

Abstract
A key process during vertebrate heart development is the migration of bilateral populations of myocardial precursors towards the midline to form the primitive heart tube. In zebrafish, signaling mediated by sphingosine-1-phosphate (S1P) and its cognate G protein-coupled receptor (S1pr2/Mil) is essential for myocardial migration, but the underlying mechanisms remain undefined. Here, we show that suppression of Gα13 signaling disrupts myocardial migration, leading to the formation of two bilaterally located hearts (cardia bifida). Genetic studies indicate that Gα13 acts downstream of S1pr2 to regulate myocardial migration through a RhoGEF-dependent pathway. Furthermore, disrupting any component of the S1pr2/Gα13/RhoGEF pathway impairs endoderm convergence during segmentation, and the endodermal defects correlate with the extent of cardia bifida. Moreover, endoderm transplantation reveals that the presence of wild-type anterior endodermal cells in Gα13-deficient embryos is sufficient to rescue the endoderm convergence defect and cardia bifida, and, conversely, that the presence of anterior endodermal cells defective for S1pr2 or Gα13 in wild-type embryos causes such defects. Thus, S1pr2/Gα13 signaling probably acts in the endoderm to regulate myocardial migration. In support of this notion, cardiac-specific expression of Gα13 fails to rescue cardia bifida in the context of global Gα13 inhibition. Our data demonstrate for the first time that the Gα13/RhoGEF-dependent pathway functions downstream of S1pr2 to regulate convergent movement of the endoderm, an event that is crucial for coordinating myocardial migration.
Keywords: S1pr2, Mil, Gα13, Myocardial migration, Endoderm convergence, Zebrafish
INTRODUCTION
During vertebrate development, two groups of myocardial precursors are specified at lateral regions of the gastrula and migrate towards the embryonic midline where they merge and form a single primitive heart tube during segmentation (Stainier, 2001; Evans et al., 2010; Bakkers, 2011). This process is essential for formation of the heart tube, the foundation for all subsequent cardiac morphogenesis. Its disruption might lead to cardiac defects that include the formation of two bilaterally located hearts - a condition known as cardia bifida (Stainier et al., 1996; Yelon, 2001), which has catastrophic consequences for cardiac function.
Studies in numerous model organisms, including Xenopus, zebrafish, chick and mouse, have shown that proper myocardial migration requires the intrinsic properties of myocardial precursors (including the proper differentiation, correct number and epithelial organization of cardiomyocytes) (Horne-Badovinac et al., 2001; Trinh and Stainier, 2004; Rohr, 2006; Garavito-Aguilar et al., 2010), as well as an environment conducive to migration. One such extracellular requirement is the presence of adjacent endoderm. Endoderm-deficient mouse and zebrafish mutants display cardia bifida (Kuo et al., 1997; Molkentin et al., 1997; Alexander et al., 1999; Reiter et al., 1999; David and Rosa, 2001). Similarly, in chick and Xenopus embryos, surgical removal of anterior endoderm impairs the migration of myocardial precursors, as well as heart formation (Rosenquist, 1970; Nascone and Mercola, 1995; Withington et al., 2001). It has been proposed that the endoderm serves as a substrate that allows cardiac mesoderm to migrate, and that it releases either cues that guide the migration of cardiac progenitor cells or signaling molecules that promote cardiac differentiation (Schultheiss et al., 1997; Andree et al., 1998; Lough and Sugi, 2000; David and Rosa, 2001; Alsan and Schultheiss, 2002; Nijmeijer et al., 2009). Additionally, the endoderm might provide a mechanical force that drives the migration of myocardial cells (Varner and Taber, 2012). Thus, despite the extensive evidence that the endoderm is important for cardiomyocyte movement during heart tube formation, its exact roles remain to be determined.
The zebrafish mutant miles apart (mil), which lacks a functional the G protein-coupled receptor S1pr2 [which responds to sphingosine-1-phosphate (S1P), a bioactive lysophospholipid], displays defects in myocardial migration that lead to cardia bifida (Kupperman et al., 2000; Osborne et al., 2008; Kawahara et al., 2009). Although the s1pr2 transcript was detected in the lateral plate mesoderm, which is where the myocardial cells are located, transplantation experiments indicate that S1pr2/Mil may function outside cardiomyocytes to make the environment permissive for myocardial precursor migration (Kupperman et al., 2000). This notion is supported by the findings that S1pr2 regulates fibronectin (Fn) expression in the local environment, to facilitate the cell-Fn interaction needed for myocardial migration (Matsui et al., 2007; Osborne et al., 2008). Furthermore, embryos defective for S1pr2 signaling have defects in endoderm morphogenesis, and this may affect myocardial migration (Kupperman et al., 2000; Osborne et al., 2008). However, which downstream effectors of S1pr2 regulate endoderm development and how endoderm defects impair myocardial migration, remain unknown.
Here, we demonstrate that specifically the G protein isoform Gα13 acts downstream of S1pr2, and that it regulates myocardial migration through a RhoGEF-dependent pathway. Moreover, our analyses using a transgene that expresses Gα13 specifically in cardiomyocytes and endoderm transplantation provide evidence that the S1pr2/Gα13/RhoGEF signaling pathway functions within the endoderm to regulate convergent movement of this tissue, and that defects in this movement impair proper myocardial migration. Together, our studies uncover, for the fist time, the signaling pathway by which S1P regulates myocardial migration, and establish S1pr2/Gα13-regulated endoderm convergence as having a crucial role in myocardial migration.
MATERIALS AND METHODS
Zebrafish strains and maintenance
AB*/Tuebingen, transgenic Tg(myl7:EGFP) (Huang et al., 2003), and Tg(sox17:EGFP) (Mizoguchi et al., 2008) and milm93 zebrafish strains (Kupperman et al., 2000) were used. Embryos were obtained by natural mating and staged according to morphology, or hours post fertilization (hpf) at 28.5°C or 32°C unless otherwise specified, as described previously (Kimmel et al., 1995).
RNA expression and morpholinos
mRNA and MOs were injected at the one-cell stage at the doses indicated. Capped mRNA was synthesized using the mMessage mMachine Kit (Ambion). The RNAs encoding the following genes were used: gna13a (100 pg) and GNA13 (100 pg) (Lin et al., 2005); the RGS domain of PDZ RhoGEF (arhgef11RGS) (500 pg) or a dominant-negative mutant Arhgef11 lacking the DHPH domain (arhgef11ΔDHPH) (500 pg) (Panizzi et al., 2007; Lin et al., 2009); s1pr2/mil (200 pg) (Osborne et al., 2008); and sox32 (400 pg) (Stafford et al., 2006). The previously validated morpholino antisense oligonucleotides (MOs) targeting the following genes were used: gna12 (4 ng), gna13a and gna13b (2 ng each) (Lin et al., 2005), and s1pr2/mil (15 ng) (Kawahara et al., 2009).
Whole-mount in situ hybridization and immunofluorescence assay
Digoxigenin-labeled antisense RNA probes were synthesized by in vitro transcription. In situ hybridization was performed as described (Lin et al., 2005; Thisse and Thisse, 2008). After in situ hybridization, the embryos were re-fixed in 4% PFA and sectioned at 10 μm, as described previously (Barthel and Raymond, 1990). An immunofluorescence assay was performed as previously described (Trinh and Stainier, 2004) using an anti-GFP antibody (1:200, Santa Cruz).
Endoderm transplantation
Endoderm transplantation was performed as described previously (Stafford et al., 2006; Chung and Stainier, 2008). Wild-type embryos injected with sox32 RNA (confers an endodermal identity to all cells) and 1% rhodamine-dextran (70,000 MW, lysine fixable, Invitrogen), with or without gna13ab MOs or s1pr2 MO served as the donors. Tg(sox17:EGFP) or Tg(sox17:EGFP)/(myl7:EGFP) embryos, a subset of which were injected with gna13ab MOs, served as hosts. At mid-blastula stage, two independent groups of donor cells were transplanted into the host embryos along the blastoderm margin, at an angle of ∼120-180° from one another (to increase the chances that they would contribute to the anterior region of the endoderm).
Microscopy, time-lapse imaging and image processing
For still images, live or fixed embryos were photographed using a Leica DMI 6000 microscope. For time-lapse experiments, embryos were embedded in a dorsal-mount imaging mold as previously described (Megason, 2009) and images were taken in the anterior region of endoderm of Tg(sox17:EGFP) at 25°C, at 5-minute intervals with a 5×/NA 0.15 objective on a Leica DMI 6000 microscope. The images were processed using the Metamorph software and cell tracking was analyzed by ImageJ. Data was exported to Excel where cell migration speed, paths and direction were determined as previously reported (Lin et al., 2005).
Statistical analysis
Data were compiled from two or three independent experiments, and are presented as the mean ± s.e.m. The number of embryos analyzed is indicated. Statistical analyses were performed using unpaired Student's t-tests with two tails and unequal variance.
RESULTS
Gα13 signaling is required for myocardial migration
S1pr2 has been shown to couple with diverse G proteins, in particular Gα12 and Gα13, to mediate a variety of cellular responses (Skoura and Hla, 2009; Suzuki et al., 2009). We previously identified one Gα12 and two Gα13 (Gα13a and Gα13b) isoforms in zebrafish, and demonstrated that they are essential during gastrulation (Lin et al., 2005; Lin et al., 2009). Here, we show that Gα13 is also required for myocardial migration. Injection of morpholinos (MOs) that inhibit the translation of gna13a and gna13b genes (Lin et al., 2005), but neither alone, caused cardia bifida, a lack of circulation and pericardial edema (Fig. 1C-D). Notably, the two hearts in gna13ab MO-injected embryos (morphants) beat and expressed the myocardial markers nkx2.5 (Serbedzija et al., 1998) and myl7 (formerly known as cardiac myosin light chain 2, cmcl2), as well as the chamber-specific markers atrial myosin heavy chain (amhc; myh6 - Zebrafish Information Network) and ventricular myosin heavy chain (vmhc) (Yelon et al., 1999) (Fig. 1G-J; not shown). Together, these data indicate that Gα13 is required for the migration, but not the differentiation, of cardiomyocytes. In addition, the morphants exhibited blistering in the tailfin (Fig. 1C,C′, arrowheads), a feature unique to embryos defective for S1P signaling and not observed in other cardia bifida mutants (Kupperman et al., 2000; Osborne et al., 2008; Kawahara et al., 2009).
Fig. 1.
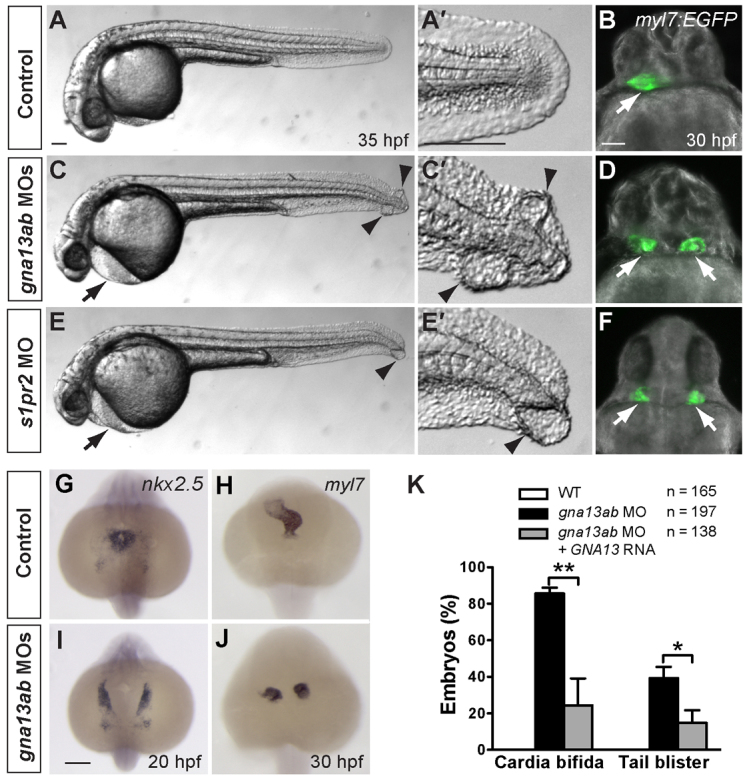
Gα13 signaling is required for myocardial migration. (A-F) Comparison of control embryos (A,B), embryos injected with MOs against both gna13a and gna13b (C,D, gna13ab MOs), and embryos injected with a MO against s1pr2/mil (E,F) at 35 hpf. (A,C,E) Lateral bright-field images of embryos. Anterior is towards the left; arrows indicate pericardial edema; arrowheads indicate tail blisters. (A′,C′,E′) High-magnification images of the tail region of each embryo. (B,D,F) Overlay of epifluorescence and bright-field images of Tg(myl7:EGFP) embryos at 30 hpf. Ventral view; white arrows indicate hearts. (G-J) Expression of nkx2.5 and myl7, as detected by whole-mount in situ hybridization, in control and gnb13ab MO-injected embryos. Dorsoanterior view with anterior upwards. (K) Frequencies of cardia bifida and tail blistering in 35 hpf embryos injected with gna13ab MOs, alone or together with an RNA encoding the human Gα13 (GNA13). *P<0.05; **P<0.01. Data are mean±s.e.m. Scale bars: 100 μm.
Both the severity and penetrance of cardia bifida and tail blistering increased with the MO dose (not shown). Expressing either human Gα13 or a zebrafish Gα13a form that is insensitive to the MOs significantly suppressed cardia bifida and tail blistering in gna13ab morphants (Fig. 1K; not shown). These findings demonstrate that the observed phenotypes are specific consequences of interference with Gα13 function, and indicate that the functions of the zebrafish and human Gα13 proteins are conserved (Lin et al., 2005).
Interestingly, although Gα12 belongs to the Gα12/13 subfamily of G proteins and exhibits functions similar to those of Gα13 in mammalian cell culture (Buhl et al., 1995; Lin et al., 2005), we found that suppression of Gα12 alone caused neither cardia bifida nor tail blistering (supplementary material Fig. S1; not shown). However, inhibiting Gα12 significantly enhanced the defects resulting from Gα13 inhibition, as judged by increases in the frequency of cardia bifida and the distance between the two hearts (supplementary material Fig. S1). These data indicate that Gα12 and Gα13 play partially redundant roles in myocardial migration.
Gα13 acts downstream of S1pr2 to regulate myocardial migration
The striking phenotypic similarities between embryos depleted of Gα13 and those deficient for S1P signaling (Fig. 1C,D versus 1E,F) led us to hypothesize that Gα13 may act downstream of S1pr2 to regulate myocardial migration. To test this, we first examined whether Gα13 overexpression can suppress the defects resulting from the S1pr2 deficiency. We found that when embryos were co-injected with s1pr2 MO and an RNA encoding either zebrafish Gα13a (Fig. 2E) or human Gα13a (not shown), the frequencies of cardia bifida and tail blistering were significantly reduced. Examination of myl7 expression revealed that, in the co-injected embryos, the bilateral populations of cardiomyocytes migrated to a point much closer to the midline (Fig. 2C versus 2B). Thus, Gα13 overexpression can partially compensate for S1pr2 deficiency, suggesting that Gα13 acts downstream of S1pr2 to regulate myocardial migration.
Fig. 2.
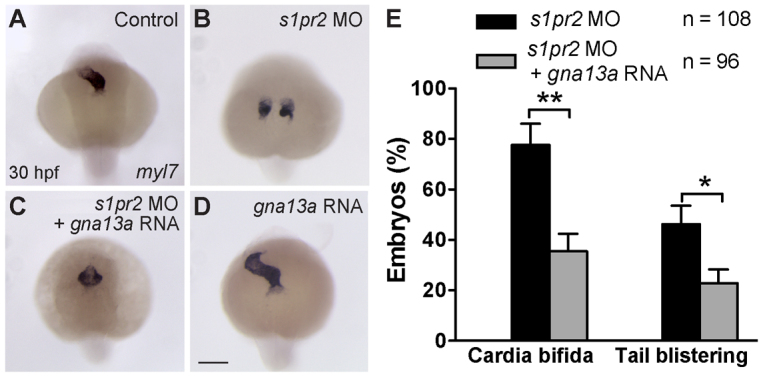
Gα13 acts downstream of S1pr2 to regulate myocardial migration. (A-D) myl7 expression detected by in situ hybridization in the indicated embryos at 30 hpf. Dorsoanterior view with anterior upwards. (E) Frequencies of cardia bifida and tail blistering in 35 hpf embryos injected with the s1pr2 MO, alone or together with the gna13a RNA. *P<0.05. Data are mean±s.e.m. Scale bar: 100 μm.
To gain further support for the hypothesis that S1pr2 and Gα13 function in the same pathway, we carried out co-injection experiments and tested for synergistic effects. Specifically, we injected MOs targeting s1pr2 or gna13ab at suboptimal doses, alone and together, and evaluated the location of the heart and the morphology of the tailfin. At the doses used, defects caused by injecting the individual MOs were very mild (supplementary material Table S1); those caused by co-injection of the two MOs were more severe than the additive effects of each manipulation alone (higher frequencies of both cardia bifida and tail blistering, as well as increased distance between the two populations of cardiomyocytes) (supplementary material Table S1; not shown). These data are consistent with the notion that S1pr2 and Gα13 function in a common genetic pathway.
We reasoned that if Gα13 acts downstream of S1pr2, its function should be required for S1P signaling. To directly test this hypothesis, we examined whether Gα13 inhibition blocks the gastrulation defects resulting from S1pr2 overexpression (Osborne et al., 2008). S1pr2 overexpression induced defects in convergence and extension (C&E) movements (revealed by broadening of the axial mesoderm, Fig. 3A versus 3B, red lines), as well as in epiboly [evident from increase in the distance between the deep-cell margin (dcm) and the vegetal pole (VP), Fig. 3A versus 3B, blue lines with double arrows]. At 35 hpf, these embryos displayed a shortened body axis and frequently also cyclopia (Fig. 3D), phenotypes often associated with impaired C&E movements (Heisenberg et al., 2000; Jessen et al., 2002; Lin et al., 2005). Remarkably, the gastrulation defects induced by S1pr2 overexpression were largely suppressed by Gα13 inhibition, as judged by morphological observation and analysis of ntl expression (Fig. 3C,F). These findings indicate that S1pr2 signals through Gα13 to regulate gastrulation movements.
Fig. 3.
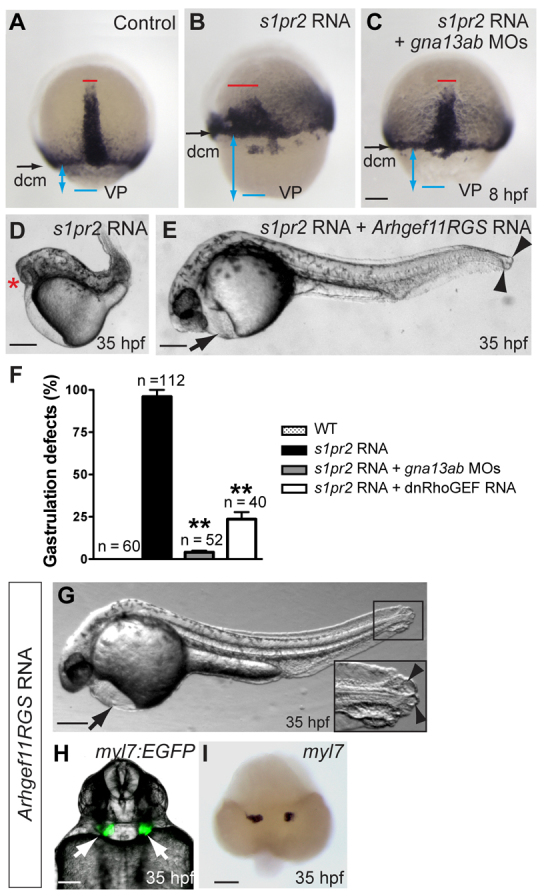
Gα13 and RhoGEF act downstream of S1pr2. (A-F) Disruption of either Gα13 or RhoGEF suppresses the gastrulation defects resulting from overexpression of S1pr2. (A-C) ntl expression detected by in situ hybridization in control embryos and in embryos injected with s1pr2 RNA, alone or together with gna13ab MOs, at 8 hpf. Dorsal view, with vegetal pole (VP, blue line) towards the bottom; black arrows indicate positions of the deep-cell margin (dcm); double-headed blue arrows indicate distance from the dcm to VP; red lines indicate axial mesoderm. (D,E) Bright-field images of 35 hpf embryos injected with the s1pr2 RNA, alone or together with the Arhgef11RGS RNA. Lateral view; red asterisk indicates cyclopia; arrow indicates pericardial edema; arrowheads indicate tail blisters. (F) Frequencies of embryos exhibiting gastrulation defects. **P<0.01 versus s1pr2 RNA-injected embryos. Data are mean±s.e.m. (G-I) Interference with RhoGEF function disrupts myocardial migration. Embryo injected with the Arhgef11RGS RNA at 35 hpf. (G) Lateral bright-field image showing pericardial edema (arrow) and tail blisters (arrowheads). The inset shows a high-magnification image of the tail region in the boxed area. (H) Overlay of epifluorescence and bright-field images, showing the locations of EGFP-expressing cardiomyocytes (arrows) in Tg(myl7:EGFP) embryos. Ventral view. (I) myl7 expression, as detected by in situ hybridization in control embryos and in embryos injected with s1pr2 RNA, alone or together with gna13ab MOs, at 8 hpf. Dorsoanterior view. Scale bars: 100 μm.
S1pr2/Gα13 signaling regulates myocardial migration via a RhoGEF/Rho-dependent pathway
Next, we investigated the molecular effectors of S1pr2/Gα13-regulated myocardial migration. Gα12 and Gα13 function primarily by activating RhoGEF and then RhoA (Suzuki et al., 2009). We have previously shown that Gα13a binds a PDZRhoGEF known as Arhgef11, via its regulator of G protein-coupled signaling (RGS) domain, and that Gα12/13 regulate zebrafish epiboly through a RhoGEF-dependent pathway. Specifically, overexpression of dominant-negative forms of Arhgef11 (Arhgef11RGS - the RGS domain of Arhgef11 that competes for Gα13 binding but contains no functional RhoA-activating DH and PH domains; and Arhgef11ΔDHPH - which lacks the DH and PH domains) significantly suppressed the epiboly defects resulting from Gα13 overexpression (Lin et al., 2009). In addition, it has been reported that disrupting RhoGEF or RhoA function causes cardia bifida (Matsui et al., 2005; Panizzi et al., 2007). Thus, we reasoned that Gα13 may signal through Arhgef11/RhoA to effect S1pr2-triggered myocardial migration.
To test this possibility, we first re-evaluated the functions of RhoGEF in myocardial migration and tailfin development by overexpressing dominant-negative forms of Arhgef11. We found that 60% of the embryos (n=73) expressing Arhgef11RGS displayed cardia bifida and pericardial edema (Fig. 3G-I). In addition, these embryos exhibited tail blistering (Fig. 3G, arrowheads), a phenotype that had not been appreciated previously (Panizzi et al., 2007). Moreover, the bilateral hearts observed in these embryos were properly differentiated, as evident from the expression of nkx2.5 and myl7 (Fig. 3I; not shown). Similar results were obtained in embryos expressing Arhgef11ΔDHPH (not shown). Based on the similar cardia bifida and tail blistering phenotypes that result from disrupted RhoGEF function and depleted S1P signaling, we postulated that RhoGEF and S1pr2 probably function in the same genetic pathway. Accordingly, a synergistic effect was observed when RhoGEF and S1pr2 were simultaneously inhibited. Partial inhibition of both proteins (by injecting suboptimal doses of the s1pr2 MO and the Arhgef11RGS RNA) resulted in cardia bifida and tail blistering that were much more severe than that produced by partial inhibition of either protein individually (supplementary material Table S1). We could not overexpress Arhgef11 to assess whether it can rescue the defects in s1pr2 morphants, as such expression caused severe morphological defects during early development (not shown) (Panizzi et al., 2007). However, we found that Arhgef11 is required for S1pr2 signaling. Similar to inhibition of Gα13 expression, interference with Arhgef11 function (by expressing Arhgef11RGS) suppressed the gastrulation defects otherwise caused by S1pr2 overexpression, as judged by ntl expression at 8 hpf and shortening of the body axis at 35 hpf (Fig. 3D-F; not shown). Specifically, embryos co-expressing S1pr2 and Arhgef11RGS had a body axis of nearly normal length and two eyes (Fig. 3E), in contrast to S1pr2-overexpressing embryos, which featured a shortened body axis and cyclopia (Fig. 3D). However, the co-injected embryos displayed cardia bifida, pericardial edema and tail blistering (Fig. 3E; not shown), features that were not observed in S1pr2-overexpressing embryos; these defects presumably resulted from the expression of Arhgef11RGS. Taken together, these results support the model that S1pr2 signals through Gα13 to activate RhoGEF/Rho, and to thereby regulate myocardial migration.
S1pr2/G α13 signaling regulates convergent movement of the anterior endoderm
Next, we examined through which tissue S1pr2/Gα13 signaling controls myocardial migration. Previous studies have suggested that S1P signaling regulates morphogenesis of the anterior endoderm, thereby indirectly affecting the migration of myocardial precursors (Osborne et al., 2008). However, how this signaling pathway regulates endoderm development and how the endodermal defects relate to those in myocardial migration is unclear. As the first step in investigating the role of S1pr2/Gα13 signaling in the endoderm, we tested for the expression of s1pr2 and gna13 in this tissue at the 14-somite (14s, 16 hpf) and 18s (18 hpf) stages, during which myocardial precursors migrate toward the midline (Matsui et al., 2005; Holtzman et al., 2007). We visualized the endoderm using transgenic Tg(sox17:EGFP) embryos, in which specifically the endodermal cells are labeled with EGFP (Mizoguchi et al., 2008). Performing a combination of in situ hybridization (to detect s1pr2 and gna13 transcripts) and immunofluorescence (to identify GFP-labeled endoderm) analyses, we found that gna13a, gna13b and s1pr2 transcripts are present in the GFP-expressing endodermal cells at both the 14s (data not shown) and 18s stages (Fig. 4A,B; the expression of gna13a is not shown, but is identical to that of gna13b). Interestingly, the s1pr2 transcript is highly enriched in the endoderm (Fig. 4A, arrowheads), whereas the gna13 transcripts are present uniformly in all tissues including the endoderm (Fig. 4B). These results suggest that S1pr2 and Gα13 may function within the endoderm.
Fig. 4.

s1pr2 and gna13 are expressed in the endoderm and cardiomyocytes during segmentation. (A-D′) Transverse sections of Tg(sox17:EGFP) (A,B) and Tg(myl7:EGFP) (C,D) embryos at 18- and 20-somite stages. Shown are s1pr2 (A-A‴,C-C‴) and gna13a (B-B‴,D-D‴) transcript, as detected by in situ hybridization (A-D), and EGFP expression in endodermal cells (A′,B′) and cardiomyocytes (C′,D′) as detected by immunofluorescence staining (anti-GFP antibody). (A″-D″) Merged images of A-D and A′-D′. (A‴-D‴) High-magnification images of the boxed regions in A″-D″. Yellow arrows, endoderm; white arrowheads, cardiomyocytes. Scale bars: 100 μm.
We further evaluated how S1pr2/Gα13 signaling affects endoderm development using Tg(sox17:EGFP) embryos. We found that the anterior endodermal sheet was significantly widened in S1pr2/Gα13-deficient embryos during segmentation, which was not reported previously (Osborne et al., 2008). By the end of gastrulation, endoderm morphology in s1pr2 or gna13ab morphants was indistinguishable from that in control embryos, as assessed by the sox17 expression pattern (supplementary material Fig. S2) and sox17:EGFP distribution (not shown). As development proceeded, however, differences became obvious. By mid-segmentation (8s stage), the anterior endodermal sheet in mil(-/-) embryos (in sox17:EGFP background) or gna13ab morphants was significantly wider than that in their controls (Fig. 5A-C,J). The endodermal sheet remained widened throughout late segmentation, the period during which the myocardial precursors migrate towards the midline to fuse at the 20-somite stage (Fig. 5D-I,J). These data indicate that S1pr2/Gα13 signaling is required for endoderm convergence.
Fig. 5.

S1pr2/Gα13 signaling is required for convergent movement of the anterior endoderm during segmentation. (A-I) Epifluorescence images of the anterior endoderm in Tg(sox17:EGFP) control, milm93 mutant or gnb13ab MOs-injected embryos raised at 25°C at the indicated stages. Yellow lines (equivalent length among embryos at the same stage) indicate the width of the anterior endodermal sheet; red asterisks indicate endodermal holes. (J) Quantification of endoderm width in each group. *P<0.05, **P<0.01 versus control. Data are mean±s.e.m. (K) The cell-transplantation procedure. Sox32-overexpressing and rhodamine-dextran labeled wild-type or s1pr2 morphant donor cells were transplanted into Tg(sox17:EGFP) host embryos. (L-O) Still images from time-lapse movies, showing the anterior endoderm in wild-type Tg(sox17:EGFP) hosts transplanted with rhodamine-labeled donor cells (L,M are wild type; N,O are s1pr2 morphant) at 3 and 10 somites. Green and red lines indicate the width of the host and donor endodermal sheets, respectively. (P) Convergence velocity of the wild-type and s1pr2 morphant endoderm populations. **P<0.001; #P=0.96 versus the control. Data are mean±s.e.m. Scale bars: 100 μm.
To further evaluate the role of S1pr2 signaling in endoderm migration, we performed endoderm transplantation (Stafford et al., 2006; Chung and Stainier, 2008) as outlined schematically in Fig. 5K, assessing how S1pr2-deficient endodermal cells migrate in the wild-type endodermal environment. We selected embryos in which donor endoderm cells had been transplanted into one side of the host endoderm, and performed time-lapse experiments on the anterior endoderm region from the 3-somite to the 10-somite stage. We then compared the extent of convergent movement in the transplanted donor versus host endodermal cells. Our analyses revealed that transplanted wild-type cell populations migrated to the midline at a speed similar to that of the wild-type host cells and that, at the end of the time-lapse experiment, the width of the transplanted endoderm populations was similar to that of the host endoderm populations (Fig. 5L,M,P; supplementary material Movie 1). By contrast, the transplanted s1pr2-deficient cells moved towards the midline at a slower speed, and the width of the sheet they formed at the end of the time-lapse experiment was wider than on the wild-type side of the embryo (Fig. 5N-P; supplementary material Movie 2). Together, these results suggest that S1pr2/Gα13 signaling is required autonomously for convergent movement of the anterior endoderm.
In addition to the impaired endoderm convergence, gna13ab morphants or mil mutants featured ‘holes’, i.e. regions devoid of endodermal cells, within the most anterior endoderm (Fig. 5A-I), a phenotype similar to that reported previously for embryos defective for S1P signaling (Osborne et al., 2008). Notably, the endodermal holes became enlarged during development (Fig. 5A-I). To identify cell behaviors that potentially contribute to the development of these holes, time-lapse experiments were performed from the 3- to 10-somite (when holes appear) stages. The analysis of cell migration revealed that in wild-type embryos, endodermal cells in the anterior region migrated mostly anteriorly, with little lateral migration (consistent with participation in the process of extension), whereas those in the posterior region migrated medially along fairly straight paths (consistent with participation in the process of convergence) (Fig. 6; supplementary material Movie 3). Although in mil mutants the cells in the anterior region likewise migrated predominantly in the anterior direction, they did so at reduced speed (82% of wild-type total speed). Strikingly, analysis of the paths and directions of cell migration revealed that mil cells tended to migrate away from the midline laterally. Thus, their net anterior migration (extension) was especially compromised, reaching only 50% of the wild-type net anterior speed, whereas the convergence velocity towards the lateral was increased (Fig. 6B-G; supplementary material Movie 4). Similarly, mil cells in the posterior region migrated less directly towards the midline and tended to move in the posterior direction, as reflected by a significantly reduced net speed of convergence and an increase in extension towards the posterior (Fig. 6; supplementary material Movie 4). Interestingly, the total migration velocity of the posterior mil cells was similar to that of control cells. Together, these data showed that S1pr2/Mil function is required for the directed cell migration that underlies C&E movements during segmentation.
Fig. 6.
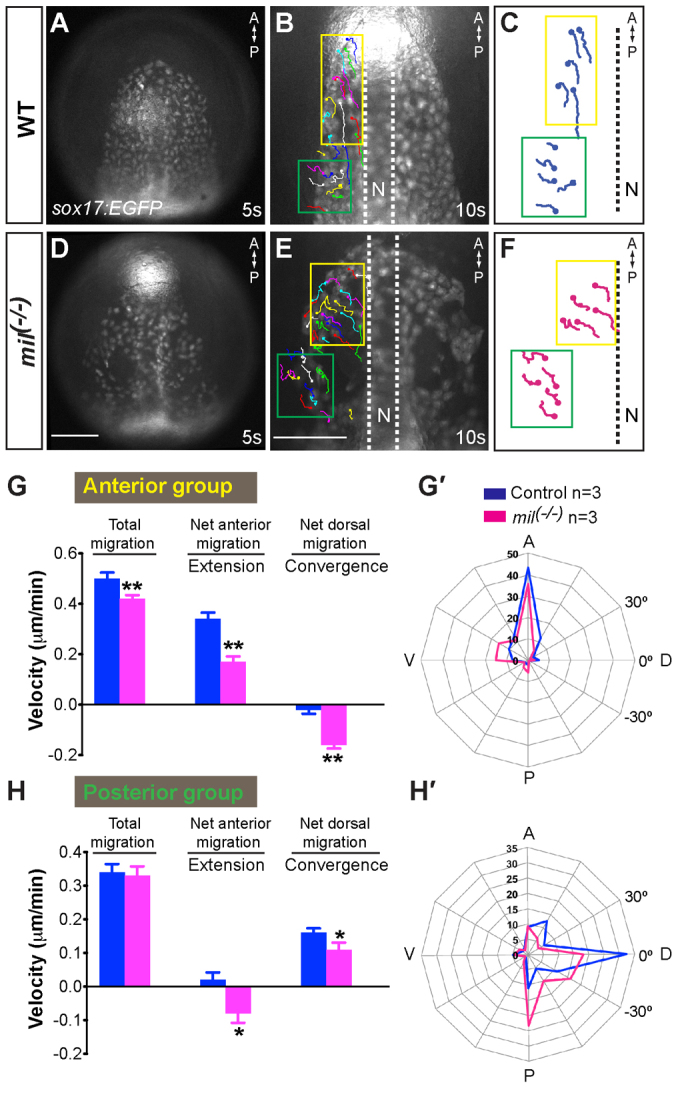
S1pr2 is required for efficient convergence and extension movements of the anterior endoderm. Epifluorescence time-lapse experiments performed on Tg(sox17:EGFP) control (A-C) and mil mutant (E,F) embryos from the 5- to 10-somite stages (supplementary material Movies 3, 4). (A,D) Still images at the 5-somite stage. (B,E) Still image at the 10-somite stage, with the migration tracks of endodermal cells from the 5- to 10-somite stage superimposed. (C,F) Representative tracks delineate routes of the two populations of anterior endodermal cells (yellow is the anterior group, cells migrate anteriorly; green is the posterior group, cells migrate dorsally). (G,H) Total, net anterior (extension) and net dorsal (convergence) migration velocity of the anterior- and posterior-group endodermal cells in wild-type and mil mutant embryos. Data are mean±s.e.m. (G′,H′) Direction of cell migration throughout the time-lapse period (5-minute intervals), shown as the percentage of cells in 30° sectors. *P<0.05 versus control; **P<0.01. Scale bar: 100 μm.
Interestingly, despite the defects in endodermal C&E in S1pr2/Gα13-deficient embryos, the C&E movements of the mesoderm and ectoderm appeared normal, as judged by the length of the body axes (Fig. 1C,E) and the expression patterns of various tissue markers (supplementary material Fig. S3). These findings suggest that S1pr2/Gα13 signaling is required to control the migration of endodermal, but not ectodermal and mesodermal, cells.
Endoderm differentiation appeared to be unaffected in embryos defective for S1pr2/Gα13 signaling, as the endodermal marker sox17 was expressed normally during gastrulation (supplementary material Fig. S2). Moreover, analyses by TUNEL assay indicated that the S1pr2/Gα13 deficiency did not lead to excessive apoptosis in the anterior endoderm (supplementary material Fig. S4). Taken together, these results demonstrate that during segmentation, S1pr2/Gα13 signaling regulates the migration, but not differentiation or apoptosis, of endodermal cells.
The endoderm defects resulting from S1pr2/Gα13 deficiency impair myocardial migration
Given that the endoderm converges as the myocardial precursors migrate towards the embryonic midline, and both processes were defective in S1pr2/Gα13-deficient embryos, we reason that these phenotypes are causally linked. To test this, we simultaneously analyzed endoderm morphology and cardiomyocyte location in Tg(sox17:EGFP/myl7:EGFP) embryos. We found that disrupting any component of the S1pr2/Gα13/RhoGEF pathway resulted in a similarly deformed and widened endodermal sheet, as well as cardia bifida (Fig. 7B,E,F versus 7A). Quantitation of endodermal defects and cardia bifida revealed that in embryos defective for S1pr2/Gα13 signaling both the frequency of endodermal holes and widening of the anterior endodermal sheet corresponded to the severity of cardia bifida (Fig. 7H,I). Notably, some of these embryos displayed cardia bifida even in the absence of an endodermal hole (not shown), and the frequency of holes in the endoderm was lower than that of cardia bifida (Fig. 7H). These findings suggest that the endoderm deformity may not be the sole defect underlying cardia bifida, as speculated previously (Osborne et al., 2008). Rather the impairment in endoderm convergence could contribute to cardiac bifida. Whereas overexpression of human Gα13 in wild-type embryos did not induce visible defects in the endoderm (Fig. 7D,I), its expression in the gna13ab morphants significantly reduced the frequency of cardia bifida and the incidence of endodermal holes, and decreased endoderm width and the distance between the two populations of myocardial precursors (Fig. 7C,H,I). Thus, there is a strong correlation between the endodermal and myocardial defects.
Fig. 7.

Disrupting the S1pr2/Gα13/RhoGEF signaling pathway causes defects in endoderm morphogenesis, as well as in myocardial migration. (A-F) Epifluorescence images of the anterior regions of the endoderm in 26 hpf, double-transgenic Tg(sox17:EGFP)/(myl7:EGFP) embryos. (A) Control uninjected siblings. (B-F) Embryos injected with: the gnb13ab MOs alone (B); the gnb13ab MOs plus the GNA13 RNA (C); the GNA13 RNA alone (D); the s1pr2 MO (E); and the Arhgef11RGS RNA (F). Dorsoanterior views with anterior upwards; yellow dots indicate cardiomyocytes; yellow lines (same length) indicate the width of the anterior endodermal sheet; blue asterisks indicate endodermal holes. (G) A proposed model for how S1pr2 regulates myocardial migration (see text for details). (H) Frequencies of incidences of endodermal holes and cardia bifida in 26 hpf embryos injected with the indicated MOs and RNAs. (I) Endodermal width and the distance between two populations of cardiomyocytes in 26 hpf embryos (obtained from the same pair of fish) injected with the indicated MOs and RNAs. *P< 0.01 versus control; **P<0.01 versus gna13ab MOs; #P>0.05 versus control. Data are mean±s.e.m. Scale bar: 100 μm.
Importantly, although we detected low levels of s1pr2 and gna13ab expression in cardiomyocytes at the 20-somite stage (Fig. 4C,D), expression of Gα13 specifically in cardiomyocytes (using the myl7 promoter) failed to rescue cardia bifida caused by global inhibition of Gα13 (injection of gna13ab MOs; supplementary material Fig. S5). These data suggest that S1pr2/Gα13 signaling in the endoderm, but not cardiomyocytes, is important for myocardial migration. Considering that the defects in endoderm convergence in S1pr2/Gα13-deficient embryos occur at mid-segmentation (8 somites, 13 hpf, Fig. 5), which is well before myocardial migration takes place (16-19 hpf) (Stainier, 2001; Holtzman et al., 2007), we hypothesize that the endodermal defects contribute to the myocardial defects. To assess directly the effects of endodermal defects on myocardial migration, we performed transplantation experiments as described in Fig. 5K, except that Tg(sox17:EGFP/myl7:EGFP) embryos were used as hosts, and endoderm morphology and cardiomyocyte locations were analyzed at 26-28 hpf (Fig. 8A). High-resolution 3D reconstruction of confocal images confirmed that donor endodermal cells were not incorporated into the myocardium (supplementary material Fig. S6); thus, the phenotypes resulting from endoderm transplantation were not due to the presence of donor cells in the myocardium.
Fig. 8.

S1pr2/Gα13 signaling functions autonomously within the endoderm to regulate myocardial migration. (A) The cell transplantation procedures. Sox32-overexpressing and rhodamine-dextran labeled wild-type or gna13ab or s1pr2 morphant donor cells were transplanted into Tg(sox17:EGFP)/(myl7:EGFP) host embryos. In the case of hosts to be injected with wild-type donor cells, the embryos were injected with gna13ab MOs. (B-G″) Epifluorescence images of the anterior region of Tg(sox17:EGFP)/(myl7:EGFP) hosts at 26-28 hpf, showing the morphology of the anterior endoderm and cardiomyocytes. (B-C″) Wild-type donor cells were transplanted into gna13ab morphant hosts. (D-E″) gna13ab morphant donor cells were transplanted into wild-type hosts. (F-G″) s1pr2 morphant donor cells were transplanted into wild-type hosts. (B-G) Rhodamine-dextran labeling donor cells. (B′-G′) GFP-expressing anterior endoderm and cardiomyocytes. (B″-G″) Merged images of B′-G′ and B-G. Dorsoanterior view with anterior upwards; yellow dots indicate cardiomyocytes; white asterisks indicate endodermal holes; the number of embryos is indicated at the bottom left of the ‘donor’ panel; average endodermal width is shown at the bottom right of the ‘merge’ panel; #P<0.001 versus the corresponding control groups. Scale bars: 100 μm.
When wild-type endodermal cells were transplanted into gna13ab morphants, all the embryos (n=46) in which the endoderm did not include donor wild-type cells displayed cardia bifida (not shown), whereas the 21.4% of embryos (n=15/70) in which the endoderm included wild-type donor cells had a single heart (Fig. 8B). In the rescued embryos (15/70), the majority of the anterior endoderm was replaced with wild-type donor cells (red cells), and the endodermal sheet lacked holes and was significantly narrower than that in non-rescued embryos. In the remaining embryos with cardia bifida (55/70), the anterior endoderm was not, or was only partially, populated by wild-type donor cells (Fig. 8C versus 8B). Strikingly, whereas all wild-type embryos (n=42) in which endoderm transplanted with wild-type donor cells exhibited single hearts (supplementary material Fig. S6; not shown), 60.5% of those transplanted with cells defective for gna13ab (n=23/38; Fig. 8D) and 61% of those transplanted with cells defective for s1pr2 (n=11/18; Fig. 8F; supplementary material Fig. S6) displayed cardia bifida. Notably, in the wild-type hosts with cardia bifida, a large proportion of the anterior endoderm contained morphant cells, and the overall width of the anterior endoderm was significantly greater than that in embryos with one heart (Fig. 8D,F versus 8E,G). In addition, in some of these wild-type hosts, holes were present in regions highly populated by transplanted morphant cells (Fig. 8F, white asterisks), suggesting that the endodermal phenotypes resulted from defects within the endoderm rather than from defects in the surrounding tissues. Overall, these experiments indicate that S1pr2 and Gα13 function autonomously in the endoderm, and that endodermal defects are sufficient to impair myocardial migration.
DISCUSSION
Gα13 acts downstream of S1pr2 to regulate myocardial migration through a RhoGEF-dependent pathway
In this study, we have elucidated the signaling mechanism by which S1pr2 regulates myocardial migration: S1pr2 activates a Gα13/RhoGEF-dependent pathway. We show that depletion of Gα13 results in cardia bifida and tail blistering (Fig. 1), phenotypes that are reminiscent of those in embryos deficient for either S1pr2 or the S1P transporter Spns2 (Kupperman et al., 2000; Osborne et al., 2008; Kawahara et al., 2009), suggesting that Gα13 is involved in the S1P signaling pathway. Indeed, overexpression of Gα13 can significantly rescue both cardia bifida and tail blistering that results from MO-mediated inhibition of S1pr2 (Fig. 2). Furthermore, our epistasis experiments demonstrate that inhibiting Gα13 can largely suppress the gastrulation defects induced by S1pr2 overexpression (Fig. 3), and that Gα13 and S1pr2 have synergistic effects on myocardial migration and tail fin development (supplementary material Table S1). Together, these results indicate that Gα13 and S1pr2 function in the same genetic pathway, and that Gα13 acts downstream of S1pr2 to regulate myocardial migration.
S1pr2 belongs to the S1P family of G protein-coupled receptors (S1P1-S1P5), which are activated by S1P (Skoura and Hla, 2009). In vitro studies in mammalian cultured cells show that S1pr2 can couple to various G protein isoforms, including Gαq, Gαi and Gα12/13, but that its coupling to Gα12/13 is the most efficient (Ancellin and Hla, 1999; Windh et al., 1999). However, we found that only interference with Gα13 results in cardia bifida (not shown), indicating that Gα13 is the predominant G-protein isoform that mediates S1pr2-regulated myocardial migration. Interestingly, the finding that Gα12 is only partially redundant with Gα13 with respect to regulating myocardial migration suggests that the functions of these two G protein isoforms are tissue specific, as our previous findings on gastrulation movements of zebrafish indicated that functions of Gα12 and Gα13 are fully redundant in that context. These findings are consistent with those from mouse studies: Gα13-deficient mice die at E9.5 owing to defective angiogenesis, whereas mice lacking Gα12 have no overt phenotypes (Offermanns et al., 1997), but mice doubly deficient for Gα12 and Gα13 die at an earlier stage (E8.5), and mice that are deficient for Gα12 and carry onlyone allele of Gα13 also die in utero (Offermanns, 2001; Gu et al., 2002). Thus, Gα12 and Gα13 appear to have differential functions in developmental processes.
In cultured mammalian cells, Gα12/13 function primarily through a RhoGEF-regulated RhoA signaling pathway (Kozasa et al., 1998; Hart et al., 2000; Worzfeld et al., 2008). We showed previously that Gα12/13 also signal through a RhoGEF/Rho-dependent pathway to regulate epiboly in zebrafish (Lin et al., 2009). Our current data indicate that Gα13 additionally employs this conserved signaling pathway to control myocardial migration. We show that interference with Arhgef11 function leads to cardia bifida and tail blistering, as do defects in S1pr2 and Gα13 signaling (Fig. 3). Moreover, simultaneous partial inhibition of the functions of S1pr2 and Arhgef11 had synergistic effects on myocardial migration (supplementary material Table S1). Finally, interference with Arhgef11 function suppresses S1pr2-induced gastrulation defects (Fig. 3E), suggesting that S1pr2 signals through Arhgef11 to regulate the development of multiple tissues in zebrafish.
S1pr2/Gα13/PDZRhoGEF signaling is required for convergent movement of the anterior endoderm during segmentation
Previous work suggests that S1pr2 may function within the endoderm to regulate myocardial migration (Osborne et al., 2008). Consistent with this notion, we found the s1pr2 transcript is highly enriched in the endoderm (Fig. 4), suggesting that S1pr2 has specific roles in the endoderm. Indeed, in addition to the appearance of endodermal holes in S1P-deficient embryos, as previously reported (Osborne et al., 2008), our analyses of the endoderm morphology of S1pr2/Gα13-deficient embryos and transplanted embryos indicate that S1pr2/Gα13 signaling is autonomously required for convergent movement of the endoderm, a defect that was not identified and appreciated previously (Figs 5, 6, 7, 8).
Consistent with previous findings in mil (Kupperman et al., 2000) and toh (Kawahara et al., 2009) mutants, we found that S1P/Gα13 signaling is not required for the C&E movements of mesodermal cells, suggesting that this signaling pathway functions specifically in the endoderm. During zebrafish embryonic development, the endodermal cells display unique migratory characteristics distinct from those of mesodermal cells, and require different signaling pathways (Warga and Nüsslein-Volhard, 1999; Pezeron et al., 2008). During gastrulation, endoderm migration is controlled by chemokine signaling (mediated by Cxcl12b and its receptor Cxcr4a), and this is not required for the migration of mesodermal cells (Mizoguchi et al., 2008; Nair and Schilling, 2008). Although Wnt/PCP and Vegfc signaling have been shown to regulate morphology of the anterior endoderm during segmentation (Ober et al., 2004; Matsui et al., 2005), how these pathways influence endoderm development has not been defined. Thus, the mechanisms whereby endodermal cells migrate during segmentation remain uncharacterized. Additionally, defective Wnt/PCP signaling severely impairs the C&E of mesodermal cells (Matsui et al., 2005), and depletion of Vegfc function affects endoderm morphogenesis and specification (Ober et al., 2004). Thus, our findings that S1pr2/Gα13 signaling is required for endoderm migration at segmentation represent a novel regulatory pathway.
Our time-lapse experiments indicate that S1pr2 signaling is required for the efficient directed migration that underlies the C&E movements of the endoderm (Fig. 6). Further studies are needed to dissect the mechanisms by which S1pr2/Gα13 signaling controls the migratory behaviors of endodermal cells. The presence of holes lacking endodermal cells in S1pr2/Gα13-deficient embryos is intriguing. Given that neither dying cells nor an excessive number of apoptotic cells were found in the endoderm of S1pr2/Gα13-deficient embryos (supplementary material Fig. S4; not shown), this phenomenon is unlikely to be a consequence of cell death/apoptosis. Time-lapse experiments suggest that abnormal migration of the cells (away from the midline) could contribute to the formation of holes (Fig. 6; supplementary material Movies 3, 4). Furthermore, given that the endoderm developed as a cohesive sheet from mid-segmentation (not shown), defects in cell-cell adhesion may also contribute. Further studies will be required to address this possibility.
The role of endoderm convergence in myocardial migration
Endoderm convergence and the migration of myocardial precursors to the midline both occur during segmentation, leaving open the possibility that these two events are linked. Indeed, we found that the extent of endodermal convergence defects correlates with the severity of cardia bifida, and that the cardiac and endodermal defects in gna13ab morphants are simultaneously rescued by the overexpression of Gα13a (Fig. 7). Moreover, our endoderm transplantation assays reveal that while the replacement of anterior endoderm in gna13ab morphants with wild-type endodermal cells simultaneously rescues the defects of both endoderm and myocardial migration, the transplantation of endoderm defective for either S1pr2 or Gα13 into wild-type embryos leads to endoderm defects as well as to cardia bifida (Fig. 8). These data support the notion that S1pr2 and Gα13 function within the endoderm to influence myocardial migration during heart-tube formation, and that impaired endoderm convergence is likely to be the root cause of the myocardial defects observed in the context of S1pr2/Gα13 deficiency. In support of a role for S1pr2/Gα13 in the microenvironment, we found that stably overexpressing Gα13a specifically in cardiomyocytes fails to rescue either cardia bifida or the endodermal defects in gna13ab morphants (supplementary material Fig. S5; not shown). Interestingly, a recent study indicates that S1pr2 signaling in the endoderm is crucial for jaw development in zebrafish (Balczerski et al., 2012), further supporting the roles of this signaling pathway in endoderm development.
The fact that the endoderm is crucial for regulating myocardial-precursor migration in a variety of organisms, including Xenopus, zebrafish, chick and mouse, is well recognized, yet the underlying mechanisms remain elusive (Lough and Sugi, 2000; Stainier, 2001). The endoderm has been shown to produce growth factors such as BMP2 and FGF8, and thereby to control the specification and differentiation of cardiac mesoderm, and indirectly influence myocardial migration (Nascone and Mercola, 1995; Schultheiss et al., 1995; Schultheiss et al., 1997; Andree et al., 1998; Lough and Sugi, 2000; Alsan and Schultheiss, 2002). However, it is unlikely that this mechanism accounts for the cardia bifida in embryos defective for S1pr2/Gα13 signaling because cardiomyocytes differentiate normally in these embryos (Fig. 1G-J) (Kupperman et al., 2000). Based on the fact that the endoderm is adjacent to the myocardial mesoderm and that myocardial precursors fail to migrate in the absence of the endoderm, the endoderm has been proposed to serve as a physical substrate that enables the myocardial cells to migrate (David and Rosa, 2001). Alternatively, it may produce migratory cues to guide the migration of cardiomyocytes towards midline (David and Rosa, 2001), although such factors have not been identified. Recent studies in chick suggest that endoderm migration may provide an active mechanical force that pulls the myocardial cells towards the midline, and that cardiomyocytes exhibit minimal autonomous migration during this process (Cui et al., 2009; Varner and Taber, 2012). Our finding of a correlation between impaired endoderm convergence and cardia bifida in zebrafish embryos defective for S1pr2/Gα13 signaling is consistent with such a mechanical role for the anterior endoderm in myocardial migration. Future work is required to determine whether the endoderm indeed exerts such an effect on cardiomyocytes, and how it does so.
In summary, the present study delineates the signaling pathway whereby S1pr2 regulates myocardial migration (Fig. 7G). Specifically, we show that S1pr2 functions through a Gα13/RhoGEF-dependent pathway to regulate myocardial migration. Moreover, we demonstrate that S1pr2/Gα13 signaling is required autonomously for the convergence and integrity of the anterior endoderm, and that convergent movement of the endoderm is crucial for the medial migration of myocardial precursors. This work represents an important step towards understanding the crucial roles of the endoderm in promoting myocardial migration during vertebrate heart development. Studies are under way to further elucidate how S1pr2/Gα13 signaling regulates endoderm morphogenesis, and how the endoderm in turn influences the migration of myocardial precursor cells.
Supplementary Material
Acknowledgments
We thank Stephanie Woo (University of California at San Francisco) for providing reagents; Sean Megason and Fengzh Xiong (Harvest Medical School) for providing mounting mold; and Hui Xu for generating the myl7:gna13a-IRES-nlsGFP construct. We thank Debbie Yelon (University of California at San Diego), Lila Solnica-Krezel (Washington University School of Medicine), and Songhai Chen, Robert Cornell and Tina Tootle (University of Iowa) for critical comments on the manuscript.
Footnotes
Funding
This study is supported by grants from the National Institutes of Health [NIH/NCRRK99R00RR024119] and American Heart Association [AHA12GRNT11670009], and by a 2012 Carver Medical Research Initiative grant, University of Iowa (to F.L.). Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.085340/-/DC1
References
- Alexander J., Rothenberg M., Henry G. L., Stainier D. Y. (1999). casanova plays an early and essential role in endoderm formation in zebrafish. Dev. Biol. 215, 343-357 [DOI] [PubMed] [Google Scholar]
- Alsan B. H., Schultheiss T. M. (2002). Regulation of avian cardiogenesis by Fgf8 signaling. Development 129, 1935-1943 [DOI] [PubMed] [Google Scholar]
- Ancellin N., Hla T. (1999). Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J. Biol. Chem. 274, 18997-19002 [DOI] [PubMed] [Google Scholar]
- Andree B., Duprez D., Vorbusch B., Arnold H. H., Brand T. (1998). BMP-2 induces ectopic expression of cardiac lineage markers and interferes with somite formation in chicken embryos. Mech. Dev. 70, 119-131 [DOI] [PubMed] [Google Scholar]
- Bakkers J. (2011). Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 91, 279-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczerski B., Matsutani M., Castillo P., Osborne N., Stainier D. Y., Crump J. G. (2012). Analysis of sphingosine-1-phosphate signaling mutants reveals endodermal requirements for the growth but not dorsoventral patterning of jaw skeletal precursors. Dev. Biol. 362, 230-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel L. K., Raymond P. A. (1990). Improved method for obtaining 3-microns cryosections for immunocytochemistry. J. Histochem. Cytochem. 38, 1383-1388 [DOI] [PubMed] [Google Scholar]
- Buhl A. M., Johnson N. L., Dhanasekaran N., Johnson G. L. (1995). Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631-24634 [DOI] [PubMed] [Google Scholar]
- Chung W. S., Stainier D. Y. (2008). Intra-endodermal interactions are required for pancreatic beta cell induction. Dev. Cell 14, 582-593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C., Cheuvront T. J., Lansford R. D., Moreno-Rodriguez R. A., Schultheiss T. M., Rongish B. J. (2009). Dynamic positional fate map of the primary heart-forming region. Dev. Biol. 332, 212-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David N. B., Rosa F. M. (2001). Cell autonomous commitment to an endodermal fate and behaviour by activation of Nodal signalling. Development 128, 3937-3947 [DOI] [PubMed] [Google Scholar]
- Evans S. M., Yelon D., Conlon F. L., Kirby M. L. (2010). Myocardial lineage development. Circ. Res. 107, 1428-1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavito-Aguilar Z. V., Riley H. E., Yelon D. (2010). Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development 137, 3215-3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J. L., Muller S., Mancino V., Offermanns S., Simon M. I. (2002). Interaction of Gα12 with Gα13 and Gαq signaling pathways. Proc. Natl. Acad. Sci. USA 99, 9352-9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart M. J., Roscoe W., Bollag G. (2000). Activation of Rho GEF activity by Gα13. Methods Enzymol. 325, 61-71 [DOI] [PubMed] [Google Scholar]
- Heisenberg C. P., Tada M., Rauch G. J., Saude L., Concha M. L., Geisler R., Stemple D. L., Smith J. C., Wilson S. W. (2000). Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 405, 76-81 [DOI] [PubMed] [Google Scholar]
- Holtzman N. G., Schoenebeck J. J., Tsai H. J., Yelon D. (2007). Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development 134, 2379-2386 [DOI] [PubMed] [Google Scholar]
- Horne-Badovinac S., Lin D., Waldron S., Schwarz M., Mbamalu G., Pawson T., Jan Y., Stainier D. Y., Abdelilah-Seyfried S. (2001). Positional cloning of heart and soul reveals multiple roles for PKC lambda in zebrafish organogenesis. Curr. Biol. 11, 1492-1502 [DOI] [PubMed] [Google Scholar]
- Huang C. J., Tu C. T., Hsiao C. D., Hsieh F. J., Tsai H. J. (2003). Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 228, 30-40 [DOI] [PubMed] [Google Scholar]
- Jessen J. R., Topczewski J., Bingham S., Sepich D. S., Marlow F., Chandrasekhar A., Solnica-Krezel L. (2002). Zebrafish trilobite identifies new roles for Strabismus in gastrulation and neuronal movements. Nat. Cell Biol. 4, 610-615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara A., Nishi T., Hisano Y., Fukui H., Yamaguchi A., Mochizuki N. (2009). The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323, 524-527 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310 [DOI] [PubMed] [Google Scholar]
- Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998). p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 280, 2109-2111 [DOI] [PubMed] [Google Scholar]
- Kuo C. T., Morrisey E. E., Anandappa R., Sigrist K., Lu M. M., Parmacek M. S., Soudais C., Leiden J. M. (1997). GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 11, 1048-1060 [DOI] [PubMed] [Google Scholar]
- Kupperman E., An S., Osborne N., Waldron S., Stainier D. Y. (2000). A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature 406, 192-195 [DOI] [PubMed] [Google Scholar]
- Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P., Chien C. B. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088-3099 [DOI] [PubMed] [Google Scholar]
- Lin F., Sepich D. S., Chen S., Topczewski J., Yin C., Solnica-Krezel L., Hamm H. (2005). Essential roles of Gα12/13 signaling in distinct cell behaviors driving zebrafish convergence and extension gastrulation movements. J. Cell Biol. 169, 777-787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F., Chen S., Sepich D. S., Panizzi J. R., Clendenon S. G., Marrs J. A., Hamm H. E., Solnica-Krezel L. (2009). Gα12/13 regulate epiboly by inhibiting E-cadherin activity and modulating the actin cytoskeleton. J. Cell Biol. 184, 909-921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lough J., Sugi Y. (2000). Endoderm and heart development. Dev. Dyn. 217, 327-342 [DOI] [PubMed] [Google Scholar]
- Matsui T., Raya A., Kawakami Y., Callol-Massot C., Capdevila J., Rodriguez-Esteban C., Izpisua Belmonte J. C. (2005). Noncanonical Wnt signaling regulates midline convergence of organ primordia during zebrafish development. Genes Dev. 19, 164-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Raya A., Callol-Massot C., Kawakami Y., Oishi I., Rodriguez-Esteban C., Izpisua Belmonte J. C. (2007). miles-apart-Mediated regulation of cell-fibronectin interaction and myocardial migration in zebrafish. Nat. Clin. Pract. Cardiovasc. Med. 4 Suppl. 1, S77-S82 [DOI] [PubMed] [Google Scholar]
- Megason S. G. (2009). In toto imaging of embryogenesis with confocal time-lapse microscopy. Methods Mol. Biol. 546, 317-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T., Verkade H., Heath J. K., Kuroiwa A., Kikuchi Y. (2008). Sdf1/Cxcr4 signaling controls the dorsal migration of endodermal cells during zebrafish gastrulation. Development 135, 2521-2529 [DOI] [PubMed] [Google Scholar]
- Molkentin J. D., Lin Q., Duncan S. A., Olson E. N. (1997). Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 11, 1061-1072 [DOI] [PubMed] [Google Scholar]
- Nair S., Schilling T. F. (2008). Chemokine signaling controls endodermal migration during zebrafish gastrulation. Science 322, 89-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascone N., Mercola M. (1995). An inductive role for the endoderm in Xenopus cardiogenesis. Development 121, 515-523 [DOI] [PubMed] [Google Scholar]
- Nijmeijer R. M., Leeuwis J. W., DeLisio A., Mummery C. L., Chuva de Sousa Lopes S. M. (2009). Visceral endoderm induces specification of cardiomyocytes in mice. Stem Cell Res. 3, 170-178 [DOI] [PubMed] [Google Scholar]
- Ober E. A., Olofsson B., Makinen T., Jin S. W., Shoji W., Koh G. Y., Alitalo K., Stainier D. Y. (2004). Vegfc is required for vascular development and endoderm morphogenesis in zebrafish. EMBO Rep. 5, 78-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S. (2001). In vivo functions of heterotrimeric G-proteins: studies in Galpha-deficient mice. Oncogene 20, 1635-1642 [DOI] [PubMed] [Google Scholar]
- Offermanns S., Mancino V., Revel J. P., Simon M. I. (1997). Vascular system defects and impaired cell chemokinesis as a result of Gα13 deficiency. Science 275, 533-536 [DOI] [PubMed] [Google Scholar]
- Osborne N., Brand-Arzamendi K., Ober E. A., Jin S. W., Verkade H., Holtzman N. G., Yelon D., Stainier D. Y. (2008). The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr. Biol. 18, 1882-1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzi J. R., Jessen J. R., Drummond I. A., Solnica-Krezel L. (2007). New functions for a vertebrate Rho guanine nucleotide exchange factor in ciliated epithelia. Development 134, 921-931 [DOI] [PubMed] [Google Scholar]
- Pezeron G., Mourrain P., Courty S., Ghislain J., Becker T. S., Rosa F. M., David N. B. (2008). Live analysis of endodermal layer formation identifies random walk as a novel gastrulation movement. Curr. Biol. 18, 276-281 [DOI] [PubMed] [Google Scholar]
- Reiter J. F., Alexander J., Rodaway A., Yelon D., Patient R., Holder N., Stainier D. Y. (1999). Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 13, 2983-2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr S. (2006). Heart and soul/PRKCi and nagie oko/Mpp5 regulate myocardial coherence and remodeling during cardiac morphogenesis. Development 133, 107-115 [DOI] [PubMed] [Google Scholar]
- Rosenquist G. C. (1970). Cardia bifida in chick embryos: anterior and posterior defects produced by transplanting tritiated thymidine-labeled grafts medial to the heart-forming regions. Teratology 3, 135-142 [DOI] [PubMed] [Google Scholar]
- Schultheiss T. M., Xydas S., Lassar A. B. (1995). Induction of avian cardiac myogenesis by anterior endoderm. Development 121, 4203-4214 [DOI] [PubMed] [Google Scholar]
- Schultheiss T. M., Burch J. B., Lassar A. B. (1997). A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11, 451-462 [DOI] [PubMed] [Google Scholar]
- Serbedzija G. N., Chen J. N., Fishman M. C. (1998). Regulation in the heart field of zebrafish. Development 125, 1095-1101 [DOI] [PubMed] [Google Scholar]
- Skoura A., Hla T. (2009). Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc. Res. 82, 221-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford D., White R. J., Kinkel M. D., Linville A., Schilling T. F., Prince V. E. (2006). Retinoids signal directly to zebrafish endoderm to specify insulin-expressing beta-cells. Development 133, 949-956 [DOI] [PubMed] [Google Scholar]
- Stainier D. Y. (2001). Zebrafish genetics and vertebrate heart formation. Nat. Rev. Genet. 2, 39-48 [DOI] [PubMed] [Google Scholar]
- Stainier D. Y., Fouquet B., Chen J. N., Warren K. S., Weinstein B. M., Meiler S. E., Mohideen M. A., Neuhauss S. C., Solnica-Krezel L., Schier A. F., et al. (1996). Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 123, 285-292 [DOI] [PubMed] [Google Scholar]
- Suzuki N., Hajicek N., Kozasa T. (2009). Regulation and physiological functions of Gα12/13-mediated signaling pathways. Neurosignals 17, 55-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 [DOI] [PubMed] [Google Scholar]
- Trinh L. A., Stainier D. Y. (2004). Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev. Cell 6, 371-382 [DOI] [PubMed] [Google Scholar]
- Varner V. D., Taber L. A. (2012). Not just inductive: a crucial mechanical role for the endoderm during heart tube assembly. Development 139, 1680-1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villefranc J. A., Amigo J., Lawson N. D. (2007). Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn. 236, 3077-3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warga R. M., Nüsslein-Volhard C. (1999). Origin and development of the zebrafish endoderm. Development 126, 827-838 [DOI] [PubMed] [Google Scholar]
- Windh R. T., Lee M. J., Hla T., An S., Barr A. J., Manning D. R. (1999). Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the Gi, Gq, and G12 families of heterotrimeric G proteins. J. Biol. Chem. 274, 27351-27358 [DOI] [PubMed] [Google Scholar]
- Withington S., Beddington R., Cooke J. (2001). Foregut endoderm is required at head process stages for anteriormost neural patterning in chick. Development 128, 309-320 [DOI] [PubMed] [Google Scholar]
- Worzfeld T., Wettschureck N., Offermanns S. (2008). Gα12/13-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci. 29, 582-589 [DOI] [PubMed] [Google Scholar]
- Yelon D. (2001). Cardiac patterning and morphogenesis in zebrafish. Dev. Dyn. 222, 552-563 [DOI] [PubMed] [Google Scholar]
- Yelon D., Horne S. A., Stainier D. Y. (1999). Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev. Biol. 214, 23-37 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.