

Abstract
Of the over 22 million protein sequences in the nonredundant TrEMBL database, fewer than 1% have experimentally confirmed functions. Structure-based methods have been used to predict enzyme activities from experimentally determined structures; however, for the vast majority of proteins, no such structures are available. Here, homology models of a functionally uncharacterized amidohydrolase from Agrobacterium radiobacter K84 (Arad3529) were computed based on a remote template structure. The protein backbone of two loops near the active site was remodeled, resulting in four distinct active site conformations. Substrates of Arad3529 were predicted by docking of 57672 high-energy intermediate (HEI) forms of 6440 metabolites against these four homology models. Based on docking ranks and geometries, a set of modified pterins were suggested as candidate substrates for Arad3529. The predictions were tested by enzymology experiments, and Arad3529 deaminated many pterin metabolites (substrate, kcat/Km [M−1s−1]): formylpterin, 5.2 × 106; pterin-6-carboxylate, 4.0 × 106; pterin-7-carboxylate, 3.7 × 106; pterin, 3.3 × 106; hydroxymethylpterin, 1.2 × 106; biopterin, 1.0 × 106; D-(+)-neopterin, 3.1 × 105; isoxanthopterin, 2.8 × 105; sepiapterin, 1.3 × 105; folate, 1.3 × 105, xanthopterin, 1.17 × 105; 7,8-dihydrohydroxymethylpterin, 3.3 × 104. While pterin is a ubiquitous oxidative product of folate degradation, genomic analysis suggests that the first step of an undescribed pterin degradation pathway is catalyzed by Arad3529. Homology model-based virtual screening, especially with modeling of protein backbone flexibility, may be broadly useful for enzyme function annotation and discovering new pathways and drug targets.
INTRODUCTION
With increasing availability of genomic sequences, a pressing challenge in biology is a reliable assignment of function to the proteins encoded by these genomes. Functional annotation of an uncharacterized protein can be conveniently accomplished by matching its sequence to that of a characterized protein1,2. However, this strategy is often inaccurate and imprecise3,4. Conservatively, over 50% of the sequences in the public databases have uncertain, unknown, or incorrectly annotated functions5. Annotation of enzymes in functionally diverse superfamilies is particularly challenging6.
A promising method to functional assignment is the identification of the substrate by docking potential substrates against the binding site of an experimentally determined enzyme structure7,8. This method was used to predict the substrates of Tm0936 from Thermotoga maritima9. High-energy intermediate forms of thousands of candidate metabolites were docked to the X-ray structure of Tm0936, and those highly ranked by docking energy score were tested experimentally, confirming a significant deaminase activity against S-adenosylhomocysteine (SAH). This approach has been subsequently applied to three other enzymes for activity determination10–12. These studies suggest that structure-based docking might be a useful tool for enzyme function annotation, when an experimentally determined atomic structure of the enzyme is available.
Often, however, an enzyme of unknown function has no experimentally determined three-dimensional structure. In such situations, a three-dimensional model of the target sequence can be computed by homology modeling if a template structure of a related protein is known13. Currently, the total fraction of protein sequences in a typical genome for which reliable homology models can be obtained varies from 20–75%, increasing the number of structurally characterized protein sequences by more than two orders of magnitude relative to the PDB14. Therefore, homology models can, in principle, greatly extend the applicability of virtual screening for ligand discovery15–20. For example, this strategy was successfully employed, in the prediction of function for Bc0371 from Bacillus cereus as an N-succinyl arginine/lysine racemase by docking dipeptides and N-succinyl amino acids to a homology model of Bc0371 based on the X-ray structure of the closest structural homologue, L-alanine-D/L-glutamate epimerase21.
The accuracy of a homology model can be estimated from the target-template sequence identity. When the sequence identity exceeds 30%, a reliable alignment can typically be constructed, and the resulting models may be useful for virtual screening. When the sequence identity decreases much below 30%, the target structure often deviates significantly from that of the template, resulting in large errors in side chain packing, loop conformations, core backbone conformations, alignment, and even fold assignment. Unfortunately, for many proteins from newly sequenced genomes, only distantly related template structures are available. Hence, it is of pressing interest to develop modeling and docking methods that account for backbone variation between homologues, so that homology models based on distant templates can be used for annotation of protein function.
The functionally diverse amidohydrolase superfamily (AHS) of enzymes provides a test case for developing robust computational methods for function identification of uncharacterized enzymes. Enzymes within this superfamily possess a mononuclear or binuclear metal center embedded within a (β/α)8-barrel structural fold22, and catalyze diverse reactions, including ester and amide hydrolysis, nucleic acid deamination, double bond hydration, carbohydrate isomerization, and decarboxylation23. To date, more than 24,000 unique bacterial proteins have been assigned to this superfamily, segregating into 24 clusters of orthologous groups (COGs)24,25.
One of these COGs, cog0402, contains approximately 1400 distinct proteins. At BLAST26 E-value of 10−70, cog0402 can be divided into 14 major groups of similar sequences27,28 (Figure 1). Although 40% of the proteins in cog0402 can be reliably annotated as catalyzing the deamination of an aromatic base or a similar functional group11,29–34, the rest remain uncharacterized. Here, we have attempted to predict the substrate profile for enzymes of unknown function represented by Group 14 of cog0402, containing approximately 140 proteins. In particular, Arad3529 from Agrobacterium radiobacter K84 was cloned, expressed, and purified to homogeneity. Substrates of Arad3529 were predicted by docking high-energy intermediate forms of candidate metabolites to the homology models constructed for Arad3529. High-ranking predictions were acquired and tested as candidate substrates by enzymology. The sequence identity between Arad3529 and its closest structurally characterized homologue, cytosine deaminase from E. coli, is only 26%, making the construction of the homology model and subsequent computational docking highly challenging.
Figure 1.

Sequence Similarity Network for cog0402. Protein sequences for cog0402 were retrieved from NCBI, subjected to an All-by-All BLAST to determine overall sequence similarity to all of the other proteins within this network27. Each dot (node) represents an enzyme, and each connection between two nodes (an edge) represents those enzyme pairs that are more closely related than the arbitrary E-value cutoff (10−70). Groups are arbitrarily numbered; those groups with experimentally determined substrate profiles are as follows: (1) S-adenosylhomocysteine/5'-deoxy-5'-methylthioadenosine deaminase (red nodes); (2) guanine deaminase (orange nodes); (4) 8-oxoguanine/isoxanthopterin deaminase (green nodes); (6) cytosine deaminase (light blue nodes); and (8), N-formimino-L-glutamate deiminase (purple nodes).
MATERIALS and METHODS
General
All chemicals were obtained from Sigma-Aldrich unless otherwise specified. 7,8-Dihydro-L-biopterin was purchased from Santa Cruz Biotechnology. Formylpterin, pterin-7-carboxylate, hydroxymethylpterin, xanthopterin, 7,8-dihydrohydroxymethylpterin, and 7,8-dihydroneopterin were purchased from Schirks Laboratories.
Cloning, Expression, and Purification of Arad3529
The gene for Arad3529 was amplified from Agrobacterium radiobacter Strain K84 genomic DNA using 5'-TTAAGAAGGAGATATACCATGTCATACAGTTTCATGTCCCCGCC-3' as the forward primer and 5'-GATTGGAAGTAGAGGTTCTCTGCTGCGCCAATCACGGTGTCCAG-3' as the reverse primer. PCR was performed using KOD Hot Start DNA Polymerase (Novagen). The amplified fragment was cloned into the C-terminal TEV cleavable StrepII-6x-His-tag containing vector, CHS30, by ligationAindependent cloning35.
The Arad3529-CHS30 vector was used to transform BL21(DE3) E. coli containing the pRIL plasmid (Stratagene), which was used to inoculate a 5 mL 2xYT culture containing 25 μg/mL kanamycin and 34 μg/mL chloramphenicol. The culture was allowed to grow overnight at 37 °C in a shaking incubator. The overnight culture was used to inoculate 1 L of PASM-5052 auto-induction media36 containing 150 mM 2-2-bipyridyl, 1.0 mM ZnCl2, and 1.0 mM MnCl2. The culture was placed in a LEX48 airlift fermenter and incubated at 37 °C for 5 hours and then at 22 °C overnight. The culture was harvested and pelleted by centrifugation.
Cells were resuspended in lysis buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, and 10% glycerol) and lysed by sonication. The lysate was clarified by centrifugation at 35,000 × g for 30 minutes. The clarified lysate was loaded onto a 5 mL Strep-Tactin column (IBA), washed with 5 column volumes of lysis buffer, and then eluted in StrepB buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, 10% glycerol, and 2.5 mM desthiobiotin). The eluent was loaded onto a 1.0 mL HisTrap FF column (GE Healthcare), washed with 10 column volumes of lysis buffer, and eluted in buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl, 500 mM imidazole, and 10% glycerol. The purified sample was loaded onto a HiLoad S200 16/60 PR gel filtration column which was equilibrated with SECB buffer (20 mM HEPES pH7.5, 150 mM NaCl, 10% glycerol, and 5 mM DTT). Peak fractions were collected, analyzed by SDS-PAGE, snap frozen in liquid nitrogen, and stored at −80 °C. The purified protein was submitted to ICP-MS for metal content analysis and found to contain 0.8 eq Mn, 0.1 eq Fe, 0.1 eq Ni, and 0.1 eq Zn.
Homology Modeling of Arad3529
The amino acid sequence of Arad3529 from Agrobacterium radiobacter K84 (gi|222086854) was retrieved from the Structure Function Linkage Database (SFLD)37. Homology models of Arad3529 were generated in four steps. First, the primary sequence was submitted to the PSI-BLAST server at NCBI to search for suitable template structures38. Cytosine deaminase (CDA) from E. coli (PDB id: 1K70) is the most closely related sequence of known structure. Cytosine deaminase is from group 6 of cog0402 and shares a 26% sequence identity to Arad3529 (Figure 1). In this structure, the enzyme is complexed with a mechanism-based inhibitor, 4-(S)-hydroxyl-3,4-dihydropyrimidine. Second, a sequence alignment between Arad3529 and cystosine deaminase was computed by MUSCLE39 (Multiple Sequence Comparison by Log-Expectation) (Figure S1). Third, 500 homology models were generated with the standard “automodel” class in MODELLER40, and the model with the best DOPE41 score was selected (Model-1) (Figure 2). Finally, two loop regions (residues 83–89, 174–186) in Model-1 were refined with the “loopmodel” class in MODELLER, resulting in Models-2, 3, and 4 (Figure 3). The “loopmodel” class in MODELLER includes several loop optimization methods, which all rely on scoring functions and optimization protocols adapted for loop modeling42. Side chains in these two loops were optimized using the “side chain prediction” protocol in PLOP43. The Fe2+ ion was included in all modeling steps. The co-crystallized inhibitor from the template structure was included in the third step for the construction of the initial homology model, but was removed from the modeled active site in the last step.
Figure 2.

The active site of E. coli CDA and residues predicted to be important for Arad3529. Residues drawn in white are conserved across all members of cog0402. Thr-66, Gln-156, and Asp-314 in CDA, displayed in green, differed significantly in the sequence of Arad3529 and may be important for substrate binding. Thr-66 corresponds to Lys-85, Gln-156 corresponds to Leu-177, and Asp-314 corresponds to Asn-332. In the X-ray structure of CDA, Gln-156 forms hydrogen bonds with the bound inhibitor; Asp-314 is found within 4 Å of the CDA inhibitor but only forms steric interactions with it; Thr-66 is more distant from the active site. The conversion of a large lysine in Arad3529 suggests that the Lys-85 may reach into the active site and play a role in substrate binding.
Figure 3.

The active site conformations of 4 representative models of Arad3529 used in virtual screening. Model-1 is ranked the best by DOPE score among 500 homology models generated automatically by MODELLER. Model-2, Model-3 and Model-4 are different from Model1 in Loop-1, Loop-2, and both Loop-1 and Loop-2, respectively.
Docking Screen against Arad3529
A high-energy intermediate (HEI) library44,45 that contains 57672 different stereoisomers generated by hydroxide attacking from the re and si faces of prochiral molecules and by exploring all possibilities of protonation state of 6440 KEGG (Kyoto Encyclopedia of Genes and Genomes) molecules46, was screened against the X-ray structure of the cytosine deaminase template (PDB id 1K70) as a control, and each of the four homology models of Arad3529, using DOCK 3.647. The computed poses were subjected to a distance cutoff to ensure that the O− moiety of the HEI portion of the molecule is found within 4 Å of the metal ion in the active site. The top 500 compounds ranked by the docking score (the sum of van der Waals, Poisson–Boltzmann electrostatic, and ligand desolvation penalty terms) were inspected visually to ensure the compatibility of the pose with the amidohydrolase reaction mechanism. The details of the HEI docking library preparation, the molecular docking procedure, and the protocol for analyzing docking results have been previously described9,48–50.
Chemoinformatic analysis of the docking hit-lists
The top 200 highest ranked compounds for each model were combined into ligand sets and used to calculate self-consistency expectation values (E-value) within each set using the Similarity Ensemble Approach (SEA)51,52. Briefly, each ligand was broken down into molecular fingerprints, here ChemAxon path fingerprints. The similarity between molecules is quantified by the number of bits they have in common divided by the total number of bits using the Tanimoto coefficient (Tc). The sum of all pairwise Tc values over a defined cutoff is calculated and compared to what is expected at random between two ligand sets of the same size. The ratio of the calculated sum of Tc values over the sum of Tc values expected at random is divided by the standard deviation of the random similarity to give a Z-score and plotted against an extreme value distribution to yield an E-value. The self similarity between the ligands of the same set provide a metric to how diverse or similar the ligands are based on the E-value. The more significant the E-value (lower) the tighter the chemical space is in the ligand set with likely fewer numbers of distinct chemotypes. Each docking model's self consistency E-value was then compared to the number of true substrates found from each ligand set.
Physical Library Screening
The initial screen of catalytic activity monitored changes in the UV-visible spectrum from 240 – 400 nm after the addition of 1.0 μM Arad3529 to a solution containing 100 μM of the target compound in 20 mM HEPES, pH 7.7 in a 96-well quartz plate using a Molecular Devices Spectramax 384 Plus spectrophotometer. The compounds that exhibited observable spectral change were subjected to more quantitative kinetic assays. On the basis of the docking ranks and compound availability, 11 compounds prioritized by docking were tested as potential substrates, including melamine, cytosine, thioguanine, guanine, adenine, 7-methylguanine, cytidine, 2'-deoxycytidine, pterin, biopterin, and neopterin. In addition, 104 compounds that were substrates of annotated amidohydrolases were tested as potential substrates (see supporting information). After the pterins were identified as substrates, 11 compounds composed of a pteridine ring were added to the screening.
Determination of Kinetic Constants
Once candidate substrates had been identified in parallel by docking and plate-based screening, quantitative kcat and kcat/Km were determined in 20 mM HEPES buffer at pH 7.7. For each compound, the change in the extinction coefficient between the substrate and the deaminated product was determined experimentally by subtraction of the absorbance spectrum of the product from the spectrum of the substrate. The wavelengths (nm) and the differential extinction coefficients, Δε (M−1 cm−1) for each of the compounds utilized in this investigation are as a follows: pterin-6-carboxylate (271 nm, 6211 M−1 cm−1); pterin (255 and 314 nm, 7021 and 1877 M−1 cm−1); biopterin (260 nm, 6527 M−1 cm−1); D-neopterin (320 nm, 4431 M−1 cm−1); isoxanthopterin (350 nm, 2223 M−1 cm−1); sepiapterin (260 nm, 3137 M−1 cm−1); folate (364 nm, 4312 M−1 cm−1); formylpterin (316 nm, 2963 M−1 cm−1), pterin-7-carboxylate (264 nm, 3706 M−1 cm−1); hydroxymethylpterin (264 nm, 7356 M−1 cm−1); xanthopterin (282 nm, 9704 M−1 cm−1); 7,8-dihydrohydroxymethylpterin (536 nm, 3220 M−1 cm−1); and 7,8-dihydroneopterin (282 nm, 9424 M−1 cm−1). The values of kcat and Km were determined by fitting the parameters in equation 1 to the experimental data, using SigmaPlot 11, where v is the initial velocity, Et is enzyme concentration, and A is the substrate concentration.
| (1) |
Confirmation of product formation
100 μM pterin-6-carboxylate was incubated with 150 nM Arad3529 in 50 mM NH4CO3H for 1 hour at 25C. The reaction mixture was filtered with a 30,000 MW cutoff spin column and the flow-through was collected. This flow-through and a sample of 100 μM pterin-6-carboxylate was submitted to ESI- mass spectrometry at the Texas A&M Laboratory for Biological Mass Spectrometry. The reaction showed an m/z change from 206.04 to 207.02, consistent with the product 2,4-dihydroxypteridine-6-carboxylate from the deamination of pterin-6-carboxylate.
RESULTS
Sequence Comparison of Arad3529 with Cytosine Deaminase from E. coli
Cytosine deaminase from E. coli is currently the closest structurally characterized protein to Arad3529. The four metal binding residues in cytosine deaminase (H61, H63, H214, and D313) align well with H80, H82, H231, and D331 from Arad3529, along with two additional residues (H246 and E217 aligned with H263 and E234 in Arad3529, respectively) that function as proton shuttles in cytosine deaminase (Figure S1). Because these active site residues to perform a deamination were all intact in Arad3529, it is very likely also a deaminase. However, in Arad3529 there are three notable changes in the amino acid sequence, relative to that found in most other cytosine deaminases from cog0402. In Arad3529, Thr66, Gln156, and Asp314 from CDA are replaced by Lys85, Leu177, and Asn332, respectively (Figure 2). In CDA from E. coli, Thr66 located near the active site does not appear to hydrogen bond with cytosine. Conversely, Gln156 forms a pair of hydrogen bonds to the carbamoyl moiety of cytosine, while Asp314 is located immediately after the invariant Asp313 and within 4 Å of the bound inhibitor in the CDA structure. The substitutions to the active site residues thus suggest that Arad3529 deaminates a substrate different from cytosine.
Molecular Docking
In the initial homology model of Arad3529 (Model-1), which overall adopts the same backbone conformation as the template, changes are located in two unstructured loop regions that help define the active site (Loop-1: residues 83–89; Loop-2: residues 174–186). Therefore, Loop-1 and Loop-2 were remodeled separately, resulting in Model-2 and Model-3 (Figure 3). Finally, the Loop-1 conformation in Model-2 and the Loop-2 conformation in Model-3 were combined in Model-4. Thus, four homology models were obtained that are distinct from each other in their active site conformations.
Because homology models are constructed from template structures, the models often share some structural features with the templates, and thus recognize similar ligands. Here, however, small molecules that are highly ranked by the template structure (PDB id: 1K70) are unlikely to be the true substrate of the remote homologue. Therefore, before targeting the homology models of Arad3529, the template structure 1K70 was docked against as a negative control. 57672 high-energy intermediates (HEI) were docked into the template structure. As expected, cytosine – the natural substrate of the template – was ranked highly (10th out of 57672), as were several other nucleosides (Table S1). These molecules were deprioritized for testing against Arad3529; we instead looked for molecules with high differential ranks between the models and the template docking screens.
The same HEI database was docked to each of the four homology models of Arad3529. From each model, putative substrates were selected from the top 500 scored molecules (Table 1). The compound sets selected using different homology models are overlapping but distinct. For Model-1, the docking hits were dominated by relatively small compounds with a 1,3,5-triazine skeleton. This pattern is also observed in the docking hits from Model-2. However, compounds composed of the purine or pteridine ring were also favored by Model-2. For Model-3, the 1,3,5-triazine skeleton completely disappeared and the docking hits were dominated by compounds composed of the pteridine ring. Model-4 shared the pteridine pattern with Model-3, but also favored nucleosides having a ribose group.
Table 1.
Virtual screening of the HEI database against four homology models of Arad3529
| Name | KEGG ID | Docking rank |
|||
|---|---|---|---|---|---|
| Model-1 | Model-2 | Model-3 | Model-4 | ||
| melamine | C08737 | 10 | |||
| cyclopropylmelamine | C14147 | 13 | 18 | ||
| diethylatrazine | C06559 | 28 | 32 | ||
| Diisopropylatrazine | C06556 | 29 | 33 | ||
| Diisopropylhydroxyatrazine | C06557 | 31 | |||
| cytosine | C00380 | 52 | 45 | 185 | 126 |
| 2-amino-4-hydroxy-6-hydroxymethyl-7,8-dihydropteridine | C01300 | 108 (N1) | 135 (N1) | 10 (N1) | 12 (N3) |
| thioguanine | C07648 | 11 | |||
| Guanine | C00242 | 20 | |||
| pterin | C00715 | 31 (N3) | 27 (N1) | 102 (N3) | |
| 127 (N1) | |||||
| phenazopyridine | C07429 | 50 | |||
| biopterin | C06313 | 66 (N3) | 14 (N1) | 51 (N1) | |
| 71 (N3) | |||||
| adenine | C00147 | 99 | |||
| neopterin | C05926 | 15 (N1) | 143 (N1) | ||
| 7-methylguanine | C02242 | 23 | |||
| deoxycytidine | C00881 | 49 | |||
| cytidine | C00475 | 75 | |||
| 5'-deoxy-5' -fluorocytidine | C16635 | 132 | |||
| 2-aminoadenosine | C00939 | 206 | |||
The names of three molecules that were found in the control are written in italic. The 2-aminopteridine-4(3H)-one and the 2-aminopteridine-4(1H)-one tautomers were attacked by the hydroxide from the re-face and the si-face, and have the N-1 ring nitrogen and N-3 ring nitrogen protonated by Glu234, resulting in two intermediates named as N1 and N3, respectively.
Chemoinformatic analysis of the docking hit-lists
Based on docking energies alone, it is difficult to prioritize one of these overall hit-lists over another, although the geometry of the interactions does provide guidance (below). One can, however, pose extra-thermodynamic criteria to judge among them. One such is to expect that in a well-behaved docking hit-list the highly ranked molecules will resemble one another, and thus that the hit-list will resembles itself by chemical similarity. We therefore compared the top-ranked 200 molecules for each of the four models of Arad3529 against themselves using the Similarity Ensemble Approach (SEA)51,52. This was quantified by an expectation value (E-value) of the likelihood the self-similarity of each hit list would occur at random. Of the four models, model 3 and model 4 had more self-consistent hit lists (SEA E-value 1.5 × 10−100 and 4.9 × 10−98, respectively) compared to model 1 and model 2 (SEA E-value 2.7 × 10−89 and 1.2 × 10−85, respectively). Comfortingly, the structures of what turned out to be the true pterin substrates of the enzyme were docked in catalytically competent geometries in model 3 and model 4. Naturally, this criterion has nothing to recommend it other than consistency, but it may merit further study as a metric to select multiple possible hit lists for targets adopting different structures.
Substrate Profile for Arad3529
On the basis of commercial availability, 8 compounds predicted to be potential substrates for Arad3529 were tested as enzyme substrates, as were three “negative control” nucleosides that also ranked well against the CDA template (Table 1). Arad3529 was found to deaminate three of the eight: pterin, biopterin, and neopterin with kcat/Km values of greater than 106 M−1 s−1. We docked the ground states of three substrates, pterin, biopterin, and neopterin, to model3. The docking scores of these ground states (−19.29, −27.30, and −23.08, respectively) were substantially higher (worse) than the corresponding HEI states (−53.83, −58.45, and −58.26, respectively), consistent with the catalytic mechanism. The pterin deaminase (PDA) reaction is illustrated in Scheme 1. To investigate the mechanism further, the enzyme was screened with other substituted pterins and was found to deaminate several pterins (Scheme S1). Various substitutions on C6 of the pteridine ring are allowed (Table 2), and with the exception of sepiapterin, the fully aromatic pteridine ring is preferred. 7,8-Dihydroneopterinand 7,8-dihydrobiopterin were deaminated very slowly and the deamination of dihydrofolate or tetrahydrofolate could not be measured (< .0008 s−1). 2,4-Diamino-6-hydroxymethylpteridine was not deaminated.
Scheme 1.
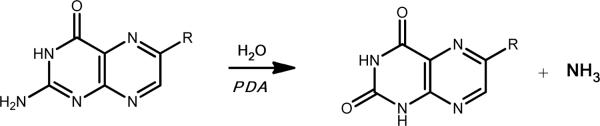
Table 2.
Catalytic Constants for Substrates of Arad3529
| Substrate | kcat/Km (M−1 s−1) | kcat (s−1) | Km (μM) | Docking rank* |
|---|---|---|---|---|
| formylpterin | 5.2 (0.5) × 106 | 64 ± 12 | 12 ± 2 | |
| pterin-6-carboxylate | 4.0 (0.2) × 106 | 110 ± 3 | 27 ± 2 | |
| pterin-7-carboxylate | 3.7 (0.2) × 106 | 48 ± 2 | 13 ± 1 | |
| pterin | 3.3 (0.3) × 106 | 131 ± 4 | 39 ± 4 | 27 |
| hydroxymethylpterin | 1.2 (0.1) × 106 | 28 ± 1 | 23 ± 3 | |
| biopterin | 1.0 (0.1) × 106 | 46 ± 4 | 47 ± 9 | 14 |
| D-(+)-neopterin | 3.1 (0.1) × 105 | 19 ± 1 | 61 ± 7 | 15 |
| isoxanthopterin | 2.8 (0.2) × 105 | 1.6 ± 0.1 | 5.7 ± 0.5 | |
| sepiapterin | 1.3 (0.4) × 105 | 2.9 ± 0.3 | 22 ± 9 | |
| folate | 1.3 (0.2) × 105 | 6.4 ± 0.5 | 50 ± 10 | |
| xanthopterin | 1.2 (0.1) × 105 | 0.46 ± 0.01 | 40 ± 1 | |
| 7,8-dihydro-hydroxymethylpterin | 3.3 (0.4) × 104 | 1.2 ± 0.1 | 37 ± 7 | |
| 7,8-dihydroneopterin | 2.6 (0.3) × 102 | 0.036 ± 0.008 | 200 ± 100 | |
| 7,8-dihydrobiopterin | 9.3 (0.5) × 102 | 0.090 ± 0.007 | 93 ± 13 |
In docking against Model-3 (Table 1), compounds were ranked out of the 57672 high-energy intermediates in the virtual library, which only contains pterin, biopterin, and neopterin among the pterin substrates.
Variations on Binding Orientation of Pterin
In all of the structurally characterized members of the amidohydrolase superfamily, the re-face of the carbonyl group of amide and ester substrates is presented to the attacking hydroxide; the same orientation was observed for the amidine or guanidine moieties of substrates in the deamination reactions. In docking screens against Arad3529, we observed two chiral intermediates of pterin (named as N1 and N3), that were generated by hydroxide re- and si-face attacking two pterin tautomers 2-aminopteridin-4(3H)-one and 2-aminopteridin-4(1H)-one, and by Glu234 protonating the N-1 and N-3 nitrogen in the pteridine ring, respectively (Scheme 2). In particular, N3 was selected by Model-2, N1 was selected by Model-3, and both intermediates were selected by Model-4 (Table 1). In Model-2, the docking pose of N3 had a stranded C-4 carbonyl group, which in N1 forms hydrogen bonds with Asn332 in Model-3 (Figure 4). This C-4 carbonyl group and the N-5 ring nitrogen in N1 also hydrogen bonded with Lys85 in Model-3. Furthermore, it is known that the 2-aminopteridin-4(1H)-one tautomer is less stable than the 2-aminopteridin-4(3H)-one tautomer (in ab initio and free energy perturbation calculations the ratio is 1:6)53. It seems clear that the N1 orientation is the true binding motif, and that an important feature for recognition of a pterin deaminase is the DN dyad (D331, N332) found on β-strand 8 and the Lys85 found on β-strand 1.
Scheme 2.

Figure 4.

(Top) the binding pose of pterin intermediate N3 (yellow stick) formed by the activated hydroxide attacking the C-2 atom on the si-face of the aromatic ring of the tautomer 2-aminopteridin-4(1H)-one, in the active site of Model-2 of Arad3529 (white stick). (Bottom) the binding pose of pterin intermediate N1 (yellow stick) formed by the activated hydroxide attacking the C-2 atom on the re-face of the aromatic ring of the tautomer 2-aminopteridin-4(3H)-one, in the active site of Model-3 of Arad3529 (white stick). Polar interactions between the docked pterin and the modeled active site are shown by red dashed lines.
DISCUSSION
Three techniques were crucial for what turned out to be the correct prediction of pterin deaminase activity for Arad3529. First, to leverage a distantly-related template, detailed modeling of two active-site loops was required to find models that were at once structurally sensible and catalytically competent, and that could discriminate new substrates from these from the template. Second, the selectivity of models was enhanced by negative control docking screens against the template structure; we sought substrates ranked highly by the models and poorly by the template. Finally, a close cycle of bioinformatics, biophysical modeling, and enzymology made this approach pragmatic. Identifying substrates for Arad3529 allows the functional annotation of about 140 previously uncharacterized amidohydrolase enzymes in Group 14 of cog0402. These proteins may be used by bacteria to salvage oxidized pterins, and form the pterin degradation pathway with neighboring genes.
Enzyme specificity recapitulated by loop modeling
The substrate specificity of enzymes comes from the corresponding compositions of binding-site residues. Remarkable changes in the binding-site residues between two enzyme sequences lead to differences in their binding-site structures. However, a homology model tends to inherit the backbone conformations of aligned regions from the template. Therefore, a homology model of the target enzyme often contains errors in the packing of side chains, and backbone conformations of structurally undefined regions (loops), especially when computed based on a remote template. Accurate substrate prediction by docking against homology models of unknown enzymes requires precise positioning of the binding site residues that interact with the new substrates. In this study, we attempted to capture binding-site features in Arad3529 by remodeling two binding-site loops in the initial homology model. Because no target ligand is known, four models that are structurally diverse were selected and docked against independently, followed by combining the screening results. True substrates of Arad3529, pterins, were not captured by the initial homology model but were captured by the models with refined loops, illustrating the necessity of the directed refinement of binding-site loops that contain residues different from the template.
Docking against the template as a negative control
When a homology model of the target enzyme is computed based on a distantly related template structure, on one side, the target enzyme very likely has different substrates from those of the template; on the other side, however, the homology model of the target even after refinement could still share some structural features with the template and thus prioritize similar compounds as the template structure in docking screens. Here, we first docked to the template as a negative control, and compared these hits to those against the homology models. Compounds that were ranked highly by the template and by the homology models were discounted, and indeed none of them was confirmed as substrates of Arad3529. This suggests that, for enzyme substrate prediction by homology Model-based docking, it may be useful to account for compounds that are recognized by the template as negative control, especially when there is only a remote relationship between the target and the template sequence.
Additional Enzymes in cog0402 that catalyze pterin deamination
Figure 1 depicts 22 sequences in Group 14. To identify a more comprehensive list of proteins in the current databases that share the same substrate profile as Arad3529, a BLAST search was conducted with the sequence of Arad3529. Of the top 200 hits, 165 proteins contained the characteristic β-strand 8 “DN” dyad and a total of 139 of these would populate Group 14 if included in the network diagram. The remaining 26 sequences cluster with the currently uncharacterized Group 13. Group 13 is closely related to cytosine deaminase and also possesses a mixture of enzymes that have either a “DN” or “NN” dyad. While the enzymatic function of Group 13 is unknown, all members of Group 14 are predicted to deaminate pterins (Table S2). Of course, this remains just a prediction, since even proteins with high sequence identity sometimes have different activities54.
Biological relevance of substrates, Genomic context in Agrobacterium radiobacter
Pterin rings form the backbone of the coenzymes folate and tetrahydrobiopterin. The 7,8-dihydro forms of hydroxymethylpterin and neopterin are intermediates in folate synthesis, whereas sepiapterin is an intermediate in the synthesis of biopterin. During the biosynthesis of these cofactors, the pterin substructure remains reduced in the 7,8-dihydro form. Fully oxidized pterins, formed by oxidation of 7,8-dihydropterins55, are not known to be salvaged by reduction56. Isoxanthopterin is formed by oxygenation of pterin by xanthine dehydrogenase57, while pterin-6-carboxylate, formylpterin and pterin can be formed by photolysis of folate58,59. It seems possible that Arad3529 and its orthologs are used by the bacteria to degrade oxidized pterins.
The gene for Arad3529 is situated in an apparent operon along with the genes TauC, GlcD and MdaB, which is flanked by TauA, TauB, LysR, and a protein from cog1402 (Figure S2). TauA, TauB and TauC resemble ATP-cassette binding transporters and LysR is a transcription factor. MdaB, GlcD and cog1402 are uncharacterized proteins resembling quinone oxidase, glycolate oxidase D, and creatininase, respectively. MdaB is predicted to contain an NAD(P)H binding domain, GlcD is predicted to be a flavin dependent redox enzyme, and the cog1402 protein will likely catalyze the hydrolysis of an amide bond. These enzymes may form a degradative pathway for catabolizing pterin rings. To our knowledge, the only known instance for the degradation of a pterin ring is by the soil bacteria Alcaligenes faecalis60. In this bacterium, isoxanthopterin is deaminated to form 7-oxylumazine, which is further oxidized to 6,7-dioxylumazine (tetraoxypteridine). An isomerase then cleaves the C6/C7 bond, and reattaches forming xanthine-8-carboxylate61. Xanthine-8-carboxylate is then decarboxylated, forming xanthine62. Unfortunately, the recently sequenced genome of A. faecalis does not possess an Arad3529 homolog or any of its operon-related genes, and the protein that deaminates isoxanthopterin remains to be discovered.
Certain caveats merit mentioning. Here, homology models of Arad3529 were predicted from a cytosine deaminase structure in complex with a mechanism-based inhibitor. This likely gave us an advantage over modeling from a ligand-free template. When such an apo- template is the only choice, enhanced sampling may be needed.19 Inevitably, this will lead to more models. To judge among these possible structures, and the docked molecules to which they lead, the criterion of a self-similar hit list may be useful, as we found it to be. Admittedly, this metric remains largely untested, and its only theoretical virtue is that it quantifies an expectation that a well-behaved docking calculation should find putative ligands that resemble one another. From a docking standpoint, we have continued to use high-energy intermediate forms of substrates, rather than their ground states. This seems well-justified for amidohydrolases, where we have a good understanding of the structures that such intermediates adopt, and confidence that the chemical step is rate determining for the reactions the enzymes catalyze. For other families of enzymes, our ability to anticipate high-energy structures, and confidence that the chemical step is rate limiting, may be more limited. Finally, docking screens, in our hands, retain a human element as a final arbiter of which among the tens-to-hundreds of high-ranking metabolites to actually test experimentally. We have argued that human inspection integrates aspects of docking that are captured by the experience of the trained modeler, enzymologist or medicinal chemist that are difficult to fully capture algorithmically.63
CONCLUSION
Members of the amidohydrolase superfamily have been found in every sequenced genome; the functions of most of these proteins remain unknown. Here we focused on one amidohydrolase, Arad3529 from Agrobacterium radiobacter K84, which represents proteins from Group 14 in cog0402. Proteins in Group 14 were uncharacterized, with sequence similarity and knowledge of the mechanistically related chemistries within the superfamily wrongly suggesting roles as chlorohydrolases or cytosine deaminases. To determine the function of Arad3529 we used a multidisciplinary approach that integrated homology modeling, molecular docking screens of a metabolite library, and physical library screening by kinetic assays. This led to what we believe are the true substrates of Arad3529, a set of modified pterins. Based on the conservation of characteristic residues that interact with substrates in the docked structure, about 140 other previously unannotated amidohydrolases from different species may now be assigned as pterin deaminases. This approach may be useful in the discovery in vitro enzymatic and in vivo metabolical functions of unknown enzymes discovered in genome projects, especially for those targets with marginal sequence identities to template structures of know function.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Patricia Babbitt and Dr. Sunil Ojha for discussion about amidohydrolse superfamily and sequence analysis. We thank Dr. Peter Kolb and Dr. Magdalena Korczynska for discussion about the high-energy intermediate (HEI) docking database.
Footnotes
This work was supported by NIH grants (GM071790 and U54 GM093342; R01 GM054762 to AS) and the Robert A. Welch Foundation (A-840) to FMR.
REFERENCES
- (1).Whisstock JC, Lesk AM. Q Rev Biophys. 2003;36:307. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- (2).Gerlt JA, Babbitt PC. Genome Biol. 2000;1 doi: 10.1186/gb-2000-1-5-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brenner SE. Trends Genet. 1999;15:132. doi: 10.1016/s0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- (4).Devos D, Valencia A. Trends Genet. 2001;17:429. doi: 10.1016/s0168-9525(01)02348-4. [DOI] [PubMed] [Google Scholar]
- (5).Gerlt JA, Allen KN, Almo SC, Armstrong RN, Babbitt PC, Cronan JE, Dunaway-Mariano D, Imker HJ, Jacobson MP, Minor W, Poulter CD, Raushel FM, Sali A, Shoichet BK, Sweedler JV. Biochemistry-Us. 2011;50:9950. doi: 10.1021/bi201312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schnoes AM, Brown SD, Dodevski I, Babbitt PC. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Glasner ME, Fayazmanesh N, Chiang RA, Sakai A, Jacobson MP, Gerlt JA, Babbitt PC. J Mol Biol. 2006;360:228. doi: 10.1016/j.jmb.2006.04.055. [DOI] [PubMed] [Google Scholar]
- (8).Favia AD, Nobeli I, Glaser F, Thornton JM. J Mol Biol. 2008;375:855. doi: 10.1016/j.jmb.2007.10.065. [DOI] [PubMed] [Google Scholar]
- (9).Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Nature. 2007;448:775. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cummings JA, Nguyen TT, Fedorov AA, Kolb P, Xu CF, Fedorov EV, Shoichet BK, Barondeau DP, Almo SC, Raushel FM. Biochemistry-Us. 2010;49:611. doi: 10.1021/bi901935y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hall RS, Fedorov AA, Marti-Arbona R, Fedorov EV, Kolb P, Sauder JM, Burley SK, Shoichet BK, Almo SC, Raushel FM. J Am Chem Soc. 2010;132:1762. doi: 10.1021/ja909817d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xiang DF, Kolb P, Fedorov AA, Xu CF, Fedorov EV, Narindoshivili T, Williams HJ, Shoichet BK, Almo SC, Raushel FM. Biochemistry-Us. 2012;51:1762. doi: 10.1021/bi201838b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Annu Rev Bioph Biom. 2000;29:291. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- (14).Pieper U, Eswar N, Webb BM, Eramian D, Kelly L, Barkan DT, Carter H, Mankoo P, Karchin R, Marti-Renom MA, Davis FP, Sali A. Nucleic Acids Res. 2009;37:D347. doi: 10.1093/nar/gkn791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Evers A, Klebe G. J Med Chem. 2004;47:5381. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- (16).Kairys V, Fernandes MX, Gilson MK. J Chem Inf Model. 2006;46:365. doi: 10.1021/ci050238c. [DOI] [PubMed] [Google Scholar]
- (17).Katritch V, Byrd CM, Tseitin V, Dai DC, Raush E, Totrov M, Abagyan R, Jordan R, Hruby DE. J Comput Aid Mol Des. 2007;21:549. doi: 10.1007/s10822-007-9138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A. J Chem Inf Model. 2009;49:2512. doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Nat Chem Biol. 2011;7:769. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A. P Natl Acad Sci USA. 2011;108:15810. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Song L, Kalyanaraman C, Fedorov AA, Fedorov EV, Glasner ME, Brown S, Imker HJ, Babbitt PC, Almo SC, Jacobson MP, Gerlt JA. Nat Chem Biol. 2007;3:486. doi: 10.1038/nchembio.2007.11. [DOI] [PubMed] [Google Scholar]
- (22).Holm L, Sander C. Proteins: Structure, Function, and Bioinformatics. 1997;28:72. [PubMed] [Google Scholar]
- (23).Seibert CM, Raushel FM. Biochemistry. 2005;44:6383. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- (24).Tatusov RL, Koonin EV, Lipman DJ. Science. 1997;278:631. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- (25).Pieper U, Chiang R, Seffernick JJ, Brown SD, Glasner ME, Kelly L, Eswar N, Sauder JM, Bonanno JB, Swaminathan S, Burley SK, Zheng X, Chance MR, Almo SC, Gerlt JA, Raushel FM, Jacobson MP, Babbitt PC, Sali A. Journal of Structural and Functional Genomics. 2009;10:107. doi: 10.1007/s10969-008-9056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- (27).Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. PLoS ONE. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang P-L, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, Bader GD. Nat. Protocols. 2007;2:2366. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Maynes JT, Yuan RG, Snyder FF. J. Bacteriol. 2000;182:4658. doi: 10.1128/jb.182.16.4658-4660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yuan G, Bin JC, McKay DJ, Snyder FF. J. Biol. Chem. 1999;274:8175. doi: 10.1074/jbc.274.12.8175. [DOI] [PubMed] [Google Scholar]
- (31).Ireton GC, McDermott G, Black ME, Stoddard BL. J Mol Biol. 2002;315:687. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- (32).Danielsen S, Kilstrup M, Barilla K, Jochimsen B, Neuhard J. Mol Microbiol. 1992;6:1335. doi: 10.1111/j.1365-2958.1992.tb00854.x. [DOI] [PubMed] [Google Scholar]
- (33).Hall RS, Agarwal R, Hitchcock D, Sauder JM, Burley SK, Swaminathan S, Raushel FM. Biochemistry-Us. 2010;49:4374. doi: 10.1021/bi100252s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Marti-Arbona R, Xu CF, Steele S, Weeks A, Kuty GF, Seibert CM, Raushel FM. Biochemistry-Us. 2006;45:1997. doi: 10.1021/bi0525425. [DOI] [PubMed] [Google Scholar]
- (35).Aslanidis C, Dejong PJ. Nucleic Acids Res. 1990;18:6069. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Studier FW. Protein Expres Purif. 2005;41:207. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- (37).Pegg SCH, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Biochemistry-Us. 2006;45:2545. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- (38).Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Edgar RC. Nucleic Acids Res. 2004;32:1792. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- (41).Shen MY, Sali A. Protein Sci. 2006;15:2507. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fiser A, Do RKG, Sali A. Protein Sci. 2000;9:1753. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. J Med Chem. 2006;49:534. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- (44).Hermann JC, Ghanem E, Li YC, Raushel FM, Irwin JJ, Shoichet BK. J Am Chem Soc. 2006;128:15882. doi: 10.1021/ja065860f. [DOI] [PubMed] [Google Scholar]
- (45).Xiang DF, Kolb P, Fedorov AA, Meier MM, Fedorov LV, Nguyen TT, Sterner R, Almo SC, Shoichet BK, Raushel FM. Biochemistry-Us. 2009;48:2237. doi: 10.1021/bi802274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. Nucleic Acids Res. 2006;34:D354. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mysinger MM, Shoichet BK. J Chem Inf Model. 2010;50:1561. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- (48).Goble AM, Fan H, Sali A, Raushel FM. Acs Chem Biol. 2011;6:1036. doi: 10.1021/cb200198c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kamat SS, Bagaria A, Kumaran D, Holmes-Hampton GP, Fan H, Sali A, Sauder JM, Burley SK, Lindahl PA, Swaminathan S, Raushel FM. Biochemistry-Us. 2011;50:1917. doi: 10.1021/bi101788n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kamat SS, Fan H, Sauder JM, Burley SK, Shoichet BK, Sali A, Raushel FM. J Am Chem Soc. 2011;133:2080. doi: 10.1021/ja110157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Nat Biotechnol. 2007;25:197. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- (52).Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK. J Chem Inf Model. 2008;48:755. doi: 10.1021/ci8000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Schwalbe CH, Lowis DR, Richards WG. J Chem Soc Chem Comm. 1993;1199 [Google Scholar]
- (54).Seffernick JL, de Souza ML, Sadowsky MJ, Wackett LP. J Bacteriol. 2001;183:2405. doi: 10.1128/JB.183.8.2405-2410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pfleiderer W. Folates and pterins. 1984;2:43. [Google Scholar]
- (56).Noiriel A, Naponelli V, Gregory JF, Hanson AD. Plant Physiol. 2007;143:1101. doi: 10.1104/pp.106.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).FORREST HS GE, MITCHELL HK. Science. 1956;124:725. doi: 10.1126/science.124.3225.725. [DOI] [PubMed] [Google Scholar]
- (58).Lowry OH, Bessey OA, Crawford EJ. Journal of Biological Chemistry. 1949;180:389. [PubMed] [Google Scholar]
- (59).Off MK, Steindal AE, Porojnicu AC, Juzeniene A, Vorobey A, Johnsson A, Moan J. J Photoch Photobio B. 2005;80:47. doi: 10.1016/j.jphotobiol.2005.03.001. [DOI] [PubMed] [Google Scholar]
- (60).McNutt WS., Jr. Journal of Biological Chemistry. 1968;238:1116. [PubMed] [Google Scholar]
- (61).McNutt WS, Damle SP. Journal of Biological Chemistry. 1964;239:4272. [PubMed] [Google Scholar]
- (62).Dairman WM, McNutt WS. Journal of Biological Chemistry. 1964;239:3407. [PubMed] [Google Scholar]
- (63).Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. P Natl Acad Sci USA. 2012;109:5517. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.