

Abstract
We report the structure of the fifth monohydrate of gallic acid and two additional anhydrate polymorphs and evidence of at least 22 other solvates formed, many containing water and another solvent. This unprecedented number of monohydrate polymorphs and diversity of solid forms is consistent with the anhydrate and monohydrate crystal energy landscapes, showing both a wide range of packing motifs and also some structures differing only in proton positions. By aiding the solution of structures from powder X-ray diffraction data and guiding the screening, the computational studies help explain the complex polymorphism of gallic acid. This is industrially relevant, as the three anhydrates are stable at ambient conditions but hydration/dehydration behavior is very dependent on relative humidity and phase purity.
Short abstract
An extensive experimental screen for solid forms, driven by computationally generated anhydrate and monohydrate crystal energy landscapes, revealed the fifth (!) monohydrate structure of gallic acid, structures of two additional anhydrate polymorphs, and evidence of at least 22 solvates. The application of computational studies in combination with other analytical techniques helps unravel the complex polymorphic system of gallic acid.
The statement, “Many people think that polymorphism and solid state chemistry is the hardest thing to get right in drug development“,1 reflects the importance of the occurrence of multiple crystalline forms (polymorphism), including solvate/hydrate formation (multicomponent systems). This is because the crystal form dictates fundamental physical properties, such as density, solubility, hardness, melting point, mechanical strength, etc., and consequently can profoundly influence the manufacturing process, long-term stability, and performance of drug products (and any other fine chemicals).2,3 The phenomenon of polymorphism affects also multiple-component systems, e.g. hydrates, albeit this is often less well established than for single components. Knowledge about the extent of hydration is essential for the development of an industrial process, where it is difficult to exclude water or moisture; thus, hydrate formation often cannot be avoided. Moreover, it is crucial to understand and control the anhydrate/hydrate balance. A hydrate may be the desired solid form, provided it displays an acceptable stability under processing and storage conditions. However, without extensive and time-consuming experimental and/or computational screens, it still remains hard to predict if multiple crystalline forms exist, let alone the possible number of solid forms and their relative thermodynamic and kinetic stability.
The most recent blind test of Crystal Structure Prediction (CSP2010)4 tested, for the first time, the ability to predict the structures of additional (unpublished) polymorphs of a hydrate. The challenge was to predict the third and fourth polymorphs of gallic acid monohydrate (Figure 1), given that two polymorphs were already in the Cambridge Structural Database with refcodes5KONTIQ6 (form MH-IV)7 and KONTIQ01(8)/029 (form MH-I°). The target structures have since been published (KONTIQ0310/0511 (form MH-II) and KONTIQ04(11) (form MH-III)), as showing remarkable hydrate polymorphism.11 Anhydrous gallic acid is also polymorphic,12 though only one solvent free structure has been published (IJUMEG11,13,14 (form AH-II°)). The industrially relevant15,16 compound exhibits biological activity and is used as an analytical standard and as starting material in synthesis of drug compounds.
Figure 1.
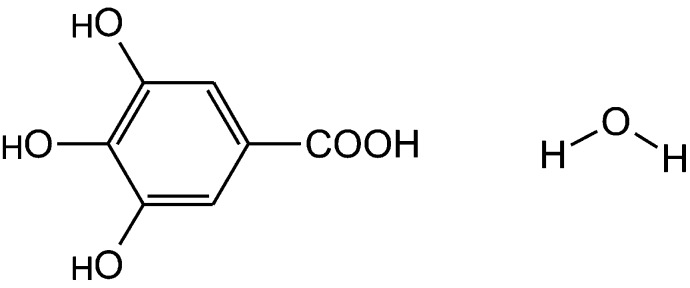
Molecular diagram of gallic acid monohydrate.
For CSP2010, we performed an extensive search17 to generate hypothetical Z′ = 1 monohydrate crystal structures (Figure 2a) by evaluating the lattice energies18 allowing for minor conformational change in the polar proton positions19 and approximating molecular polarization20 within the crystal structures, as detailed in the Supporting Information. The same methods were used to generate Z′ = 1 and Z′ = 2 anhydrate structures, so that the crystal energy landscapes could be contrasted to understand the influence of water on the crystal packing.
Figure 2.
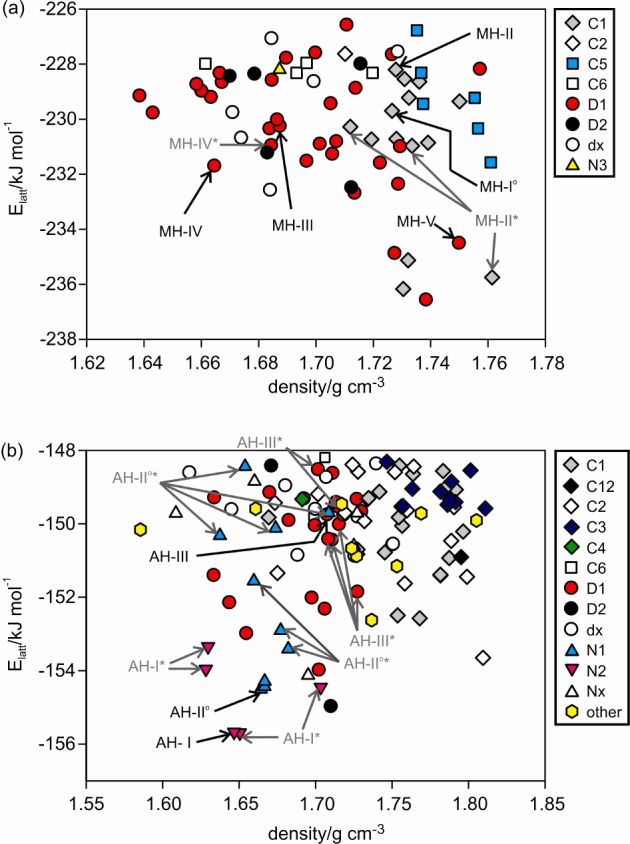
Crystal energy landscapes for (a) gallic acid monohydrate (Z′ = 1) and (b) anhydrate (Z′ = 1 and 2), classified by the hydrogen-bonding motif (Figure 3). Each symbol denotes a crystal structure. Experimental structures are picked out with black arrows; structures labeled with an asterisk (*) differ from the experimental structure in proton position(s) but not in packing.
The crystal energy landscapes (Figure 2) both exhibit numerous thermodynamically feasible structures within the energy range expected for polymorphism,21 including all experimental anhydrates and the four Z′ = 1 monohydrate structures. The MH-III structure, a Z′ = 4 structure, lies outside the scope of the calculations and cannot be found in a Z′ = 1 search. Therefore, the structure was computed starting from the experimental structure. Furthermore, the calculated and experimental structures closely match (Supporting Information).
Analysis of the packing22 and hydrogen bonding motifs23 in the low energy crystal structures showed the delicate balance of different hydrogen bonding between carboxylic acid, phenol, and water. The R22(8) carboxylic acid dimer is the most favored for the anhydrate structures (Figure 3), with interpenetrated 3D networks (N1 and N2) or a ladder motif (D1) being the most favorable extended hydrogen bonding motifs for R2(8) structures. The presence of water molecules can lead to the formation of a very stable carboxylic acid···water···phenol tape (C1, Figure 3a), in addition to the stable ladder motif (D1). An anhydrate C1 tape or a 3D interpenetrated network (N3) hydrate structure is energetically less favorable. Similarly carboxylic acid···carboxylic acid chains (C5, C6) are calculated to be less stable than the D1 and C1 motifs and are observed less frequently on both landscapes.
Figure 3.

Illustration of the monohydrate (a) and anhydrate (b) hydrogen-bonding motifs in structures on the crystal energy landscapes (Figure 2).
Several of the computed low-energy structures were found to differ only in proton positions, that is, carboxylic acid, hydroxyl, and/or water protons, resulting in structures with different directionality of the strong intermolecular interactions, e.g. O–H···O vs O···H–O, but otherwise essentially exhibit the same crystal packing. This observation has also been made for the crystal energy landscapes of dihydroxybenzoic acids24 and phloroglucinol,25 sometimes correlating with observed disorder in the crystal structures.
Driven by these crystal energy landscapes (Figure 2), suggesting that alternative monohydrate and anhydrate forms should be feasible, a manual crystallization screen was conducted using 29 solvents and employing four crystallization techniques (fast and slow solvent evaporation, cooling crystallization, and solvent drop grinding). The screen also included sublimation and desolvation experiments (Supporting Information).
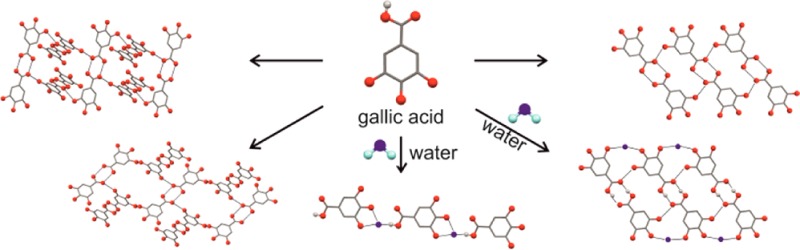
The experimental screen successfully found all five structurally characterized solid forms, allowed characterization of two additional anhydrate polymorphs (AH-I7 and AH-III), and resulted in numerous novel forms: a fifth monohydrate (MH-V) and 22 solvates, the majority of which were mixed solvates with water. Ten of these solvates were isostructural26 (Supporting Information).
Control of the solid form produced could be achieved by careful choice of the crystallization conditions. Solvent drop grinding and most cooling crystallization experiments (using solvents that do not form homosolvates; see Supporting Information) led to the thermodynamically most stable phases at room temperature (AH-II° and MH-I°). Evaporation experiments led to concomitant and varying crystallization products, favoring metastable forms. Evaporating gallic acid from water, methanol, or 2-butanol solutions resulted predominantly in MH-II, MH-IV, and MH-V. MH-III was never observed to nucleate directly in our screen but was formed by leaving wet alcohol–water solvate crystals at ambient conditions.
AH-I and AH-III (mp 225–230 °C, Lindpaintner’s unstable polymorph12) could only be obtained using sublimation or desolvation techniques. Sublimation led to the three anhydrate polymorphs, with AH-III being a minor byproduct. Very small granular crystals of AH-I were initially observed, but these disappeared because AH-I has a higher vapor pressure than AH-II° at the sublimation temperature, and spearlike crystals of AH-II° grew instead. At temperatures above 245 °C, AH-II° transformed to the high temperature, enantiotropically related, form AH-I. Thermolysis of the acid started at the transformation temperature, followed by the melting (mp 260–265 °C) of AH-I.
Thermal desolvation (without melting) of MH-I°, MH-II, the acetic acid monosolvate, or dimethyl formamide monosolvate (S-DMF4) at approximately 60–75 °C resulted in AH-I (sometimes concomitantly with AH-II°), desolvation of a dimethyl formamide–water solvate (S-DMF2, Supporting Information) gave samples with AH-III being the major product, and desolvation of MH-IV resulted in anhydrate mixtures of AH-II° and AH-III. In contrast, desolvation of the hydrates and solvates at 0% relative humidity (RH) normally resulted in AH-II°.
The structures for AH-I,27 AH-III,28 and MH-V29 were determined using powder X-ray diffraction data (Supporting Information) using computationally generated structures to generate the starting structure (AH-III) and to locate the proton positions (AH-I and MH-V). The best affordable periodic ab initio structure DFT-D calculations within CASTEP30 (Supporting Information) were used to refine and check the candidate structures from the crystal energy landscapes (Figure 2). The structure for AH-III was confirmed using the computationally generated AH-III structure as the basis for a mixed-phase Rietveld refinement, with the final structure supported by structural information derived from its infrared spectrum (Supporting Information).
All three anhydrates (AH-I, AH-III (Figure 4a–d), and AH-II°11,13,14) form centrosymmetric R22(8) acid dimers, but they represent different low energy packing motifs in Figure 3b. In AH-II° (C2/c, Z′ = 1) R2(8) dimers form linear coplanar chains, with adjacent chains being arranged in perpendicular fashion and interacting through O–H···O hydrogen bonds, forming a 3D interpenetrated net (N1). In AH-I (P1̅, Z′ = 2), one crystallographically unique molecule forms the same type of chains, but the second forms a distinct chain (Figure 4a). The two types of chains are arranged perpendicularly and interact through O–H···O hydrogen bonds, forming a different 3D interpenetrated net (N2) to that observed for AH-II°. In AH-III (P21/c, Z′ = 1) the R22(8) dimers form the D1 ladder motif (Figures 3b and 4c).
Figure 4.
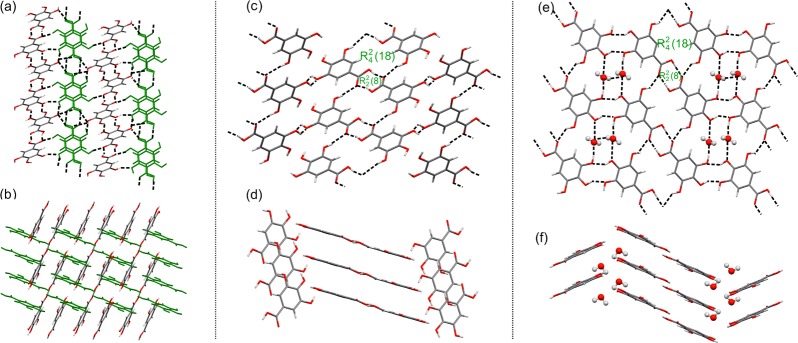
Crystal packing in the three new structures (a, b) gallic acid AH-I (viewed along (a) [100] and (b) [011]), (c, d) AH-III (viewed along (c) [010] and (d) [1–10]), and (e, f) MH-V (viewed along (e) [010] and (f) [100]). Dashed lines mark the intermolecular hydrogen bonds. The two crystallographically independent molecules in AH-I are colored differently.
The five hydrates exhibit the two most stable and most frequent hydrogen bonding motifs on the crystal energy landscape Figure 2a. MH-I° (P2/n, Z′ = 1) and MH-II (P21/c, Z′ = 1) form linear tapes of water and gallic acid molecules (C1, Figure 2b), but they differ in molecular orientation within the tapes and the crystal packing.11 The main difference between the three ladder based hydrates (D1) is the mutual arrangement of the stacked ladders: coplanar in MH-III (P1̅, Z′ = 4), twisted in MH-IV (P21/c, Z′ = 1), and tilted in MH-V (P21/c, Z′ = 1). The three lowest unobserved monohydrates in Figure 2a have major aspects of their hydrogen bonding and packing in common with observed structures and, thus, could be expected to transform easily to one of the experimental forms if nucleated. The structural similarities between AH-III and MH-V could also be inferred from their infrared spectra (Supporting Information).
All three anhydrates can be classified as storage stable at ambient conditions. No transformation of the metastable polymorphs (AH-I, AH-III) to the thermodynamically more stable room temperature form (AH-II°) or to any of the monohydrates was observed for samples stored for one year in glass vials at ambient conditions. Furthermore, AH-II° was found to be stable at RH below 80% at 25 °C by moisture sorption studies (Supporting Information). Conversely, anhydrate/MH-I° mixtures transformed to the MH-I° at RH above 50% at 25 °C, suggesting that hydrate impurities catalyze the absorption of water. All the previously known hydrates MH-I°–IV could be stored at room temperature and below, provided some moisture was present (RH > 10%) and that they were phase pure samples. In aqueous solutions all hydrates transformed to MH-I°. The new hydrate MH-V could be stored at lower temperatures (−4 °C) in a RH range of 50–60%. At higher temperatures, lower RH (below 50%), or extremely high RH, MH-V transformed to either one of the three anhydrous forms or a hydrate (MH-I, MH-III, MH-IV). Thermogravimetric measurements (Supporting Information) showed that the thermal stability of the hydrates decreases: MH-I° (most stable) > MH-II > MH-III > MH-IV > MH-V (least stable). More energy is required to release the water from the C1 based structures, where water molecules link the carboxylic acid and phenol groups, than the D1 ladder structures, where the molecules are located between ladders but do not interact with the acid groups.
The lattice energy calculations of relative stability ignore temperature and humidity effects and also depend on the methodology employed. However, they agree that all the energies are fairly close, with the DFT-D calculations suggesting that MH-V should be distinctly less stable than the other hydrates (Table 1), as observed experimentally. The calculated energy difference between AH-II° and AH-I is in excellent agreement with the transformation enthalpy of 1.9 ± 0.3 kJ mol–1 obtained by differential scanning calorimetry measurements at temperatures above 230 °C (Supporting Information).
Table 1. Relative Lattice Energies (ΔElatt/kJ mol–1) for Gallic Acid Anhydrate and Monohydrate Polymorphs Based on Periodic DFT-D ab Initio Calculations (Supporting Information).
| anhydrates | AH-I | AH-II° | AH-III | ||
| ΔElatt | +1.9 | 0 | +3.2 | ||
| monohydrates | MH-I° | MH-II | MH-III | MH-IV | MH-V |
| ΔElatt | +1.5 | +5.1 | +1.0 | 0 | +11.6 |
In conclusion, the formation of at least five polymorphic monohydrates, all structurally characterized, is unprecedented for an organic compound. This unusual complexity extends to there being at least six other structures containing gallic acid, water, and another solvent (Supporting Information). The fact that numerous structures on the computationally generated crystal energy landscapes, which are based on distinct hydrogen bonding and packing motifs, are highly competitive in energy explains why gallic acid shows this remarkable tendency toward polymorphism. Further anhydrate or monohydrate polymorphs could exist, and other solvates and cocrystals with different hydrogen bond donor/acceptor groups may well show similar complexity. This has recently been demonstrated by the structures of five gallic acid/acetamide cocrystals.31 Desolvation of the solvated/hydrated crystal forms has again provided a route to a novel polymorph which could not be obtained directly from solution,32 though the kinetics and outcome of dehydration processes are very dependent on phase purity. The experimental and computed range of hydrate and monohydrate crystal structures explains why the product phase(s) is sensitive to minor variations in conditions (temperature, relative humidity, phase purity). Hence, controlling product quality throughout processing, handling, and storage, to the standards required for pharmaceuticals, will be challenging. However, this type of study provides the information needed for designing a controlled process.
Acknowledgments
The authors would like to thank Dr. P. G. Karamertzanis for computational support and Prof. U. J. Griesser for helpful discussion. D.E.B. gratefully acknowledges funding by the Erwin-Schrödinger (FWF, project J2897-N17) and Hertha Firnberg (FWF, project T593-N19) programs of the Austrian Science Fund. Other resources used were from EPSRC funding of Control and Prediction of the Organic Solid State www.cposs.org.uk.
Supporting Information Available
Computational and experimental details, solid form screening, structure solutions from PXRD, thermal analysis, moisture (de)sorption, infrared spectroscopy, and cif data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Byrn S. R. Presented at the International School of Crystallography, Erice, Italy, 2004.
- Byrn S. R.; Pfeiffer R. R.; Stowell J. G.. Solid-State Chemistry of Drugs, 2nd ed.; SSCI Inc.: West Lafayette, IN, 1999. [Google Scholar]
- Bernstein J.Polymorphism in Molecular Crystals; Clarendon Press: Oxford, 2002. [Google Scholar]
- Bardwell D. A.; Adjiman C. S.; Ammon H. L.; Arnautova E. A.; Bartashevich E.; Boerrigter S. X. M.; Braun D. E.; Cruz-Cabeza A. J.; Day G. M.; Della Valle R. G.; Desiraju G. R.; van Eijck B. P.; Facelli J. C.; Ferrao M. D.; Grillo D.; Habgood M.; Hofmann D. W. M.; Hofmann F.; Jose J.; Karamertzanis P. G.; Kazantsev A. V.; Kendrick J.; Kuleshova L. N.; Leusen F. J. J.; Maleev A.; Misquitta A. J.; Mohamed S.; Needs R. J.; Neumann M. A.; Nikylov D.; Orendt A. M.; Pal R.; Pantelides C. C.; Pickard C. J.; Price L. S.; Price S. L.; Scheraga H. A.; van de Streek J.; Thakur T. S.; Tiwari S.; Venuti E.; Zhitkov I. Acta Crystallogr., Sect. B: Stuct. Sci. 2011, 67, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F. H. Acta Crystallogr., Sect. B: Stuct. Sci. 2002, 58, 380–388. [DOI] [PubMed] [Google Scholar]
- Jiang R. W.; Ming D. S.; But P. P. H.; Mak T. C. W. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2000, 56, 594–595. [DOI] [PubMed] [Google Scholar]
- The forms are named according to the Kofler notation (i.e., consecutive order of melting points with I exhibiting the highest melting point). Thermodynamic room temperature forms are marked (°).
- Okabe N.; Kyoyama H.; Suzuki M. Acta Crystallogr., Sect. E: Struct. Rep. Online 2001, 57, o764–o766. [Google Scholar]
- Billes F.; Mohammed-Ziegler I.; Bombicz P. Vib. Spectrosc. 2007, 43, 193–202. [Google Scholar]
- Demirtas G.; Dege N.; Buyukgungor O. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o1509–o1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke H. D.; Arora K. K.; Wojtas L.; Zaworotko M. J. Cryst. Growth Des. 2011, 11, 964–966. [Google Scholar]
- Lindpaintner E. Mikrochim. Acta 1939, 27, 21–41. [Google Scholar]
- Zhao J.; Khan I. A.; Fronczek F. R. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o316–o317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirun N.; Saithong S.; Pakawatchai C.; Tantishaiyakul V. Acta Crystallogr., Sect. E: Struct. Rep. Online 2011, 67, o787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L. J.; Zhang Z. H.; Huang J. L.; Xu H.; Zhong C. M. Shengwuzhi Huaxue Gongcheng 2010, 44, 48–52. [Google Scholar]
- Ostwald W. Z. Phys. Chem., Stoechiom. Verwandtschaftsl. 1897, 22, 289–330. [Google Scholar]
- Karamertzanis P. G.; Pantelides C. C. Mol. Phys. 2007, 105, 273–291. [Google Scholar]
- Price S. L.; Leslie M.; Welch G. W. A.; Habgood M.; Price L. S.; Karamertzanis P. G.; Day G. M. Phys. Chem. Chem. Phys. 2010, 12, 8478–8490. [DOI] [PubMed] [Google Scholar]
- Kazantsev A. V.; Karamertzanis P. G.; Adjiman C. S.; Pantelides C. C. J. Chem. Theory Comput. 2011, 7, 1998–2016. [DOI] [PubMed] [Google Scholar]
- Cooper T. G.; Hejczyk K. E.; Jones W.; Day G. M. J. Chem. Theory Comput. 2008, 4, 1795–1805. [DOI] [PubMed] [Google Scholar]
- Price S. L. Acc. Chem. Res. 2009, 42, 117–126. [DOI] [PubMed] [Google Scholar]
- Gelbrich T.; Hursthouse M. B. CrystEngComm 2005, 7, 324–336. [Google Scholar]
- Etter M. C. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar]
- Braun D. E.; Karamertzanis P. G.; Price S. L. Chem. Commun. 2011, 47, 5443–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D. E.; Tocher D. A.; Price S. L.; Griesser U. J. J. Phys. Chem. B 2012, 116, 3961–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solvates were obtained from acetic acid, formic acid, dioxane, dimethyl formamide, dimethyl sulfoxide, lower alcohols, acetone, ethyl methyl ketone, and tetrahydrofurane.
- Crystal data of AH-I: C7H6O5, Mr = 170.12, triclinic, space group P1̅, T = 25 °C, sample formulation: powder, specimen shape: 12 mm × 1.0 mm × 0.7 mm, wavelength: 1.54056 Å, a = 7.3183 (2) Å, b = 8.2536(2) Å, c = 11.7148(3) Å, α = 100.472(1)°, β = 90.234(2)°, γ = 90.984(2)°, V = 695.73(3) Å3, Z = 4, density = 1.624 g cm–3, 2θ range for data collection: 3–70°, background treatment: Chebyshev polynomial, no. of measured reflections: 580, refinement method: Rietveld, data/parameter/restraints: 580/87/0, goodness of fit: 5.227 (on Yobs), Rwp = 5.904, Rexp = 1.130, Rp = 4.818.
- Crystal data of AH-III: C7H6O5, Mr = 170.12, monoclinic, space group P21/c, T = 25 °C, sample formulation: powder, specimen shape: 12 mm × 1.0 mm × 0.7 mm, wavelength: 1.54056 Å, a = 5.2303(1) Å, b = 5.26495(12) Å, c = 24.7927(4) Å, β = 102.1116(17)°, Z = 4, density = 1.693 g cm–3, 2θ range for data collection: 3–80°, background treatment: Chebyshev polynomial, no. of measured reflections: 414, refinement method: Rietveld, data/parameter/restraints: 414/116/44, goodness of fit: 2.065 (on Yobs), Rwp = 3.074, Rexp = 1.460, Rp = 2.586.
- Crystal data of MH-V: C7H6O5·H2O, Mr = 188.13, monoclinic, space group P21/c, T = 123 °C, sample formulation: powder, specimen shape: 12 mm × 1.0 mm × 0.7 mm, wavelength: 1.54056 Å, a = 7.60719(9) Å, b = 3.64133(4) Å, c = 26.7915(4) Å, β = 98.421(1)°, V = 734.13(2)Å3, Z = 4, Z = 1, density = 1.702 g cm–3, 2θ range for data collection: 3–70°, background treatment: Chebyshev polynomial, no. of measured reflections: 358, refinement method: Rietveld, data/parameter/restraints: 358/71/0, goodness of fit: 4.298 (on Yobs), Rwp = 4.909, Rexp = 1.142, Rp = 4.179.
- Clark S. J.; Segall M. D.; Pickard C. J.; Hasnip P. J.; Probert M. J.; Refson K.; Payne M. C. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar]
- Kaur R.; Guru Row T. N. Cryst. Growth Des. 2012, 12, 2744–2747. [Google Scholar]
- Braun D. E.; Kahlenberg V.; Gelbrich T.; Ludescher J.; Griesser U. J. CrystEngComm 2008, 10, 1617–1625. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.