

Abstract
Developmental biology has long benefited from studies of classic model organisms. Recently, human pluripotent stem cells (hPSCs), including human embryonic stem cells and human induced pluripotent stem cells, have emerged as a new model system that offers unique advantages for developmental studies. Here, we discuss how studies of hPSCs can complement classic approaches using model organisms, and how hPSCs can be used to recapitulate aspects of human embryonic development ‘in a dish’. We also summarize some of the recently developed genetic tools that greatly facilitate the interrogation of gene function during hPSC differentiation. With the development of high-throughput screening technologies, hPSCs have the potential to revolutionize gene discovery in mammalian development.
Keywords: Human pluripotent stem cells, Directed differentiation, Gene targeting, High-throughput screening
Introduction
Human pluripotent stem cells (hPSCs), which include human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), can self-renew indefinitely in culture while maintaining the ability to become almost any cell type in the human body (Takahashi et al., 2007; Thomson et al., 1998; Yu et al., 2007). The potential of using hPSCs for cell replacement therapy and disease modeling has been discussed extensively (Wu and Hochedlinger, 2011). This Review focuses on an equally important, yet often overlooked, aspect: using hPSCs to gain insights into human embryonic development. Although this potential has been long recognized (Keller, 2005; Pera and Trounson, 2004), it is only beginning to be realized, owing to advances in both in vitro differentiation approaches and genetic manipulation tools in hPSCs. In this Review, we summarize the latest progress in this nascent, yet rapidly advancing, field and discuss future prospects and potential challenges of using hPSCs for studies of developmental biology. First, the strengths and limitations of classic model organisms are discussed to highlight the need for a new model. Second, we illustrate how hPSCs can be used to recapitulate defined steps of embryogenesis, and we discuss how hPSC-based studies can lead to novel insights into human development. Next, we review new genetic tools that can be applied to interrogate gene function during the in vitro differentiation of hPSCs. Finally, the potential of using hPSCs for discovery-driven research is discussed.
This Review focuses primarily on work performed using hPSCs, with occasional references to studies using mouse pluripotent stem cells when comparable studies are not yet available in human cells; work from both hiPSCs and hESCs is discussed, although more examples stem from hESCs, owing to their more-frequent use in differentiation experiments. Nonetheless, because of the high degree of similarity between hESCs and hiPSCs (Yamanaka, 2012), it is likely that the general strategies and most conclusions will apply to both hESCs and hiPSCs.
The need for a new model system
A main challenge in biology is to understand the development of the human body. This is driven not only by our basic curiosity of life, but also by the practical need to find cures for the numerous human diseases caused by developmental defects. We have accumulated an incredible amount of knowledge about the human genome, physiology and anatomy. By contrast, there is little direct information about how embryonic development is regulated. Mutation analysis in hereditary diseases is widely used to identify potential disease-associated genes, although this approach is largely limited to studies of defects presenting after birth. Furthermore, the functional validation of risk loci presents a major challenge. As we obviously cannot manipulate human development in the same way we do when experimenting on a mouse or a fruit fly, much of our knowledge about human development has been extrapolated from studies of model organisms. These studies have provided fundamental insights into the general principles of development, as well as into the genes and signaling pathways that control specific aspects of cell fate specification and tissue morphogenesis. Many of these genes and signaling pathways play conserved roles in human development. For example, genetic studies in Drosophila have demonstrated crucial roles for Hox genes in controlling body plan, and similar roles for Hox genes have been described in other organisms, including humans (Mallo et al., 2010). For practical reasons, the mouse has become the premier experimental system with which to model human organ development. Mice and humans have a similar genome size. They share 99% of their genes and exhibit vast similarities in development, anatomy and physiology. We now have a large repertoire of powerful genetic tools available for studies of murine development (Nagy, 2002). For example, fluorescent-protein reporters are widely used to visualize dynamic biological processes during embryonic development (Hadjantonakis et al., 2003), various lineage-tracing strategies have been developed to follow the progeny of individual cells (Kretzschmar and Watt, 2012) and numerous mouse mutants have been generated to study gene function in development (Capecchi, 2005). Reassuringly, mutations often cause similar phenotypes in mice and humans. For example, null mutations in pancreatic and duodenal homeobox 1 (Pdx1) or pancreas specific transcription factor 1a (Ptf1a) cause almost complete absence of the pancreas in both mice (Jonsson et al., 1994; Kawaguchi et al., 2002; Krapp et al., 1998; Offield et al., 1996) and humans (Sellick et al., 2004; Stoffers et al., 1997).
However, despite being a powerful model organism, the mouse has some notable limitations. Around 1% of human genes have no identifiable mouse homologs (Waterston et al., 2002). Although most genes are expected to play conserved roles in mice and humans, obvious species-specific differences exist in gestation period, morphology, and the spatial and temporal regulation of gene expression during embryonic development. Consequently, mouse models do not always fully replicate the features of human diseases (Elsea and Lucas, 2002). A famous example is hypoxanthine-guanine phosphoribosyl transferase (Hprt), the first gene mutated through gene targeting in mouse embryonic stem cells (mESCs) (Doetschman et al., 1988; Thomas and Capecchi, 1987). In humans, HPRT deficiency causes Lesch-Nyhan syndrome, a disorder of uric acid over-production and neurological dysfunction. However, these symptoms are not observed in Hprt mutant mice (Finger et al., 1988). In a more recent example, heterozygous inactivating mutations in GATA6 have been identified in more than half of human cases of pancreatic agenesis (Allen et al., 2012). However, Gata6 heterozygous null mice show no obvious phenotype, whereas Gata6 homozygous null mice die during gastrulation, precluding analysis of pancreatic phenotypes (Morrisey et al., 1998). Recent conditional knockout studies show that deletion of both Gata4 and Gata6 (i.e. loss of four alleles of the GATA genes) is necessary to generate the same pancreatic phenotype in the mouse (Carrasco et al., 2012; Xuan et al., 2012). There are almost certainly many other genes like GATA6 for which such mouse-human discrepancies exist, owing to several non-mutually exclusive possibilities: (1) species-specific gene requirement; (2) overlapping functions with a related gene in a species-specific manner; and/or (3) differences in gene dose sensitivity between humans and mice, as observed in the development of organs such as the heart and the limb (Bruneau, 2008; Wilkie, 2003). Therefore, it is not uncommon to miss the crucial function of a gene in human development, even after conducting extensive studies in mouse models.
Another challenge lies in the difficulty of discovery-driven research in the mouse. Since the ‘Heidelberg screen’ in fruit flies led by Christiane Nüsslein-Volhard and Eric Wieschaus (Nüsslein-Volhard and Wieschaus, 1980), genetic screens in model organisms have been instrumental in aiding our understanding of embryonic development and human diseases. Such screens are most popular in invertebrate species such as Caenorhabditis elegans and Drosophila melanogaster, although several forward genetic screens have also been conducted in the mouse (Acevedo-Arozena et al., 2008). One such screen led to the surprising discovery of the primary cilium as an essential cellular organelle for Hedgehog (HH) signal transduction (Huangfu et al., 2003). This study clearly demonstrates the necessity of conducting genetic screens in mammals, as cilia are not required for HH signaling in Drosophila. It is theoretically possible to perform saturation screens in the mouse to uncover all genes involved in a specific process of interest. However, there are practical constraints: money, time and the space to house the mice produced from a genome-wide mutagenesis screen. Consequently, a more-efficient screening platform is highly desirable for systematic studies of mammalian development. Compelling evidence suggests that in vitro differentiation of hPSCs recapitulate aspects of human development, and may be used as a new model system for human developmental studies.
Modeling embryonic development using hPSCs
Thomson and colleagues derived the first hESC lines from cultured human blastocysts (Fig. 1A) (Thomson et al., 1998). This was almost two decades after the first mESCs were derived (Evans and Kaufman, 1981; Martin, 1981). In a more recent breakthrough, Yamanaka and Takahashi reprogrammed adult mouse cells into induced pluripotent stem cells by expressing four transcription factors [Oct3/4 (Pou5f1), Sox2, Myc and Klf4] (Takahashi and Yamanaka, 2006). This time, the leap to human cells took only 1 year, and hiPSCs (Fig. 1B) were soon generated (Takahashi et al., 2007; Yu et al., 2007). The ability to generate hiPSCs from patient samples makes it possible to study crucial aspects of a disease of interest (see section below), and for autologous cell replacement therapy (Wu and Hochedlinger, 2011). Furthermore, and as we discuss below, in vitro differentiation and genetic manipulation of hPSCs also provide great opportunities to study human embryonic development.
Fig. 1.

The derivation of hESCs and hiPSCs. (A) Human embryonic stem cells (hESCs) are derived from the inner cell mass of cultured preimplantation human blastocysts. When grown on mouse embryonic fibroblast feeders, human pluripotent stem cells (hPSCs) can self-renew indefinitely in culture while maintaining the ability to become derivatives of all three germ layers. (B) Human somatic cells can be reprogrammed into human induced pluripotent stem cells (hiPSCs) by: (1) ectopic expression of transcription factors; (2) ectopic expression of transcription factors together with small molecules; and (3) ectopic expression of microRNAs. These reprograming factors can be delivered into somatic cells via viral infection, transposon transgenesis, plasmid transfection and direct delivery of cell-permeable proteins or synthetic mRNAs.
Two unique characteristics of hPSCs make them well suited for studies of human development. First, hPSCs have the potential to generate every adult cell type, offering an attractive window into understanding human development. Their in vitro culture system also provides a rapid, cost-effective way to interrogate the function of a gene during a specific developmental process. Second, hPSCs have unlimited self-renewal capacity, providing abundant material for high-throughput screening (HTS). Therefore, hPSCs may be used not only for testing hypotheses stemming from prior studies in model organisms, but also for discovery-driven research through biological and chemical screening. Such studies rely on robust in vitro differentiation platforms that faithfully mimic embryonic development.
Embryoid bodies: a potential model of early embryogenesis
When cultured in suspension without feeder layers, hPSCs spontaneously form aggregates known as embryoid bodies (EBs) (Itskovitz-Eldor et al., 2000). EBs typically start from densely packed cells, and progress to cystic structures, the center of which becomes cavitated and filled with fluids. EB formation is widely used as the initial step in many differentiation protocols; cells in EBs can be guided towards specific cell lineages through exposure to differentiation cues (discussed below) (Schuldiner et al., 2000). Interestingly, some degree of polarity and tissue regionalization have been observed during EB formation (Itskovitz-Eldor et al., 2000). Although far from recapitulating the patterning of actual embryos, this regionalization is reminiscent of gastrulation, the process responsible for the formation of the three embryonic germ layers: ectoderm, mesoderm and endoderm.
Analysis of the expression profiles in human EBs demonstrated that a cascade of genes that govern gastrulation and germ layer formation is activated sequentially, which appears to correspond to the sequential stages of embryonic development (Dvash et al., 2004). Among them are genes known to be involved in early pattern formation, supporting the use of EBs as a valuable model for understanding the molecular mechanisms that underlie early human embryogenesis. For example, studies in human EBs suggest that LEFTY and NODAL, members of the transforming growth factor β (TGFβ) family, play conserved roles in gastrulation in human embryos (Dvash et al., 2007); inhibition of the NODAL/LEFTY pathway impairs differentiation of hESCs into the mesodermal lineage. However, species-specific differences also exist. Using human EBs to recapitulate yolk sac development, TGFβ signaling was shown to inhibit endothelial differentiation, the opposite of its role in mice (Poon et al., 2006). Therefore, human EBs provide a valuable model system for studying both conserved and non-conserved mechanisms of early embryogenesis.
Compared with differentiation in adherent culture conditions, the three-dimensional (3D) structure of EBs offers the benefit of potentially recapitulating complex cell and tissue interactions. For example, cells that resemble the gastrula organizer were identified in human EBs, and these cells induced a secondary axis when transplanted into frog embryos (Sharon et al., 2011). These findings suggest that human EBs can offer insights into the establishment of body axis in human embryos. However, spontaneous differentiation in EBs often involves cellular responses to local morphogen signals. As it is difficult to control precisely the microenvironment in EBs, it is not always straightforward to interpret results from EB-based experiments. Besides, the heterogeneity in the size and developmental timing of EBs may also limit their use as a robust in vitro model of early embryogenesis. Methods are being developed to generate EBs in a more uniform and reproducible manner. For example, forced aggregation of defined numbers of hPSCs or the use of 3D microwell cultures tends to generate EBs that are more uniform in size and morphology (Mohr et al., 2010a; Ng et al., 2005). Another study employed a semi-solid 3D extracellular matrix to support the formation of EBs with more organized germ layer structures (Rust et al., 2006). These technological improvements may lead to the wider use of EBs for studies of early embryogenesis.
Modeling cell fate specification through directed differentiation
To address specific issues regarding lineage commitment, it is necessary to develop more defined differentiation conditions. Diverse methods have been developed: most involve the addition of recombinant growth factors or small-molecule compounds, with many methods using adherent culture conditions, some using EB formation and others using feeder cells (Fig. 2). We summarize below three key aspects of directed differentiation, and highlight the strong connection between hPSC differentiation and embryonic development in model organisms.
Fig. 2.
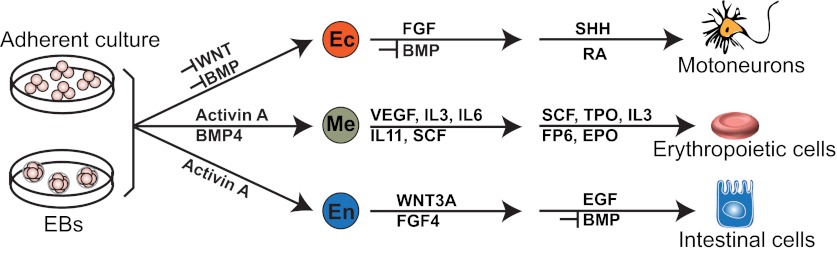
Directed differentiation of hPSCs. In vitro differentiation of human pluripotent stem cells (hPSCs) can be performed in adherent culture or in suspension culture via embryoid body (EB) formation. In both formats, differentiation can be induced by treatment with growth factors and small molecules to activate or inhibit various signaling pathways in a step-wise manner by mimicking embryonic development. Typical differentiation protocols are illustrated using three specific examples: motoneurons from the ectoderm (Li et al., 2005; Wichterle et al., 2002), erythropoietic cells from the mesoderm (Niwa et al., 2011) and intestinal cells from the endoderm (Spence et al., 2011). In each case, the signaling factors and pathways required (or those that need to be inhibited) to drive differentiation into the appropriate cell types are indicated. BMP, bone morphogenetic protein; EGF, epidermal growth factor; EPO, erythropoietin; FGF, fibroblast growth factor; FP6, interleukin 6 (IL6) and IL6 receptor fusion protein; IL, interleukin; RA, retinoic acid; SCF, Kit ligand; SHH, sonic hedgehog; TPO, thyroid peroxidase; VEGF, vascular endothelial growth factor.
First, directed differentiation typically consists of a series of defined steps that mimic the process of embryonic development. One of the first developmental events to consider is the formation of three germ layers through gastrulation; consequently, most protocols involve first directing hPSCs to ectoderm, endoderm or mesoderm, followed by a series of steps to guide the differentiation towards a particular cell type of interest (Fig. 2) (Williams et al., 2012). For example, to generate retinal epithelium or photoreceptors, hPSCs are first differentiated into neuroectoderm, then into cells representing the eye field; finally, a subset of cells undergoes advanced retinal differentiation over a time course that closely mimicks that of human retinal development (Meyer et al., 2009).
Second, each differentiation step is guided by specific differentiation cues. Recombinant growth factors and small-molecule compounds are commonly used to mimic signals that are known to instruct embryonic development. For example, guided by knowledge of germ layer formation during gastrulation (Keller, 2005), hPSCs are exposed to high concentration of activin A (a TGFβ family member) for differentiation into endoderm (D'Amour et al., 2005), to bone morphogenetic protein 4 (BMP4) and activin A for mesoderm differentiation (Yang et al., 2008), and to inhibitors of BMP and WNT signaling for ectoderm formation (Lamba et al., 2006) (Fig. 2). The same is true for later steps of differentiation. To illustrate, the generation of pancreatic progenitor cells from hPSCs (D'Amour et al., 2006) involves manipulating multiple signaling pathways known (from studies in mice, zebrafish and frogs) to play a role in pancreas specification, including activation of retinoic acid and fibroblast growth factor 10 (FGF10) signaling and inhibition of HH signaling (Cleaver and MacDonald, 2010). When prior knowledge of embryonic development is not readily available, one strategy is to recapitulate the in vivo environment by using cells isolated from the physical location where the desired cell type emerges. In an elegant recent study, for example, defined signaling cues were first used to differentiate mESCs into ectoderm, and then into otic progenitors (Oshima et al., 2010). These otic progenitors were then plated onto a layer of stromal cells from the inner ear to induce the formation of sensory hair cells. Collectively, these studies demonstrate that lineage commitment during hPSC differentiation can mimic embryonic development and involves similar signaling events.
Finally, the identity of an hPSC-derived cell type is validated through comparison with its in vivo counterpart. An ever-increasing list of cell types has now been generated from hPSCs (Williams et al., 2012). Typically, the first step of validation involves the examination of specific markers expressed by the in vivo counterpart, often using information from mouse studies. More stringent functional validation is performed based on knowledge about the physiological functions of the cell. For example, transplanted hESC-derived retinal progenitor cells were shown to differentiate into functional photoreceptors and restore some light responses in a blind mouse model (Lamba et al., 2009). Another recent study has created dopaminergic (DA) neurons that exhibit similar electrophysiological features to endogenous DA neurons (Kriks et al., 2011). When transplanted into animal models of Parkinson's disease, such hPSC-derived DA neurons completely reversed the drug-induced rotation behavior, and recipient mice and rats demonstrated improvements in tests of forelimb use and control of voluntary movements.
A complication in the functional analysis of hPSC-derived cells, however, lies in the fact that the desired cell type is present in the differentiation culture with many other cell types, and the therapeutic use of such mixed cell populations has been debated. One study, for example, used hPSCs to generate pancreatic progenitor-like cells, which appear to give rise to functional, glucose-responsive pancreatic β-cells 3 months after transplantation into immunocompromised mice (Kroon et al., 2008). However, the differentiation culture contains endodermal cells at different stages of differentiation, as well as various cell types from non-endodermal lineages. This poses a hurdle to therapeutic use of such cells, owing to concerns about teratoma formation and the unpredictable effects of a mixed population. Additionally, transplantation of such a mixed cell population and the long in vivo differentiation time also casts doubts on the origin of β-cells formed after the transplantation, and makes it difficult to identify the exact signals necessary for the specification and maturation of β-cells. A follow-up study by the same group has identified cell-surface markers that can be used to enrich pancreatic progenitor cells for transplantation assays (Kelly et al., 2011). Although the cells were not enriched to purity, this is certainly an important step towards better functional assessment of hPSC-derived cells. Another study also showed that it is possible to produce a purer target cell population through enrichment of progenitor cells at an earlier differentiation step (Cai et al., 2010). In addition, more precisely controlled culture conditions at early differentiation stages may also reduce the heterogeneity in the target cell population. For example, although treatment with both high and low doses of activin generates similar percentage of endoderm progenitors, the high dose produces a higher percentage of insulin-expressing cells at a later stage (D'Amour et al., 2006). Future functional analyses will probably benefit from improvements in differentiation protocols as well as cell enrichment methods, which can be based on the identification of specific cell-surface markers or on the development of faithful fluorescent reporters (see below).
Understanding human development: insights gained from studying hPSCs
The maintenance of pluripotency in hPSCs has been a topic of great interest to stem cell biologists. The knowledge gained from such studies, although not always directly applicable to embryonic development, has greatly improved our understanding of the interplay of signal transducers, transcription factors and epigenetic regulators during development in general. For example, transcriptional pausing after promoter binding and transcription initiation was first described in hESCs through genome-wide analysis of histone modifications (Guenther et al., 2007). Further studies in zebrafish and flies have shown that such transcriptional pausing features are also present in differentiated cell types and may contribute to cell fate determination during embryonic development (Bai et al., 2010; Zeitlinger et al., 2007).
Perhaps more importantly, hPSCs have also emerged as a model system for investigating directly the mechanisms that underlie embryonic development. It is clear that information gained from studies of model organisms has greatly facilitated the search for defined conditions to direct hPSCs to specific fates (Fig. 3). This supports the general conclusion that most developmental mechanisms are conserved. One may argue that the best way to evaluate our understanding of embryonic development is to use hPSCs to recapitulate the same process in vitro. However, human-specific transcriptional regulation and signaling pathways have also been uncovered. For example, neuroectoderm specification is one of the best-studied processes during gastrulation. A recent study shows that Pax6 is both necessary and sufficient for neuroectoderm specification from hESCs but not from mESCs (Zhang et al., 2010). Furthermore, hPSCs also offer unique advantages for studying conserved developmental mechanisms. For example, the exact role of TGFβ signaling in pancreatic development has been debated in mice (Harmon et al., 2004; Sanvito et al., 1994; Tei et al., 2005; Tulachan et al., 2007; Wandzioch and Zaret, 2009), potentially owing to the time-sensitive requirement of TGFβ signaling during pancreatic development. Like many other genes and signaling pathways, TGFβ signaling is used repeatedly during embryonic development. To circumvent limitations of pleiotropic gene functions in in vivo studies, substantial efforts are often required to generate conditional knockout or hypomorphic alleles, whereas it is relatively straightforward to manipulate TGFβ signaling (or another protein of interest) during a specific developmental time window using hPSCs. Recent hPSC-based studies show that TGFβ signaling inhibits the differentiation of pancreatic progenitors into the endocrine lineage - the lineage that gives rise to β-cells (Nostro et al., 2011; Rezania et al., 2011). Therefore, we foresee that hPSCs will become a powerful model system that can be used to uncover both conserved and non-conserved novel developmental mechanisms.
Fig. 3.
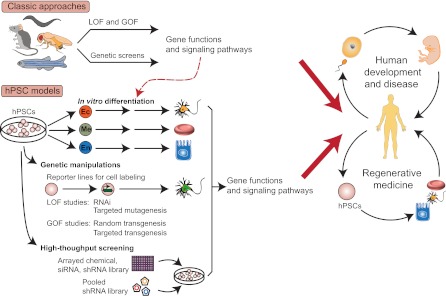
Advancing developmental biology and regenerative medicine through studies of hPSCs and model organisms. Genetic studies, including genetic screens and loss- and gain-of-function (LOF and GOF) studies, from the mouse and other model organisms have identified many genes and signaling pathways that govern various aspects of development. Such information has guided the search for defined conditions to turn hPSCs into specific cell types of all three germ layers (Ec, ectoderm; Me, mesoderm; En, endoderm). With the development of new genetic tools, it is now possible to use hPSCs as a new model system for studies of human development. The generation of desired cell types from hPSCs will also advance regenerative medicine in several aspects, including cell replacement therapy, disease modeling and drug discovery. shRNA, short hairpin RNA; siRNA, small-interfering RNA.
Prospects and potential challenges of using hPSCs as a developmental model
Through directed differentiation of hPSCs, developmental biologists now have unprecedented access to a wide variety of hPSC-derived human embryonic cell types and early developmental processes. However, several challenges remain. For example, there is marked heterogeneity between hPSC lines in their ability to differentiate into different cell lineages (Hu et al., 2010; Osafune et al., 2008). Consequently, hPSC-line-specific optimization is often necessary to adapt differentiation protocols developed by different research groups. Several factors contribute to such heterogeneity among hPSC lines, including the genetic background, and the derivation and culture conditions. Indeed, epigenetic differences have been identified among different hPSC lines or the same line after different numbers of passages (Mekhoubad et al., 2012; Nazor et al., 2012). For induced pluripotent stem cells (iPSCs), the epigenetic memory of their tissue of origin (Bar-Nur et al., 2011; Kim et al., 2010; Polo et al., 2010), and genetic and epigenetic alterations during reprogramming and the subsequent propagation of cells (Gore et al., 2011; Hussein et al., 2011; Laurent et al., 2011; Lister et al., 2011; Mayshar et al., 2010) may contribute further to the heterogeneity. The existence of heterogeneity among hPSC lines emphasizes the importance of validating findings using multiple cell lines.
Another major challenge lies in the difficulty of generating mature functional cell types. For example, in vitro differentiation of hPSC-derived pancreatic progenitors generates mostly immature β-cells with poor glucose responsiveness (D'Amour et al., 2006). The difficulty in generating mature cells may simply lie in the challenge of recapitulating the long developmental time frame within an in vitro environment; during human development, insulin-secreting cells appear around 2 months post-conception, but regulated glucose-stimulated insulin secretion occurs only after birth (Espinosa de los Monteros et al., 1970). A recent study employed a series of chemical inhibitors to deliberately accelerate the acquisition of a mature cell fate, in this case neuronal, by threefold (Chambers et al., 2012). This strategy may apply to other cell types, although it remains unclear whether ‘accelerated development’ employs the same signaling cascade used in normal embryonic development. A second hurdle is the lack of knowledge of key signals required for the final maturation step. Indeed, compared with the vast amount of knowledge about early development, we know relatively little about terminal differentiation. As most differentiation protocols are modeled on murine development, key human-specific components may be missing. Large-scale chemical and biological screening (discussed below) may help to identify some of the missing components. Finally, an often-overlooked aspect is that non-functional cells may be generated because of problems in earlier differentiation steps. For example, DA neurons can be generated through either rosette- or floor-plate-based protocols; however, only the second approach appears to recapitulate midbrain DA neuron development faithfully and to generate functional DA neurons that efficiently engraft in animal models (Kriks et al., 2011). This result highlights the necessity for recapitulating the exact ontogeny of a cell. In another example, a seemingly trivial optimization step at the first stage of differentiation (definitive endoderm) leads to a significant increase in expression of β-cell markers several stages later (Nostro et al., 2011). As the generation of a terminally differentiated cell almost certainly requires multiple steps of differentiation, these findings emphasize the importance of optimizing every single differentiation step, and verifying the identity of each intermediate cell type.
Finally, unlike lineage commitment, some aspects of development, such as tissue morphogenesis and patterning, cannot be easily recapitulated using current differentiation protocols. This may also pose a hurdle for studying lineage commitment of cells that rely on tissue-tissue interactions for their proper specification, maturation and survival. Several recent studies have successfully generated 3D organs or ‘organoids’ from hPSCs. In one example, hPSCs were guided through definitive endoderm, posterior endoderm and hindgut, before forming hindgut epithelial tubes that bud off as floating spheroids (Spence et al., 2011). When cultured in conditions that support the growth of the adult intestinal epithelium, these hPSC-derived spheroids developed further into intestinal organoids consisting of major fetal intestinal cell types. Several self-forming 3D neural and glandular tissue structures have also been reported by Sasai and colleagues (Eiraku et al., 2011; Eiraku et al., 2008; Nakano et al., 2012; Suga et al., 2011). In a visually striking example, mESC-derived retinal epithelium spontaneously formed cup-like hemispherical epithelial vesicles reminiscent of optic cups (Eiraku et al., 2011), and similar findings were recently reported using hESCs (Nakano et al., 2012). Though still in their early stages, these discoveries suggest hPSCs may be used not only to study cell fate specification, but also to analyze more complex cellular behaviors during development, such as tissue morphogenesis.
Modeling human diseases using hPSCs
In addition to studies of normal development, hPSCs also offer a way to recapitulate abnormal development and to investigate the pathogenesis of human diseases. Many disease-relevant cells, such as neurons, are not easily accessible in patients. Therefore, animal models, especially mouse models, have been widely used to understand the pathogenesis of human diseases. However, mouse models do not always recapitulate the phenotypes manifested in humans, as discussed above. hPSCs carrying disease-associated genetic modifications may overcome these limitations by providing unlimited supplies of any disease-relevant human cell type for studies of human disease and developmental toxicology (van Dartel et al., 2010).
Disease-relevant hPSCs can be generated using several approaches. Human embryos with genetic defects identified through preimplantation genetic diagnosis (PGD) can be used to create hESCs for study of diseases caused by chromosomal abnormalities and known disease-associated mutations (Maury et al., 2012). In addition to offering developmental biologists access to human embryonic development, such studies may lead to a better understanding of spontaneous abortions during early pregnancy, the underlying causes of which remain poorly understood (Macklon et al., 2002). However, PGD embryos are available only for a limited number of human diseases. This limitation has been overcome, as it is now possible to generate iPSCs from individuals with a wide ranges of diseases, including monogenic diseases such as spinal muscular atrophy (SMA) and complex diseases such as Parkinson's disease (Wu and Hochedlinger, 2011). The development of these iPSC-based disease models can allow researchers to study the roles of a specific gene in developmental cell fate decisions and the physiological functions of disease-relevant cells. In the first proof-of-principle study, iPSCs derived from an individual with SMA were differentiated into motoneurons with similar efficiencies to iPSCs from the unaffected mother of the individual. However, prolonged cultures revealed a significant reduction in the number of motoneurons, consistent with the disease phenotype of selective motoneuron loss (Ebert et al., 2009). Although this study supports the enormous potential of using iPSCs to model a specific pathological condition associated with a hereditary disease, variations among hiPSC cell lines may affect their use during disease modeling. Although it is possible to control the derivation/culture conditions, and the types of cells used for generating hiPSCs, it is impossible to completely eliminate genetic variations in hiPSC lines generated from two different individuals. A good solution to this problem is to generate isogenic hPSC lines that differ only in the disease-causing genetic modification(s). hESC lines have been generated to create the HPRT mutation for modeling of Lesch-Nyhan syndrome (Urbach et al., 2004; Zwaka and Thomson, 2003). Unlike the mouse model, mutant human cells exhibit significant accumulation of uric acid, supporting the feasibility of using genetically modified hPSCs for disease modeling. More successful examples are expected to emerge with improvements in gene targeting technologies (discussed below).
New genetic tools for hPSC research
To use hPSCs for studies of human development and disease, it is essential to develop powerful genetic tools. Below, we highlight recent advances that enable both the generation of tissue-specific fluorescent reporter lines and the perturbation of gene expression. The strengths and weaknesses of transgenic approaches have been extensively discussed elsewhere (Giudice and Trounson, 2008); of note, the main advantages are the convenience and experimental feasibility of the approach, while the drawbacks include position effects, copy number variation and transgene silencing. Instead, we focus on the gene targeting approach, which has undergone a major transformation in recent years and may revolutionize genetic studies in hPSCs.
Gene targeting mediated by ZFNs and TALENs
Gene targeting refers to the introduction of site-specific modifications into the genome by homologous recombination (HR). This approach is widely used in the mouse to generate null (complete loss-of-function) or hypomorphic (partial loss-of-function) mutations, or to create reporter genes to track the expression of an endogenous transcript. Since the first successful targeting of the mouse Hprt gene (Doetschman et al., 1987; Thomas and Capecchi, 1987), numerous murine genes have been targeted. However, only a small number of loci have been successfully targeted using conventional gene targeting methods in hPSCs, owing to the low efficiency of HR (Hockemeyer and Jaenisch, 2010). This is now poised to change.
It has long been recognized that inducing a DNA double-stranded break (DSB) at the target locus substantially increases the efficiency of HR (Puchta et al., 1993; Rouet et al., 1994; Smih et al., 1995). Based on this idea, two methods have been developed to introduce DSBs at the target site. These involve zinc-finger nucleases (ZFNs; see Box 1); and, more recently, transcription activator-like (TAL) effector nucleases (TALENs; see Box 1). These engineered chimeric proteins are composed of separate DNA-binding and DNA-cleavage domains, which enable them to act as ‘genomic scissors’ that can induce DNA breaks at specific genomic loci. In the presence of a donor DNA fragment containing homology arms to the target locus, HR-mediated DNA repair results in the insertion of the donor DNA sequence into the specific genomic locus. This approach can be used to generate point mutations, genomic deletions and insertions of reporter genes.
Box 1. Designer ZFNs and TALENs for gene targeting

Zinc-finger nucleases (ZFNs) are engineered chimeric proteins composed of a zinc-finger DNA-binding domain and a FokI DNA cleavage domain (see figure; colored blocks represent zinc-finger motifs, which are color coded for different binding specificity to DNA triplet sequences). Zinc fingers are among the most common DNA-binding motifs in eukaryotic transcription factors. When Pavletich and Pabo described the first crystal structure of zinc fingers bound to DNA (Pavletich and Pabo, 1991), they immediately recognized the potential of designing artificial DNA-binding proteins based on the simple modular zinc-finger/DNA interaction: each finger primarily interacts with three nucleotides in DNA. Chandrasegaran and colleagues went one step further, and engineered the first ZFNs to cleave DNA at a predetermined site (Kim et al., 1996). To target a specific genomic locus, a pair of ZFNs is designed to bind to DNA in opposite orientation with a defined space in between. In the presence of a donor DNA fragment containing homology arms to the target gene, homologous recombination-mediated DNA repair will result in the insertion of donor DNA sequence into the specific genomic locus.
Transcription activator-like (TAL) effector nucleases (TALENs) are built upon a new class of DNA-binding proteins - the TAL effectors - identified in Xanthomonas, a group of bacterial plant pathogens (Boch and Bonas, 2010) (see figure; colored blocks represent TAL effector repeats, which are color coded for different binding specificity to single-nucleotide targets). They have a unique DNA-binding domain consisting of repetitive units that each recognizes one specific nucleotide (Boch et al., 2009; Deng et al., 2012; Mak et al., 2012; Moscou and Bogdanove, 2009). This simple code makes it more straightforward to engineer artificial TAL DNA-binding domains than zinc fingers to recognize specific DNA sequences. Similar to ZFNs, designer TALENs can be created by fusing the TAL DNA-binding domain to the FokI DNA cleavage domain (Cermak et al., 2011; Miller et al., 2011). TALEN-mediated gene targeting has already been used in hPSC lines with minimal off-target effects (Hockemeyer et al., 2011).
The use of ZFNs or TALENs greatly improves the gene targeting efficiency over traditional methods. It is also more convenient, as only short homology arms (∼500 bp or even shorter) are needed, in contrast to typically much longer homology arms (2-6 kb or even longer) used in conventional gene targeting experiments. Using single-stranded DNA oligos (ssDNA) as the donor, the length of each homology arm can be further reduced to ∼40 bp, with the full length of donor ssDNA being only ∼100 bp (Chen et al., 2011; Liu et al., 2010); this has recently been applied to hPSCs (Soldner et al., 2011). A shorter homology arm can reduce the time needed to generate the donor DNA template and increase the transfection efficiency.
These new gene-editing technologies are expected to greatly accelerate the pace of using hPSCs lines for studies of development and disease. In a recent study, point mutations in the gene encoding α-synuclein were generated to model Parkinson's disease (Soldner et al., 2011). The generation of isogenic cell lines that differ only in disease-associated mutations will be invaluable for distinguishing disease-associated phenotypes from background noise. In addition to generating reporter lines and modifying endogenous genes, ZFNs and TALENs may also assist the targeting of transgenes into a chosen genomic locus. Compared with random transgene integration, targeted transgenesis has a number of advantages: the integration site can be chosen to allow reliable expression, and only a single copy of the transgene is introduced. Site-specific transgenesis was first used to introduce a transgene into the Hprt locus in mESCs 16 years ago (Bronson et al., 1996). Since then, the mouse ROSA26 locus, identified in a gene trap screen by Philippe Soriano and colleagues (Zambrowicz et al., 1997), has become the most widely used locus for transgene insertion. Identifying similar transgene safe-harbor loci (see Box 2) in the human genome would allow convenient interrogation of gene function during hPSC differentiation. Notably, the adeno-associated virus site 1 (AAVS1) locus appears to be a good candidate for such a locus: around half of the clones are correctly targeted in ZFN- or TALEN-mediated gene targeting experiments (Hockemeyer et al., 2009; Hockemeyer et al., 2011). Consequently, straightforward gene overexpression or knockdown studies can be performed in hPSCs by targeting cDNA or shRNAs into the AAVS1 locus. For example, using ZFNs, it is possible to target both AAVS1 alleles in trans in a single experiment for expression of M2rtTA (reverse tetracycline-controlled transactivator) and a tetracycline response element that drives the expression of a gene of interest for flexible temporal control of gene expression (DeKelver et al., 2010).
Box 2. Choosing transgene safe harbors
A transgene safe harbor must satisfy a number of criteria: (1) the locus should allow ubiquitous sustained transgene expression without affecting expression of neighboring genes; (2) integration of the transgene should not disrupt an essential endogenous gene or affect the maintenance or differentiation of hPSCs; and (3) the gene targeting efficiency should be relatively high to facilitate rapid, efficient transgenesis experiments. It is worth noting that more stringent criteria have been proposed for transgene safe harbors in therapeutic applications (Papapetrou et al., 2011). Three promising loci have been identified so far: the human ortholog of the mouse ROSA26 locus (Irion et al., 2007), the chemokine (CC motif) receptor 5 (CCR5) gene locus (Lombardo et al., 2007) and AAVS1 (also known as PPP1R12C) (Smith et al., 2008). These loci have been extensively discussed in a recent review in the context of gene therapy (Sadelain et al., 2012).
Gene targeting in hPSCs: prospects and challenges
We anticipate these technologies will have a profound impact on studies of developmental biology and disease using hPSCs. Future challenges include further improving the efficiency of ZFNs and TALENs to generate DNA breaks, minimizing off-target cleavages and increasing the transfection efficiency. Recent improvements in ZFN and TALEN backbone optimization and mutation detection methods (Bedell et al., 2012; Miller et al., 2011) are expected to make gene targeting in hPSCs more convenient, efficient and cost effective. At the same time, other technologies may further improve the gene targeting efficiency in hPSCs, such as the use of helper-dependent adenoviral vectors (HDAdVs) (Aizawa et al., 2012; Suzuki et al., 2008). Instead of creating locus-specific DSBs, HDAdVs have high cloning capacities (up to ∼25-35 kb of DNA), and allow the use of much longer homology arms, which can increase the HR efficiency. Protection of adenoviral genomes by the terminal protein may also reduce random integration in HDAdV-mediated gene targeting. It will be exciting to see how these new technologies empower researchers to use hPSCs for developmental studies.
Discovery-driven research using hPSCs
Genetic screening is a powerful approach with which to identify novel regulators of development, as manifested throughout the history of developmental biology. The unlimited self-renewing capacity of hPSCs makes them well suited for large-scale genetic screens. Recent advances in HTS technologies, including the automated robotics and imaging systems, have made it feasible to perform large-scale screens. The aforementioned genetic tools should facilitate tracking or isolating cell types of interest in such screens. Below, we summarize two main screening formats that have been used for hPSCs, and discuss the potential of using HTS with hPSCs to uncover novel mechanisms of human development (Fig. 4).
Fig. 4.
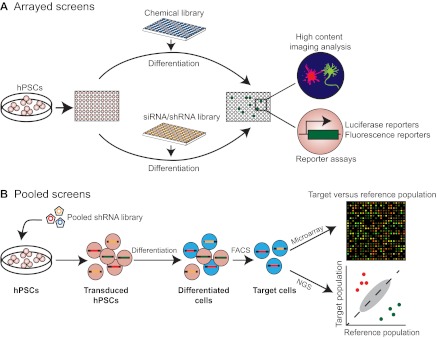
hPSC-based high-throughput screening. High throughput screening in human pluripotent stem cells (hPSCs) using chemical or RNAi libraries can be performed in arrayed (A) or pooled (B) format. (A) In arrayed screens, chemicals, small-interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) pre-arranged in multi-well plates are applied to hPSCs, and the effect can be examined by high-content imaging or by reporter assays. (B) Although pooled screens have not yet been reported using hPSCs, we envision it is possible to perform such screens using pooled shRNA viruses. Transduced cells can be further differentiated into the cell type of interest, and the target cell population can be isolated using cell surface markers or fluorescent reporters. To identify genes that inhibit or promote the lineage commitment, respectively, microarrays or next generation sequencing (NGS) can then be used to identify any over- or under-represented shRNAs (represented by red and green dots, respectively) in target cells compared with the reference cell population (e.g. the transduced cells before differentiation). FACS, fluorescence-activated cell sorting.
Chemical screens
Chemical screening (see Box 3) is an attractive approach with which to identify compounds that promote the differentiation of hPSCs into specific cell lineages. The subsequent identification of the molecular target(s) of a hit compound, though not always straightforward, may uncover novel genes and signaling pathways involved in the differentiation process. Several high-throughput screens have been performed to identify chemical compounds that influence the decision of hPSCs to either self-renew or differentiate (Barbaric et al., 2010; Desbordes et al., 2008; Zhu et al., 2009). One such screen demonstrated that protein kinase C (PKC) agonists increase the number of pancreatic progenitors derived from hPSCs (Chen et al., 2009). It remains to be seen whether PKC signaling plays similar roles in vivo, and what its mechanisms of action are. Future in vivo studies will be necessary to determine the biological relevance of PKC signaling and other biological targets identified in various chemical screens performed in hPSCs.
Box 3. Chemical screens
Chemical libraries are typically supplied in an arrayed format, such that each chemical occupies a unique well of a multi-well plate (e.g. 96- or 384-well plate). The effects of chemicals can be determined by a variety of high-throughput screen assays, such as fluorescent reporter assays, luciferase-based reporter assays and assays for cell morphology and function. Notably, high-content image-based assays allow acquisition of multiple cellular features simultaneously, thus enabling investigation of complex cellular behaviors. Successful chemical screens rely on the establishment of robust directed differentiation platform, and access to chemical libraries and high-throughput equipment for compound application and image analysis. There is also the challenge of identifying the biological target(s) of a hit compound. Finally, chemical screens are limited by the number of genes that can be effectively targeted, which may be overcome by increasing the structural diversity of compounds in chemical libraries.
RNAi screens
The discovery of RNA interference - gene silencing by double-stranded RNA - has not only transformed our understanding of gene regulation, but has also provided a powerful research tool with which to examine gene function directly. In recent years, RNAi has become increasingly popular as an effective tool for genome-scale, high-throughput analysis of gene function not only in classic model organisms such as C. elegans and D. melanogaster, but also in cultured human and mouse cells (Mohr et al., 2010b). RNAi screens are typically conducted with either small-interfering RNA (siRNA)-based transient transfection, or short hairpin RNA (shRNA)-based stable gene knockdown, in arrayed or pooled formats (see Box 4, Fig. 4).
Box 4. RNAi screens
A standard small-interfering RNA (siRNA) library is composed of chemically synthesized 21 nucleotide siRNAs supplied in an arrayed format. Transfection of siRNAs into target cells can transiently downregulate target genes, thus providing a convenient way to screen for loss-of-function phenotypes. Short hairpin RNAs (shRNAs) are typically cloned into retro- or lentiviral vectors, which allows integration of the shRNA expression cassette into the host genome for sustained expression. Several genome-wide shRNA libraries have been constructed, including the Netherlands Cancer Institute (NKI) libraries (Berns et al., 2004), the RNAi consortium (TRC) libraries (Moffat et al., 2006) and the Hannon-Elledge libraries (Paddison et al., 2004; Silva et al., 2005). More recently, Cellecta has released the Decipher libraries available to the research community free of charge.
High-throughput RNAi screens in mammalian cells can be conducted in either arrayed (siRNA and shRNA libraries) or pooled format (shRNA libraries) (Fig. 4). As the targeted gene in each well is known in the arrayed screen, identifying the target gene is straightforward once a phenotype is observed. However, the wider use of this method of screening is limited by several factors, including the high cost of reagents and robotics, and the equipment required for phenotype detection. In pooled screens, effects of individual shRNAs are identified by their enrichment or depletion in target cell populations. Microarray analyses were used in earlier screens to detect changes in shRNA abundance (Berns et al., 2004; Paddison et al., 2004). Recent advances in next generation sequencing now offers a more quantitative and cost-effective alternative. Compared with the arrayed format, the pooled format is less laborious and more feasible in a standard laboratory setting. However, pooled screens depend on methods for selection of target cells, and are not yet compatible with high-content image-based analyses.
Several RNAi screens of various scales have been conducted to study the self-renewal of mESCs (Ding et al., 2009; Fazzio et al., 2008; Hu et al., 2009; Ivanova et al., 2006; Jian et al., 2007; Zhang et al., 2006), though only one screen has been performed on hESCs (Chia et al., 2010). Using a whole-genome siRNA library, this screen successfully identified PRDM14 as a novel regulator of hPSC self-renewal. This type of screening has not yet been conducted to study specific aspects of embryonic development. With the rapid development of better directed differentiation protocols and powerful genetic tools for making faithful reporter lines, it is theoretically possible to first establish a reliable differentiation platform, and then to generate a tissue-specific fluorescent reporter, and eventually use either pooled or arrayed screens to identify genes that regulate the specification of a cell type of interest (Fig. 4). We anticipate RNAi screens will offer enormous opportunities to identify novel genes involved in human development.
One potential challenge for HTS may lie in the length of differentiation protocols for some cell types, which could last for weeks or even months. As multiple differentiation steps are likely to be involved in such cases, individual screens can be designed to interrogate each specific step of differentiation. This approach, analogous to conditional knockout studies in vivo, also circumvents challenges such as early lethality and gene pleiotropy encountered in mouse genetic screens. At the same time, other genetic screening methods may emerge that complement RNAi screens. Large-scale insertional mutagenesis has been used to generate mESCs libraries carrying mutations of all protein-coding genes (Gragerov et al., 2007; Skarnes et al., 2011). The in vitro application of these libraries has been limited so far by the fact that observation of recessive phenotypes requires homozygous mutant alleles. Recently, it has become possible to derive haploid mESCs that have the potential to generate a wide range of differentiated cell types, including germ cells both in vitro and in the embryo (Elling et al., 2011; Leeb et al., 2012; Leeb and Wutz, 2011; Li et al., 2012; Yang et al., 2012). By combining with insertional mutagenesis, haploid mESCs can provide a useful platform for in vitro genetic screening. Likewise, such a platform may be developed for human cells through the use of insertional mutagenesis and the establishment of haploid hPSC lines. We anticipate that unbiased genetic screens using RNAi or newer, yet-to-be-developed methods will revolutionize gene discovery in human development.
Conclusions
Knowledge gained from studies of model organisms has clearly furthered our understanding of human development and has guided our efforts to generate specific cell types from hPSCs. Technological advancements in recent years have now presented us with an exciting opportunity to use hPSCs to gain novel insights into human embryonic development. We anticipate that hPSC-based research, combined with studies of classic model organisms, will teach us how human embryos develop. This knowledge will not only help us understand the underlying causes of human diseases, but will also guide our efforts to develop treatments for these diseases. For example, the generation of functional mature cell types from hPSCs will be useful for cell replacement therapy, in vitro disease modeling and drug discovery. Will hPSCs become a favorite model of choice for developmental biologists? It is too early to say. Further development of this new model system will require the same kind of open-mindedness that has propelled the progress of previous model organisms in biology.
One future challenge of hPSC-based studies lies in validating the in vivo relevance of any in vitro findings. An obvious strategy is to use mouse models to investigate gene function during development, as we expect most genes will play conserved roles in mice and humans. However, there are clearly genes that do not exhibit conserved functions between mouse and human, and there is currently no clear strategy for investigating such genes. One solution is to engraft genetically modified hPSCs into animal models, such as mouse embryos or adult mice, in order to observe developmental or physiological phenotypes. Various ‘humanized’ mouse models have been explored in the past (Behringer, 2007; Eckardt et al., 2011; Shultz et al., 2007). We anticipate more powerful experimental systems will emerge in the next decade for study of gene function during human development in an in vivo environment.
Acknowledgments
We apologize to colleagues whose work could not be cited owing to space limitations. We are grateful to the members of the Huangfu lab for insightful discussions and critical reading of the manuscript.
Footnotes
Funding
Our work is supported by the National Institutes of Health (NIH), by a Basil O'Connor Starter Scholar Award from March of Dimes Birth Defects Foundation, by a Louis V. Gerstner Jr Young Investigators Award and by Memorial Sloan-Kettering Cancer Center Society Special Projects Committee. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
References
- Acevedo-Arozena A., Wells S., Potter P., Kelly M., Cox R. D., Brown S. D. (2008). ENU mutagenesis, a way forward to understand gene function. Annu. Rev. Genom. Hum. Genet. 9, 49-69 [DOI] [PubMed] [Google Scholar]
- Aizawa E., Hirabayashi Y., Iwanaga Y., Suzuki K., Sakurai K., Shimoji M., Aiba K., Wada T., Tooi N., Kawase E., et al. (2012). Efficient and accurate homologous recombination in hESCs and hiPSCs using helper-dependent adenoviral vectors. Mol. Ther. 20, 424-431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen H. L., Flanagan S. E., Shaw-Smith C., De Franco E., Akerman I., Caswell R., Ferrer J., Hattersley A. T., Ellard S. (2012). GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat. Genet. 44, 20-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X., Kim J., Yang Z., Jurynec M. J., Akie T. E., Lee J., LeBlanc J., Sessa A., Jiang H., DiBiase A., et al. (2010). TIF1gamma controls erythroid cell fate by regulating transcription elongation. Cell 142, 133-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nur O., Russ H. A., Efrat S., Benvenisty N. (2011). Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 9, 17-23 [DOI] [PubMed] [Google Scholar]
- Barbaric I., Gokhale P. J., Jones M., Glen A., Baker D., Andrews P. W. (2010). Novel regulators of stem cell fates identified by a multivariate phenotype screen of small compounds on human embryonic stem cell colonies. Stem Cell Res. 5, 104-119 [DOI] [PubMed] [Google Scholar]
- Bedell V. M., Wang Y., Campbell J. M., Poshusta T. L., Starker C. G., Krug R. G., 2nd, Tan W., Penheiter S. G., Ma A. C., Leung A. Y., et al. (2012). In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer R. R. (2007). Human-animal chimeras in biomedical research. Cell Stem Cell 1, 259-262 [DOI] [PubMed] [Google Scholar]
- Berns K., Hijmans E. M., Mullenders J., Brummelkamp T. R., Velds A., Heimerikx M., Kerkhoven R. M., Madiredjo M., Nijkamp W., Weigelt B., et al. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431-437 [DOI] [PubMed] [Google Scholar]
- Boch J., Bonas U. (2010). Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 48, 419-436 [DOI] [PubMed] [Google Scholar]
- Boch J., Scholze H., Schornack S., Landgraf A., Hahn S., Kay S., Lahaye T., Nickstadt A., Bonas U. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509-1512 [DOI] [PubMed] [Google Scholar]
- Bronson S. K., Plaehn E. G., Kluckman K. D., Hagaman J. R., Maeda N., Smithies O. (1996). Single-copy transgenic mice with chosen-site integration. Proc. Natl. Acad. Sci. USA 93, 9067-9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau B. G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943-948 [DOI] [PubMed] [Google Scholar]
- Cai J., Yu C., Liu Y., Chen S., Guo Y., Yong J., Lu W., Ding M., Deng H. (2010). Generation of homogeneous PDX1(+) pancreatic progenitors from human ES cell-derived endoderm cells. J. Mol. Cell Biol. 2, 50-60 [DOI] [PubMed] [Google Scholar]
- Capecchi M. R. (2005). Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat. Rev. Genet. 6, 507-512 [DOI] [PubMed] [Google Scholar]
- Carrasco M., Delgado I., Soria B., Martín F., Rojas A. (2012). GATA4 and GATA6 control mouse pancreas organogenesis. J. Clin. Invest. 122, 3504-3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T., Doyle E. L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J. A., Somia N. V., Bogdanove A. J., Voytas D. F. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S. M., Qi Y., Mica Y., Lee G., Zhang X. J., Niu L., Bilsland J., Cao L., Stevens E., Whiting P., et al. (2012). Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 30, 715-720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Borowiak M., Fox J. L., Maehr R., Osafune K., Davidow L., Lam K., Peng L. F., Schreiber S. L., Rubin L. L., et al. (2009). A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 5, 258-265 [DOI] [PubMed] [Google Scholar]
- Chen F., Pruett-Miller S. M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T. N., Frodin M., Davis G. D. (2011). High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 8, 753-755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia N. Y., Chan Y. S., Feng B., Lu X., Orlov Y. L., Moreau D., Kumar P., Yang L., Jiang J., Lau M. S., et al. (2010). A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316-320 [DOI] [PubMed] [Google Scholar]
- Cleaver O., MacDonald R. (2010). Developmental molecular biology of the pancreas. In Pancreatic Cancer (ed. Evans D. B., Pisters P. W. T., Abbruzzese J. L.), pp. 71-117 New York, NY: Springer; [Google Scholar]
- D'Amour K. A., Agulnick A. D., Eliazer S., Kelly O. G., Kroon E., Baetge E. E. (2005). Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 23, 1534-1541 [DOI] [PubMed] [Google Scholar]
- D'Amour K. A., Bang A. G., Eliazer S., Kelly O. G., Agulnick A. D., Smart N. G., Moorman M. A., Kroon E., Carpenter M. K., Baetge E. E. (2006). Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 24, 1392-1401 [DOI] [PubMed] [Google Scholar]
- Dahlem T. J., Hoshijima K., Jurynec M. J., Gunther D., Starker C. G., Locke A. S., Weis A. M., Voytas D. F., Grunwald D. J. (2012). Simple methods for generating and detecting locusspecific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 8, e1002861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKelver R. C., Choi V. M., Moehle E. A., Paschon D. E., Hockemeyer D., Meijsing S. H., Sancak Y., Cui X., Steine E. J., Miller J. C., et al. (2010). Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 20, 1133-1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D., Yan C., Pan X., Mahfouz M., Wang J., Zhu J. K., Shi Y., Yan N. (2012). Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335, 720-723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbordes S. C., Placantonakis D. G., Ciro A., Socci N. D., Lee G., Djaballah H., Studer L. (2008). High-throughput screening assay for the identification of compounds regulating self-renewal and differentiation in human embryonic stem cells. Cell Stem Cell 2, 602-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Paszkowski-Rogacz M., Nitzsche A., Slabicki M. M., Heninger A. K., de Vries I., Kittler R., Junqueira M., Shevchenko A., Schulz H., et al. (2009). A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 4, 403-415 [DOI] [PubMed] [Google Scholar]
- Doetschman T., Gregg R. G., Maeda N., Hooper M. L., Melton D. W., Thompson S., Smithies O. (1987). Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576-578 [DOI] [PubMed] [Google Scholar]
- Doetschman T., Maeda N., Smithies O. (1988). Targeted mutation of the Hprt gene in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 85, 8583-8587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvash T., Mayshar Y., Darr H., McElhaney M., Barker D., Yanuka O., Kotkow K. J., Rubin L. L., Benvenisty N., Eiges R. (2004). Temporal gene expression during differentiation of human embryonic stem cells and embryoid bodies. Hum. Reprod. 19, 2875-2883 [DOI] [PubMed] [Google Scholar]
- Dvash T., Sharon N., Yanuka O., Benvenisty N. (2007). Molecular analysis of LEFTY-expressing cells in early human embryoid bodies. Stem Cells 25, 465-472 [DOI] [PubMed] [Google Scholar]
- Ebert A. D., Yu J., Rose F. F., Jr, Mattis V. B., Lorson C. L., Thomson J. A., Svendsen C. N. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt S., McLaughlin K. J., Willenbring H. (2011). Mouse chimeras as a system to investigate development, cell and tissue function, disease mechanisms and organ regeneration. Cell Cycle 10, 2091-2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiraku M., Watanabe K., Matsuo-Takasaki M., Kawada M., Yonemura S., Matsumura M., Wataya T., Nishiyama A., Muguruma K., Sasai Y. (2008). Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519-532 [DOI] [PubMed] [Google Scholar]
- Eiraku M., Takata N., Ishibashi H., Kawada M., Sakakura E., Okuda S., Sekiguchi K., Adachi T., Sasai Y. (2011). Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51-56 [DOI] [PubMed] [Google Scholar]
- Elling U., Taubenschmid J., Wirnsberger G., O'Malley R., Demers S. P., Vanhaelen Q., Shukalyuk A. I., Schmauss G., Schramek D., Schnuetgen F., et al. (2011). Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell 9, 563-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsea S. H., Lucas R. E. (2002). The mousetrap: what we can learn when the mouse model does not mimic the human disease. ILAR J. 43, 66-79 [DOI] [PubMed] [Google Scholar]
- Espinosa de los Monteros A., Driscoll S. G., Steinke J. (1970). Insulin release from isolated human fetal pancreatic islets. Science 168, 1111-1112 [DOI] [PubMed] [Google Scholar]
- Evans M. J., Kaufman M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154-156 [DOI] [PubMed] [Google Scholar]
- Fazzio T. G., Huff J. T., Panning B. (2008). An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134, 162-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger S., Heavens R. P., Sirinathsinghji D. J., Kuehn M. R., Dunnett S. B. (1988). Behavioral and neurochemical evaluation of a transgenic mouse model of Lesch-Nyhan syndrome. J. Neurol. Sci. 86, 203-213 [DOI] [PubMed] [Google Scholar]
- Giudice A., Trounson A. (2008). Genetic modification of human embryonic stem cells for derivation of target cells. Cell Stem Cell 2, 422-433 [DOI] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H. L., Young J. E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M. A., Kiskinis E., et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragerov A., Horie K., Pavlova M., Madisen L., Zeng H., Gragerova G., Rhode A., Dolka I., Roth P., Ebbert A., et al. (2007). Large-scale, saturating insertional mutagenesis of the mouse genome. Proc. Natl. Acad. Sci. USA 104, 14406-14411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther M. G., Levine S. S., Boyer L. A., Jaenisch R., Young R. A. (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis A. K., Dickinson M. E., Fraser S. E., Papaioannou V. E. (2003). Technicolour transgenics: imaging tools for functional genomics in the mouse. Nat. Rev. Genet. 4, 613-625 [DOI] [PubMed] [Google Scholar]
- Harmon E. B., Apelqvist A. A., Smart N. G., Gu X., Osborne D. H., Kim S. K. (2004). GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development 131, 6163-6174 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D., Jaenisch R. (2010). Gene targeting in human pluripotent cells. Cold Spring Harb. Symp. Quant. Biol. 75, 201-209 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R. C., Katibah G. E., Amora R., Boydston E. A., Zeitler B., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851-857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Wang H., Kiani S., Lai C. S., Gao Q., Cassady J. P., Cost G. J., Zhang L., Santiago Y., Miller J. C., et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731-734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Kim J., Xu Q., Leng Y., Orkin S. H., Elledge S. J. (2009). A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 23, 837-848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B. Y., Weick J. P., Yu J., Ma L. X., Zhang X. Q., Thomson J. A., Zhang S. C. (2010). Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 107, 4335-4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., Anderson K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83-87 [DOI] [PubMed] [Google Scholar]
- Hussein S. M., Batada N. N., Vuoristo S., Ching R. W., Autio R., Närvä E., Ng S., Sourour M., Hämäläinen R., Olsson C., et al. (2011). Copy number variation and selection during reprogramming to pluripotency. Nature 471, 58-62 [DOI] [PubMed] [Google Scholar]
- Irion S., Luche H., Gadue P., Fehling H. J., Kennedy M., Keller G. (2007). Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 25, 1477-1482 [DOI] [PubMed] [Google Scholar]
- Itskovitz-Eldor J., Schuldiner M., Karsenti D., Eden A., Yanuka O., Amit M., Soreq H., Benvenisty N. (2000). Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 6, 88-95 [PMC free article] [PubMed] [Google Scholar]
- Ivanova N., Dobrin R., Lu R., Kotenko I., Levorse J., DeCoste C., Schafer X., Lun Y., Lemischka I. R. (2006). Dissecting self-renewal in stem cells with RNA interference. Nature 442, 533-538 [DOI] [PubMed] [Google Scholar]
- Jian R., Cheng X., Jiang J., Deng S., Hu F., Zhang J. (2007). A cDNA-based random RNA interference library for functional genetic screens in embryonic stem cells. Stem Cells 25, 1904-1912 [DOI] [PubMed] [Google Scholar]
- Jonsson J., Carlsson L., Edlund T., Edlund H. (1994). Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371, 606-609 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J., Wright C. V. (2002). The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 32, 128-134 [DOI] [PubMed] [Google Scholar]
- Keller G. (2005). Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev. 19, 1129-1155 [DOI] [PubMed] [Google Scholar]
- Kelly O. G., Chan M. Y., Martinson L. A., Kadoya K., Ostertag T. M., Ross K. G., Richardson M., Carpenter M. K., D'Amour K. A., Kroon E., et al. (2011). Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 29, 750-756 [DOI] [PubMed] [Google Scholar]
- Kim Y. G., Cha J., Chandrasegaran S. (1996). Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 93, 1156-1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Doi A., Wen B., Ng K., Zhao R., Cahan P., Kim J., Aryee M. J., Ji H., Ehrlich L. I., et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A., Knofler M., Ledermann B., Burki K., Berney C., Zoerkler N., Hagenbuchle O., Wellauer P. K. (1998). The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 12, 3752-3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K., Watt F. M. (2012). Lineage tracing. Cell 148, 33-45 [DOI] [PubMed] [Google Scholar]
- Kriks S., Shim J. W., Piao J., Ganat Y. M., Wakeman D. R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480, 547-551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E., Martinson L. A., Kadoya K., Bang A. G., Kelly O. G., Eliazer S., Young H., Richardson M., Smart N. G., Cunningham J., et al. (2008). Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443-452 [DOI] [PubMed] [Google Scholar]
- Lamba D. A., Karl M. O., Ware C. B., Reh T. A. (2006). Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 103, 12769-12774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba D. A., Gust J., Reh T. A. (2009). Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 4, 73-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L. C., Ulitsky I., Slavin I., Tran H., Schork A., Morey R., Lynch C., Harness J. V., Lee S., Barrero M. J., et al. (2011). Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M., Wutz A. (2011). Derivation of haploid embryonic stem cells from mouse embryos. Nature 479, 131-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M., Walker R., Mansfield B., Nichols J., Smith A., Wutz A. (2012). Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development 139, 3301-3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. J., Du Z. W., Zarnowska E. D., Pankratz M., Hansen L. O., Pearce R. A., Zhang S. C. (2005). Specification of motoneurons from human embryonic stem cells. Nat. Biotechnol. 23, 215-221 [DOI] [PubMed] [Google Scholar]
- Li W., Shuai L., Wan H., Dong M., Wang M., Sang L., Feng C., Luo G. Z., Li T., Li X., et al. (2012). Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 490, 407-411 [DOI] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Kida Y. S., Hawkins R. D., Nery J. R., Hon G., Antosiewicz-Bourget J., O'Malley R., Castanon R., Klugman S., et al. (2011). Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Majumdar A., Liu J., Thompson L. H., Seidman M. M. (2010). Sequence conversion by single strand oligonucleotide donors via non-homologous end joining in mammalian cells. J. Biol. Chem. 285, 23198-23207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A., Genovese P., Beausejour C. M., Colleoni S., Lee Y. L., Kim K. A., Ando D., Urnov F. D., Galli C., Gregory P. D., et al. (2007). Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 25, 1298-1306 [DOI] [PubMed] [Google Scholar]
- Macklon N. S., Geraedts J. P., Fauser B. C. (2002). Conception to ongoing pregnancy: the ‘black box’ of early pregnancy loss. Hum. Reprod. Update 8, 333-343 [DOI] [PubMed] [Google Scholar]
- Mak A. N., Bradley P., Cernadas R. A., Bogdanove A. J., Stoddard B. L. (2012). The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335, 716-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo M., Wellik D. M., Deschamps J. (2010). Hox genes and regional patterning of the vertebrate body plan. Dev. Biol. 344, 7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 78, 7634-7638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury Y., Gauthier M., Peschanski M., Martinat C. (2012). Human pluripotent stem cells for disease modelling and drug screening. Bioessays 34, 61-71 [DOI] [PubMed] [Google Scholar]
- Mayshar Y., Ben-David U., Lavon N., Biancotti J. C., Yakir B., Clark A. T., Plath K., Lowry W. E., Benvenisty N. (2010). Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 7, 521-531 [DOI] [PubMed] [Google Scholar]
- Mekhoubad S., Bock C., de Boer A. S., Kiskinis E., Meissner A., Eggan K. (2012). Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595-609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. S., Shearer R. L., Capowski E. E., Wright L. S., Wallace K. A., McMillan E. L., Zhang S. C., Gamm D. M. (2009). Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 106, 16698-16703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. C., Tan S., Qiao G., Barlow K. A., Wang J., Xia D. F., Meng X., Paschon D. E., Leung E., Hinkley S. J., et al. (2011). A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143-148 [DOI] [PubMed] [Google Scholar]
- Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., Grenier J. K., et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283-1298 [DOI] [PubMed] [Google Scholar]
- Mohr J. C., Zhang J., Azarin S. M., Soerens A. G., de Pablo J. J., Thomson J. A., Lyons G. E., Palecek S. P., Kamp T. J. (2010a). The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 31, 1885-1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S., Bakal C., Perrimon N. (2010b). Genomic screening with RNAi: results and challenges. Annu. Rev. Biochem. 79, 37-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey E. E., Tang Z., Sigrist K., Lu M. M., Jiang F., Ip H. S., Parmacek M. S. (1998). GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 12, 3579-3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou M. J., Bogdanove A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501 [DOI] [PubMed] [Google Scholar]
- Nagy A. (2002). Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Nakano T., Ando S., Takata N., Kawada M., Muguruma K., Sekiguchi K., Saito K., Yonemura S., Eiraku M., Sasai Y. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771-785 [DOI] [PubMed] [Google Scholar]
- Nazor K. L., Altun G., Lynch C., Tran H., Harness J. V., Slavin I., Garitaonandia I., Müller F. J., Wang Y. C., Boscolo F. S., et al. (2012). Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 10, 620-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E. S., Davis R. P., Azzola L., Stanley E. G., Elefanty A. G. (2005). Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood 106, 1601-1603 [DOI] [PubMed] [Google Scholar]
- Niwa A., Heike T., Umeda K., Oshima K., Kato I., Sakai H., Suemori H., Nakahata T., Saito M. K. (2011). A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLoS ONE 6, e22261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro M. C., Sarangi F., Ogawa S., Holtzinger A., Corneo B., Li X., Micallef S. J., Park I. H., Basford C., Wheeler M. B., et al. (2011). Stage-specific signaling through TGF family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 138, 861-871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Wieschaus E. (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795-801 [DOI] [PubMed] [Google Scholar]
- Offield M. F., Jetton T. L., Labosky P. A., Ray M., Stein R. W., Magnuson M. A., Hogan B. L., Wright C. V. (1996). PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983-995 [DOI] [PubMed] [Google Scholar]
- Osafune K., Caron L., Borowiak M., Martinez R. J., Fitz-Gerald C. S., Sato Y., Cowan C. A., Chien K. R., Melton D. A. (2008). Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313-315 [DOI] [PubMed] [Google Scholar]
- Oshima K., Shin K., Diensthuber M., Peng A. W., Ricci A. J., Heller S. (2010). Mechanosensitive hair cell-like cells from embryonic and induced pluripotent stem cells. Cell 141, 704-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison P. J., Silva J. M., Conklin D. S., Schlabach M., Li M., Aruleba S., Balija V., O'Shaughnessy A., Gnoj L., Scobie K., et al. (2004). A resource for large-scale RNA-interference-based screens in mammals. Nature 428, 427-431 [DOI] [PubMed] [Google Scholar]
- Papapetrou E. P., Lee G., Malani N., Setty M., Riviere I., Tirunagari L. M., Kadota K., Roth S. L., Giardina P., Viale A., et al. (2011). Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 29, 73-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich N. P., Pabo C. O. (1991). Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252, 809-817 [DOI] [PubMed] [Google Scholar]
- Pera M. F., Trounson A. O. (2004). Human embryonic stem cells: prospects for development. Development 131, 5515-5525 [DOI] [PubMed] [Google Scholar]
- Polo J. M., Liu S., Figueroa M. E., Kulalert W., Eminli S., Tan K. Y., Apostolou E., Stadtfeld M., Li Y., Shioda T., et al. (2010). Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 28, 848-855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon E., Clermont F., Firpo M. T., Akhurst R. J. (2006). TGFbeta inhibition of yolk-sac-like differentiation of human embryonic stem-cell-derived embryoid bodies illustrates differences between early mouse and human development. J. Cell Sci. 119, 759-768 [DOI] [PubMed] [Google Scholar]
- Puchta H., Dujon B., Hohn B. (1993). Homologous recombination in plant cells is enhanced by in vivo induction of double strand breaks into DNA by a site-specific endonuclease. Nucleic Acids Res. 21, 5034-5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A., Riedel M. J., Wideman R. D., Karanu F., Ao Z., Warnock G. L., Kieffer T. J. (2011). Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes 60, 239-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P., Smih F., Jasin M. (1994). Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 14, 8096-8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust W. L., Sadasivam A., Dunn N. R. (2006). Three-dimensional extracellular matrix stimulates gastrulation-like events in human embryoid bodies. Stem Cells Dev. 15, 889-904 [DOI] [PubMed] [Google Scholar]
- Sadelain M., Papapetrou E. P., Bushman F. D. (2012). Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 12, 51-58 [DOI] [PubMed] [Google Scholar]
- Sanvito F., Herrera P. L., Huarte J., Nichols A., Montesano R., Orci L., Vassalli J. D. (1994). TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development 120, 3451-3462 [DOI] [PubMed] [Google Scholar]
- Schuldiner M., Yanuka O., Itskovitz-Eldor J., Melton D. A., Benvenisty N. (2000). Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 97, 11307-11312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick G. S., Barker K. T., Stolte-Dijkstra I., Fleischmann C., Coleman R. J., Garrett C., Gloyn A. L., Edghill E. L., Hattersley A. T., Wellauer P. K., et al. (2004). Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat. Genet. 36, 1301-1305 [DOI] [PubMed] [Google Scholar]
- Sharon N., Mor I., Golan-lev T., Fainsod A., Benvenisty N. (2011). Molecular and functional characterizations of gastrula organizer cells derived from human embryonic stem cells. Stem Cells 29, 600-608 [DOI] [PubMed] [Google Scholar]
- Shultz L. D., Ishikawa F., Greiner D. L. (2007). Humanized mice in translational biomedical research. Nat. Rev. Immunol. 7, 118-130 [DOI] [PubMed] [Google Scholar]
- Silva J. M., Li M. Z., Chang K., Ge W., Golding M. C., Rickles R. J., Siolas D., Hu G., Paddison P. J., Schlabach M. R., et al. (2005). Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 37, 1281-1288 [DOI] [PubMed] [Google Scholar]
- Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T., et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337-342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smih F., Rouet P., Romanienko P. J., Jasin M. (1995). Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 23, 5012-5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. R., Maguire S., Davis L. A., Alexander M., Yang F., Chandran S., ffrench-Constant C., Pedersen R. A. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells 26, 496-504 [DOI] [PubMed] [Google Scholar]
- Soldner F., Laganière J., Cheng A. W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L. I., Myers R. H., Lindquist S., et al. (2011). Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J. R., Mayhew C. N., Rankin S. A., Kuhar M. F., Vallance J. E., Tolle K., Hoskins E. E., Kalinichenko V. V., Wells S. I., Zorn A. M., et al. (2011). Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers D. A., Zinkin N. T., Stanojevic V., Clarke W. L., Habener J. F. (1997). Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15, 106-110 [DOI] [PubMed] [Google Scholar]
- Suga H., Kadoshima T., Minaguchi M., Ohgushi M., Soen M., Nakano T., Takata N., Wataya T., Muguruma K., Miyoshi H., et al. (2011). Self-formation of functional adenohypophysis in three-dimensional culture. Nature 480, 57-62 [DOI] [PubMed] [Google Scholar]
- Suzuki K., Mitsui K., Aizawa E., Hasegawa K., Kawase E., Yamagishi T., Shimizu Y., Suemori H., Nakatsuji N., Mitani K. (2008). Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc. Natl. Acad. Sci. USA 105, 13781-13786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663-676 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861-872 [DOI] [PubMed] [Google Scholar]
- Tei E., Mehta S., Tulachan S. S., Yew H., Hembree M., Preuett B., Snyder C. L., Yamataka A., Miyano T., Gittes G. K. (2005). Synergistic endocrine induction by GLP-1 and TGF-beta in the developing pancreas. Pancreas 31, 138-141 [DOI] [PubMed] [Google Scholar]
- Thomas K. R., Capecchi M. R. (1987). Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51, 503-512 [DOI] [PubMed] [Google Scholar]
- Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., Jones J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145-1147 [DOI] [PubMed] [Google Scholar]
- Tulachan S. S., Tei E., Hembree M., Crisera C., Prasadan K., Koizumi M., Shah S., Guo P., Bottinger E., Gittes G. K. (2007). TGF-beta isoform signaling regulates secondary transition and mesenchymal-induced endocrine development in the embryonic mouse pancreas. Dev. Biol. 305, 508-521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A., Schuldiner M., Benvenisty N. (2004). Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells 22, 635-641 [DOI] [PubMed] [Google Scholar]
- van Dartel D. A., Pennings J. L., de la Fonteyne L. J., van Herwijnen M. H., van Delft J. H., van Schooten F. J., Piersma A. H. (2010). Monitoring developmental toxicity in the embryonic stem cell test using differential gene expression of differentiation-related genes. Toxicol. Sci. 116, 130-139 [DOI] [PubMed] [Google Scholar]
- Wandzioch E., Zaret K. S. (2009). Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science 324, 1707-1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston R. H., Lindblad-Toh K., Birney E., Rogers J., Abril J. F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520-562 [DOI] [PubMed] [Google Scholar]
- Wichterle H., Lieberam I., Porter J. A., Jessell T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385-397 [DOI] [PubMed] [Google Scholar]
- Wilkie A. O. (2003). Why study human limb malformations? J. Anat. 202, 27-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L. A., Davis-Dusenbery B. N., Eggan K. C. (2012). SnapShot: directed differentiation of pluripotent stem cells. Cell 149, 1174-1174e1. [DOI] [PubMed] [Google Scholar]
- Wu S. M., Hochedlinger K. (2011). Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 13, 497-505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan S., Borok M. J., Decker K. J., Battle M. A., Duncan S. A., Hale M. A., Macdonald R. J., Sussel L. (2012). Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J. Clin. Invest. 122, 3516-3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S. (2012). Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 10, 678-684 [DOI] [PubMed] [Google Scholar]
- Yang L., Soonpaa M. H., Adler E. D., Roepke T. K., Kattman S. J., Kennedy M., Henckaerts E., Bonham K., Abbott G. W., Linden R. M., et al. (2008). Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 453, 524-528 [DOI] [PubMed] [Google Scholar]
- Yang H., Shi L., Wang B. A., Liang D., Zhong C., Liu W., Nie Y., Liu J., Zhao J., Gao X., et al. (2012). Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell 149, 605-617 [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917-1920 [DOI] [PubMed] [Google Scholar]
- Zambrowicz B. P., Imamoto A., Fiering S., Herzenberg L. A., Kerr W. G., Soriano P. (1997). Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. USA 94, 3789-3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J., Stark A., Kellis M., Hong J. W., Nechaev S., Adelman K., Levine M., Young R. A. (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet. 39, 1512-1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Z., Gao W., Yang H. B., Zhang B., Zhu Z. Y., Xue Y. F. (2006). Screening for genes essential for mouse embryonic stem cell self-renewal using a subtractive RNA interference library. Stem Cells 24, 2661-2668 [DOI] [PubMed] [Google Scholar]
- Zhang X., Huang C. T., Chen J., Pankratz M. T., Xi J., Li J., Yang Y., Lavaute T. M., Li X. J., Ayala M., et al. (2010). Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7, 90-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S., Wurdak H., Wang J., Lyssiotis C. A., Peters E. C., Cho C. Y., Wu X., Schultz P. G. (2009). A small molecule primes embryonic stem cells for differentiation. Cell Stem Cell 4, 416-426 [DOI] [PubMed] [Google Scholar]
- Zwaka T. P., Thomson J. A. (2003). Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 21, 319-321 [DOI] [PubMed] [Google Scholar]