fragakis N, Koskinas KC, Katritsis DG, Pagourelias ED, Zografos T, Geleris P. Comparison of effectiveness of ranolazine plus amiodarone versus amiodarone alone for conversion of recent-onset atrial fibrillation. Am J Cardiol. 2012;110:673–7. doi: 10.1016/j.amjcard.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 86.Comtois P, Sakabe M, Vigmond EJ, Munoz MA, Texier A, Shiroshita-Takeshita A, et al. Mechanisms of atrial fibrillation termination by rapidly unbinding Na+ channel blockers. Insights from mathematical models and experimental correlates. Am J Physiol Heart Circ Physiol. 2008;295:H1489–504. doi: 10.1152/ajpheart.01054.2007. [DOI] [PubMed] [Google Scholar]
- 87.Burashnikov A, Belardinelli L, Antzelevitch C. Atrial-selective sodium channel block strategy to suppress atrial fibrillation. Ranolazine versus propafenone. J Pharmacol Exp Ther. 2012;340:161–8. doi: 10.1124/jpet.111.186395. [DOI] [PMC free article] [PubMed] [Google Scholar]