

Abstract
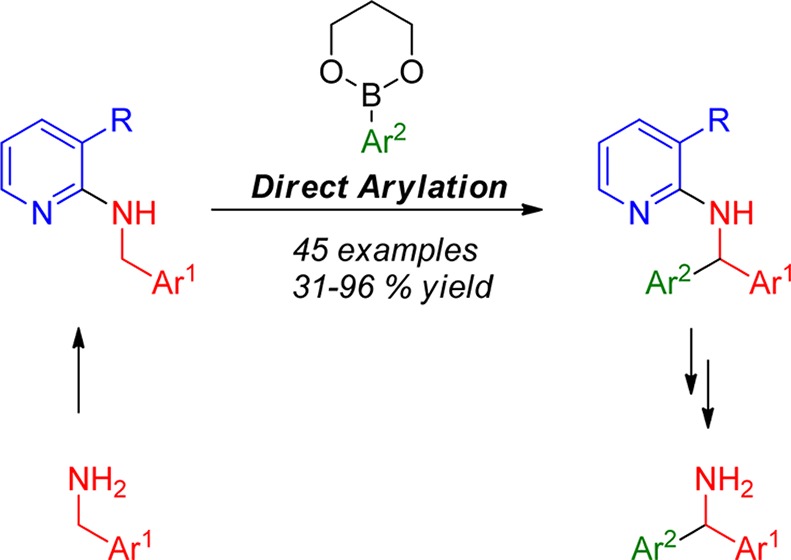
A highly efficient direct arylation process of benzylic amines with arylboronates was developed that employs Ru catalysis. The arylation takes place with greatest efficiency at the benzylic sp3 carbon. If the distance to the activating aryl ring is increased, arylation is still possible but the yield drops significantly. Efficiency of the CH activation was found to be significantly increased by use of 3-substituted pyridines as directing groups, which can be removed after the transformation in high yield. Calculation of the energy profile of different rotamers of the substrate revealed that presence of a substituent in the 3-position favors a conformation with the CH2 group adopting a position in closer proximity to the directing group and facilitating C–H insertion. This operationally simple reaction can be carried out in argon atmosphere as well as in air and under neutral reaction conditions, displaying a remarkable functional group tolerance. Mechanistic studies were carried out and critically compared to mechanistic reports of related transformations.
Introduction
Efficient synthesis of complex organic molecules is a permanent challenge for synthetic chemists. The available portfolio of chemical transformations allows for the synthesis of almost any molecule; however, the introduction of functional groups or substituents in a specific position often requires multistep synthesis, which naturally lowers atom efficiency. Additionally, purification of the intermediates is time-consuming and resource-intensive, and yields over several steps are often rather low. Therefore, one of the most important quests for synthetic chemists is the development of new, more efficient, and direct transformations that allow the elimination of synthetic detours. In this regard, the direct catalytic cleavage of C–H bonds is highly attractive and one of the most investigated but also most challenging topics in modern organic synthesis.1 Much effort is put into realizing such C–H activation reactions since they would increase the atom efficiency of transformations and, therefore, be consistent with the principles of green chemistry.
In recent years, the field of transition-metal-catalyzed C–H activation reactions has rapidly expanded, and the commitment of research groups all around the world afforded many interesting results in this area. However, most of the developed methods are focused on the direct functionalization of sp2 C–H bonds.2 The more challenging direct functionalization of sp3 C–H bonds is a highly attractive process, since regioselective functionalization of such sp3 C–H bonds still requires multistep sequences in many cases in order to address a specific C–H bond without compromising others.3 Cyclometalation is among the proposed solutions to achieve C–H bond activation by transition metal complexes utilizing nearby heteroatoms as directing/coordinating groups.4 Again, many chelation-assisted functionalizations have been reported in the context of sp2 C–H bond activation, but only a few involve sp3 C–H bonds.5 One of the first chelation-assisted transformations of sp3 C–H bonds was described by Jun in 1998.6 A few years later, Kakiuchi et al.7 demonstrated in their pioneering work the arylation of aromatic ketones with arylboronates. Afterward, Sames and co-workers8 published the arylation of pyrrolidines and piperidine directed by a cyclic imine (Figure 1A), followed by the discovery of Maes and co-workers9, where they showed a pyridine-directed arylation of piperidine derivatives (Figure 1B). However, the last two methods were limited to cyclic amines. The limitation to saturated N-heterocycles is probably related to the preferred geometrical alignment of such systems where the directing group is excellently positioned to activate the CH2 group adjacent to the heteroatom. In acyclic systems such a conformational lock is not possible; therefore, the directing group and positions to be activated rather arrange in a way of greatest distance to minimize energy. Hence, in order to successfully achieve a direct substitution via C–H activation, provisions have to be undertaken in order to bring the directing group and the C–H bond to be functionalized in close enough proximity. We reported recently10 the direct arylation of acyclic sp3 C–H bonds adjacent to a free NH group with various arenes via cyclometalation with Ru3(CO)12 (Figure 1C).
Figure 1.
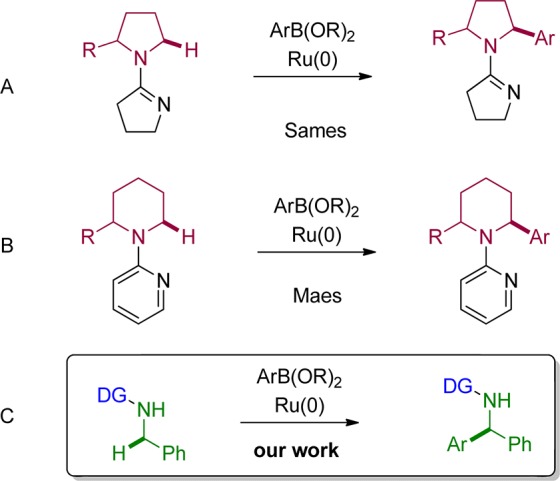
Direct arylation of sp3 C–H bonds adjacent to nitrogen.
Results and Discussion
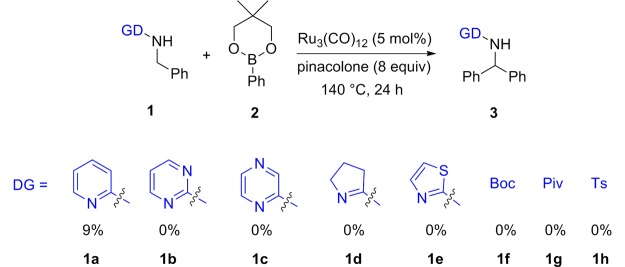
We started our project by investigating the role of the directing group. Therefore, we synthesized different benzylic amines and tested them under the specific conditions described by Sames and co-workers8 (Scheme 1). We tested different heterocyclic, (sulfon)amide, and carbamate directing groups, but only pyridine showed low activity. All other directing groups, including the carbonyl of Boc carbamate and the pivaloyl moiety, were not suitable for this protocol.
Scheme 1. Investigated Directing Groups for Ru-Catalyzed C–H Arylation Protocol.
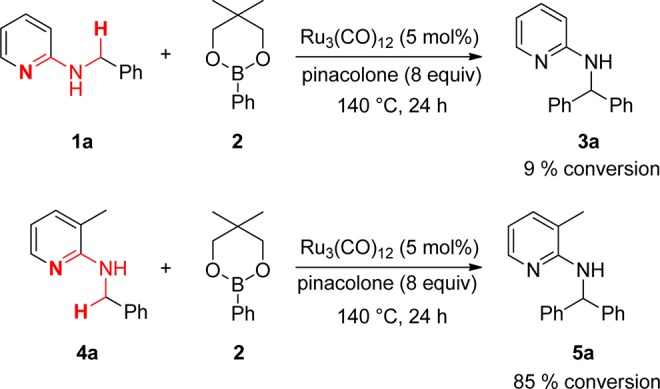
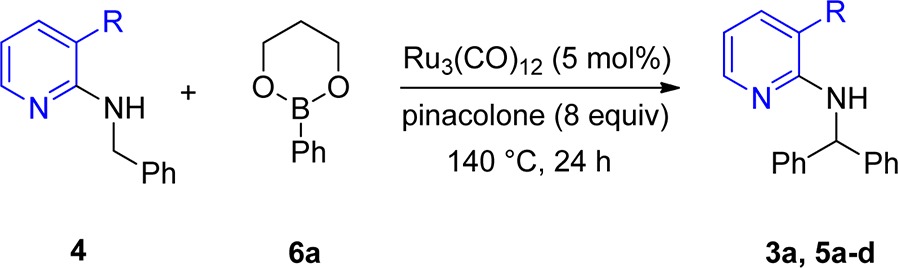
Hence, we focused on pyridine as a suitable directing group. However, intensive screening of the conditions afforded no higher conversion until we discovered that the installation of a substituent in the 3-position of pyridine enhanced the conversion significantly, which was described in our previously published study containing preliminary results (Scheme 2).10 The crucial role of the substituent in 3-position was also described by Jun and co-workers.11 When the direct arylation was carried out with 4a, a much higher conversion (85%) could be achieved compared to the reaction with 1a. We hypothesized that the installation of a sterically demanding group in the 3-position of the pyridine ring forces the benzyl group into a position where the metal and CH2 group are in much closer proximity, which facilitates the C–H insertion of the metal significantly. This Ru(0)-catalyzed arylation of benzylic amines with arylboronates works most efficiently (but is not limited to) when benzylic C–H bonds are arylated. Notably, this operationally simple method does not require the proximity of a heteroatom. In subsequent work we could develop a method for the reaction with aryl halides as well by using a Ru(II) catalyst with the same directing group.12
Scheme 2. Ru-Catalyzed Arylation of sp3 C–H Bond.
Within this paper, we report a systematic evaluation of directing groups, substrate scope exploration, and mechanistic investigations of the Ru(0)-catalyzed direct arylation process.
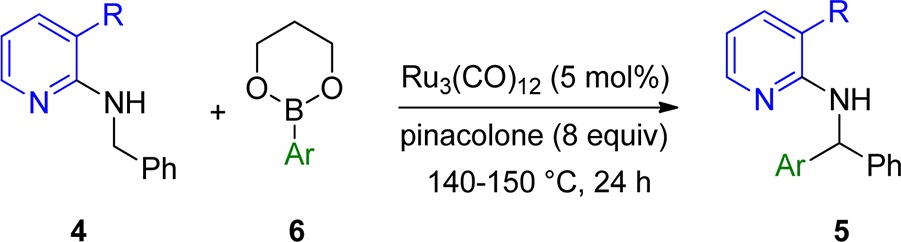
Having identified the presence of a bulky group in the 3-position as a key element of the directing group, we tested which other groups in the 3-position would be tolerated and give similar results as the methyl group in 4a. Therefore, substrates carrying chlorine (4b), CF3 (4c), and phenyl (4d) in the 3-position were prepared. The 3-phenyl substrate 4d could be efficiently prepared from 4b via Suzuki–Miyaura cross-coupling (Scheme 3).13 In the case of chlorine, conversion to the product was detected but only to an extent of ∼10% (Table 1, entry 2). Trifluoromethyl was also a suitable bulky group, and an improved yield of 78% was obtained (Table 1, entry 4). The phenyl group gave even better results, and 90% yield of product was obtained (Table 1, entry 5). This indicates that the size of the bulky group in the 3-position has an influence on the effectiveness of the arylation process. It should be mentioned that the overall process of benzylamine attachment, phenyl introduction, and finally direct arylation to 5d is very efficient and gave 80% overall yield over three steps (Scheme 3).
Scheme 3. Sequential Coupling of 2,3-Dichloropyridine (7) to the Arylated Product (5d).
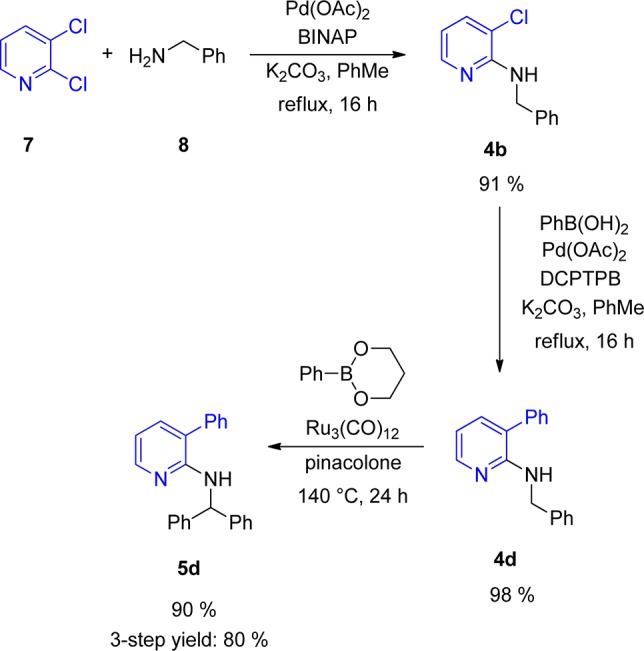
Table 1. Influence of Substituent at the 3-Position of Pyridine on the Direct Arylation Processa.
| entry | reactant 4 | R | product 5 | convb (%) | yieldc (%) |
|---|---|---|---|---|---|
| 1 | 1a | H | 3a | 9 | ni |
| 2 | 4b | Cl | 5b | 10 | ni |
| 3 | 4a | CH3 | 5a | 85 | 64 |
| 4 | 4c | CF3 | 5c | 90 | 78 |
| 5 | 4d | Ph | 5d | 100 | 90 |
Reaction conditions: 4 (0.5 mmol), 6a (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion based on GC analysis with respect to 4 (dodecane as internal standard).
ni = not isolated.
The reaction was further optimized regarding the aryl source, revealing that the 1,3-propanediol boron ester 6 gave the best result.10 This ester is also most convenient in the reaction workup since it can be hydrolyzed easily to the boronic acid, facilitating chromatographic purification of the product.
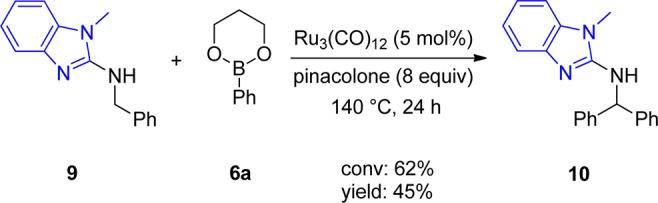
Taking into account the mandatory features of a suitable directing group, we expected N-substituted benzimidazole 9 to perform in a similar way as 4a due to the analogous geometry of the directing group compared to 3-methylpyrid-2-yl. Indeed, this group showed activity, albeit with comparatively lower conversion and yield (Scheme 4). The lower conversion can be rationalized by the differences in geometry of a five- compared to a six-membered ring; this leads to less interference of the N-methyl group with the CH2 group to be arylated as compared to the methyl group in 4a and the benzylic position. Hence the energy difference between the two conformers will be lower and the “right” conformer is less preferred, resulting in lower conversion and yield.
Scheme 4. Ru-Catalyzed Arylation of 9.
Hence, 3-substituted pyridines were chosen as directing groups for further investigations. Such directing groups can be introduced easily via Buchwald–Hartwig amination, starting from commercially available 2-chloro-3-substituted pyridine derivatives and benzylic amines (see Supporting Information). This operationally simple, high-yielding reaction provides an applicable entry to the starting materials.
With the optimized conditions in hand, we wanted to investigate the scope and limitations of the presented transformation. This catalytic method was found to be compatible with arene donors carrying a variety of different functional groups. With 3-methyl substituted pyridine as directing group, simple phenylboronic acid ester gave a good yield of 64% (Table 2, entry 1). Sterically demanding aryls (2-methylphenyl, 1-naphthyl) were not tolerated and led to unsatisfactory yields (Table 2, entries 2 and 3), but meta (3-Me, 3-Cl) and para phenyl substituents showed good reactivity (Table 2, entries 4 and 5). Aryls containing electron-donating alkyl substituents (Me, t-Bu) also gave good yields (Table 2, entries 6 and 7), but 4-methoxy substitution led to a decreased yield of 39% (Table 2, entry 8). Also, a 4-fluoro substituent was well tolerated (Table 2, entry 9); however, 4-Cl (33%) and 4-CF3 (41%) phenyl substituents led to significantly lower yields (Table 2, entries 10 and 11), indicating that electron-withdrawing substituents are unfavorable. Still, halides are tolerated and can be used for further manipulations, for example, cross-coupling reactions. Strong electron-withdrawing substituents were not tolerated (Table 2, entries 12–14). Also, heterocyclic boronic esters were inefficient (Table 2, entries 15 and 16).
Table 2. Ru(0)-Catalyzed Arylation of Pyridine Derivativesa.
| entry | reactant 4 | R | Ar | product 5 | convb (%) | yieldc (%) |
|---|---|---|---|---|---|---|
| 1 | 4a | Me | Ph | 5a | 86 | 64 |
| 2 | 4a | Me | 2-Me-Ph | 5e | 55 | nid |
| 3 | 4a | Me | 1-naph | 5f | 8 | ni |
| 4 | 4a | Me | 3-Me-Ph | 5g | 87 | 61 |
| 5 | 4a | Me | 3-Cl-Ph | 5h | 59 | 38 |
| 6 | 4a | Me | 4-Me-Ph | 5i | 88 | 62 |
| 7 | 4a | Me | 4-t-Bu-Ph | 5j | 87 | 64 |
| 8 | 4a | Me | 4-OMe-Ph | 5k | 50 | 39 |
| 9 | 4a | Me | 4-F-Ph | 5l | 89 | 66 |
| 10 | 4a | Me | 4-Cl-Ph | 5m | 49 | 33 |
| 11 | 4a | Me | 4-CF3-Ph | 5n | 61 | 41 |
| 12 | 4a | Me | 4-Ac-Ph | 5o | 11 | ni |
| 13 | 4a | Me | 4-NO2-Ph | 5p | 0 | 0 |
| 14 | 4a | Me | 4-CN-Ph | 5q | 0 | 0 |
| 15 | 4a | Me | 3-pyridyl | 5r | 0 | 0 |
| 16 | 4a | Me | 2-thienyl | 5s | 0 | 0 |
| 17 | 4c | CF3 | Phe | 5c | 90 | 78 |
| 18 | 4c | CF3 | 4-Me-Phe | 5t | 92 | 77 |
| 19 | 4c | CF3 | 4-t-Bu-Phe | 5u | 84 | 70 |
| 20 | 4c | CF3 | 4-OMe-Phe | 5v | 76 | 61 |
| 21 | 4c | CF3 | 4-F-Phe | 5w | 65 | 51 |
| 22 | 4d | Ph | Phe | 5d | 100 | 90 |
| 23 | 4d | Ph | 4-Me-Phe | 5x | 100 | 85 |
| 24 | 4d | Ph | 4-t-Bu-Phe | 5y | 100 | 96 |
| 25 | 4d | Ph | 4-F-Phe | 5z | 100 | 72 |
| 26 | 4d | Ph | 4-Cl-Phe | 5aa | 87 | 64 |
| 27 | 4d | Ph | 4-CF3-Phe | 5ab | 65 | 31 |
| 28 | 4d | Ph | 4-Ac-Phe | 5ac | 89 | 52 |
| 29 | 4d | Ph | 4- NO2-Phe | 5ad | 0 | 0 |
| 30 | 4d | Ph | 4-CN-Phe | 5ae | 0 | 0 |
Reaction conditions: 4 (0.5 mmol), 6 (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion based on GC analysis with respect to 4 (dodecane as internal standard).
ni = not isolated.
Could not be isolated because of side products.
150 °C.
We replaced the 3-methyl group with the 3-trifluoromethyl group in order to test whether stereoelectronic effects of the 3-substituent had an influence on the reaction outcome. Interestingly, the 3-trifluoromethyl group showed even better yields but also required a higher reaction temperature (Table 2, entries 17–21). For instance, the yield could be increased from 64% to 78% for Ph (Table 2, entry 1 vs entry 17) and from 62% to 77% for 4-Me-Ph (Table 2, entry 6 vs entry 18). In one case, namely, 4-fluoro-substituted boronic acid ester, the yield was lower in the CF3 case as compared to the CH3 case (Table 2, entry 9 vs entry 21).
Additionally, we tested the phenyl group as an even bulkier substituent in 3-position. Indeed, the arylation yield could be significantly increased when the substitutent at the 3-position of the pyridine directing group was changed to a phenyl group (Table 2, entries 22–30). The yield could be increased in almost all cases [e.g., 64% (entry 1) → 90% (entry 22) for Ph; 64% (entry 7) → 96% (entry 24) for 4-t-Bu-Ph]. Even the more problematic electron-withdrawing substituents now gave better yields [e.g., 33% (entry 10) → 64% (entry 26) for 4-Cl-Ph; 52% (entry 28) for 4-Ac-Ph]. However, as expected, strong coordinating substituents such as NO2 and CN showed again no conversion at all (Table 2, entries 29 and 30).
After exploration of the substrate scope of the boronic acid ester coupling partner, the influence of substituents on the benzylic position was investigated by exercising different electronic effects. Model substrates were synthesized with different substituents in the para position of the benzylic substituent, and these precursors were submitted to the optimized conditions with phenylboronic acid ester 6a as coupling partner. In general, it appears that the reaction is very sensitive to the electronic properties of the benzylic amine. Strong electron-donating (Table 3, entries 1 and 2) and electron-withdrawing (Table 3, entries 6 and 7) substituents diminished the conversion. Best results could again be achieved with weak electron-donating or -withdrawing substituents (Table 3, entries 3–5). In the case of the methyl ester substrate 4j, we could also detect decarboxylated compound 5a as a side product. Even though Ru3(CO)12 has been used in decarboxylative coupling reactions,14 this was unexpected since a directing group is lacking to bring the catalyst in proximity to the ester functionality and the pyridine is too far away in order to act as directing group for the decarboxylation process.
Table 3. Competitive Experiments for the Ru(0)-Catalyzed Reactiona.
| entry | reactant 4 | R | X | Y | product 5 | convb (%) | yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 4e | Me | OiPr | H | 5af | 35 | 25 |
| 2 | 4f | Me | OMe | H | 5k | 58 | 32 |
| 3 | 4g | Me | Me | H | 5i | 99 | 76 |
| 4 | 4a | Me | H | H | 5a | 86 | 64 |
| 5 | 4h | Me | F | H | 5l | 73 | 44 |
| 6 | 4i | Me | CF3 | H | 5n | 24 | 15 |
| 7 | 4j | Mec | CO2Me | H | 5ag | 43 | 26 |
| 8 | 4g | Me | Me | Cl | 5ah | 69 | 50 |
| 9 | 4g | Me | Me | CF3 | 5ai | 48 | 33 |
| 10 | 4k | CF3 | Me | H | 5t | 95 | 80 |
| 11 | 4m | Phc | Me | H | 5x | 98 | 90 |
| 12 | 4m | Phc | Me | Me | 5aj | 88 | 73 |
| 13 | 4m | Phc | Me | t-Bu | 5ak | 76 | 67 |
| 14 | 4m | Phc | Me | F | 5al | 71 | 60 |
| 15 | 4m | Phc | Me | CF3 | 5am | 47 | 33 |
Reaction conditions: 4 (0.5 mmol), 6 (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion based on GC analysis with respect to 4 (dodecane as internal standard).
150 °C.
Since the p-methyl substrate 4g gave the best conversion and yield so far, a series of reactions was performed with substrates 4g, 4k, and 4m, all carrying this p-methylbenzyl substituent and different blocking groups in the 3-position of pyridine. In the introduction of the phenyl substituent, a significantly higher yield was obtained with 4g as substrate (Table 3, entry 3, 76%) as compared to 4a (Table 2, entry 1, 64%). In cases where 4a gave low conversions and yield (Table 2, entries 10 and 11) the difference compared to 4g as substrate was negligible (Table 3, entries 8 and 9). For p-methylbenzyl-substituted substrates with either trifluoromethyl (4k) or phenyl (4m) as a bulky substituent in the 3-position of pyridine (Table 3, entries 10 and 11), the yields were almost identical to those of substrates 4c and 4d. Substrate 4m was also reacted with different aryl donors (Table 3, entries 12–15). The trend of decrease of conversion from electron-donating to -withdrawing substituents is again similar to the previous observed results.
In order to investigate the relative reactivity of differently substituted starting materials, we performed a series of intermolecular competition experiments. For this purpose, 1 equiv each of two different starting materials and 1 equiv of phenylboronic acid ester 6a were reacted under standard conditions (Scheme 5). The reaction was monitored via gas chromatography (GC), and the product distribution was also determined by GC analysis. The obtained results indicate that substrates with electron-neutral or weakly electron-donating substituents react fastest, which also goes in line with the finding that those starting materials give the highest yields. The 4-methyl-substituted starting material 4g reacts at the same rate as unsubstituted 4a (Scheme 5, H:X = 1). The substrates carrying electron-donating (4e and 4f) and electron-withdrawing substituents (4h and 4i) react comparatively more slowly. In the case of the methyl ester 4j, again we observed decarboxylation to 5a as a side reaction. Hence, the high H:X ratio of 5.3 does not correlate to the difference in reaction rate in this case.
Scheme 5. Competitive Experiments for the Ru(0)-Catalyzed Reaction.

Finally, we investigated whether the reaction is limited to C–H bonds adjacent to nitrogen. We found that the NH group can be replaced by a CH2 group as in 11, which even gives an improved yield in the arylation step (Scheme 6, 75% yield of 14). However, extending the reaction time to 36 h was required. Interestingly, the reaction did not work when NH was replaced with oxygen as in 12 (Scheme 6, 0% yield of 15). Furthermore, the transformation also works with nonbenzylic sp3 C–H bonds as in 13, however, again with longer reaction time and lower yield (Scheme 6, 39% yield of 16). Overall, this suggests that an adjacent NH has a significantly lower activating influence (if any) on the CH2 group as compared to the adjacent phenyl group.
Scheme 6. Direct Transformation of C–H Bond Adjacent to CH2 Group (11) and Nonbenzylic C–H Bond (13).
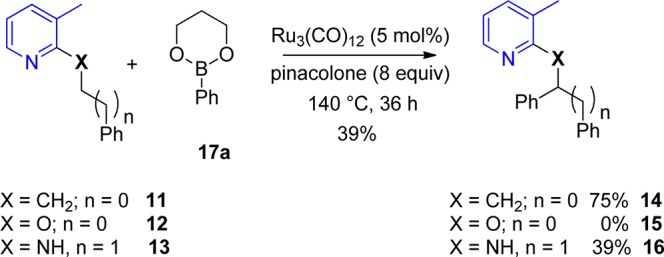
To strengthen our hypothesis that a bulky group in position 3 of pyridine favors the desired conformation of the substrate, we calculated the structures of two stable rotamers of N-benzyl-3-methylpyridine-2-amine 4a and N-benzylpyridine-2-amine 1a and the corresponding transition states for their interconversion by means of density functional theory (DFT) calculations (Gaussian 03/PBE1PBE; see Supporting Information).15 The energy profiles, optimized structures, and transition states obtained are presented in Figure 2 (results for the 1a system are given in parentheses). The methyl substituent in the 3-position of the pyridine moiety stabilizes rotamer A over rotamer B by 4.4 kcal/mol, while in the case of parent amine the energies of both rotamers are essentially the same, differing merely by 0.5 kcal/mol. The energy barrier for interconversion of A to B via 180° rotation around the C–N bond is slightly higher in the case of 4a by 1.6 kcal/mol. Accordingly, chelate-assisted C–H bond activation at the benzylic C–H bonds by transition metals is facilitated in the case of 4a and most likely generally by derivatives with bulky substituents in the 3-position.
Figure 2.
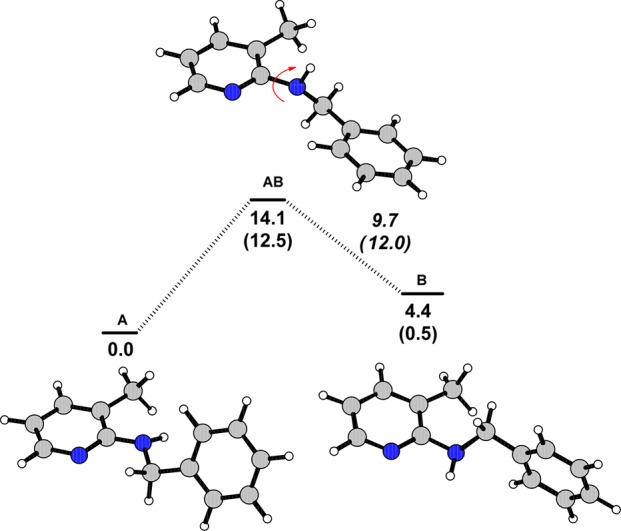
Energy profile (PBE1PBE) for interconversion of the stable N-benzyl-3-methylpyridine-2-amine 4a rotamers A and B via rotation about the C–N bond. The numbers in parentheses refer to parent N-benzylpyridine-2-amine 1a. Energy values (in kilocalories per mole) are referred to the more stable rotamer A.
So far, only substrates with a free NH group had been tested (with the exception of 11 and 12). Now we tried to introduce a second substituent on the amino nitrogen to see whether such substituents would be tolerated. Use of compound 17 as starting material, which carries an N-methyl group but lacks the pyridine methyl group, gave very low conversion of 8% (Table 4, entry 3). Furthermore, compound 18 led also to a decreased conversion of only 17% (Table 4, entry 4). Other substituents on the amine nitrogen such as acetyl, pivalyl, or benzoyl (substrates 19–24 in the Experimental Section) did not give any conversion, whether the methyl group at the 3-position of pyridine was present or not.
Table 4. Direct Arylation of N-Substituted Benzylic Aminesa.
| entry | starting material | R1 | R2 | convb (%) |
|---|---|---|---|---|
| 1 | 1a | H | H | 9 |
| 2 | 4a | Me | H | 86 |
| 3 | 17 | H | Me | 8 |
| 4 | 18 | Me | Me | 17 |
Reaction conditions: Benzylic amine (0.5 mmol), 6a (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion determined by GC analysis with respect to benzylic amine.
Further DFT calculations showed that the N-Me substitution leads to a decreased energy difference between rotamer B, which has the preferred conformation for C–H insertion of the metal, over rotamer A (Figure 3). As shown in Figure 2, rotamer B of N-benzyl-3-methylpyridine-2-amine 4a is stabilized by 4.4 kcal/mol over rotamer A. After N-methylation, rotamer B of N-benzyl-N-3-dimethylpyridin-2-amine 18 shows only 0.2 kcal/mol stability over A (the stability of rotamer B of compound 17 over rotamer A was 0.9 kcal/mol). We hypothesize that this decreased stability causes a lower energy barrier for rotation of the benzylic amine around the N–C bond, which hinders the metal insertion into the C–H bond. This calculation explains the lower conversion (17%) of 18 compared to the unsubstituted compound 4a (86%).
Figure 3.
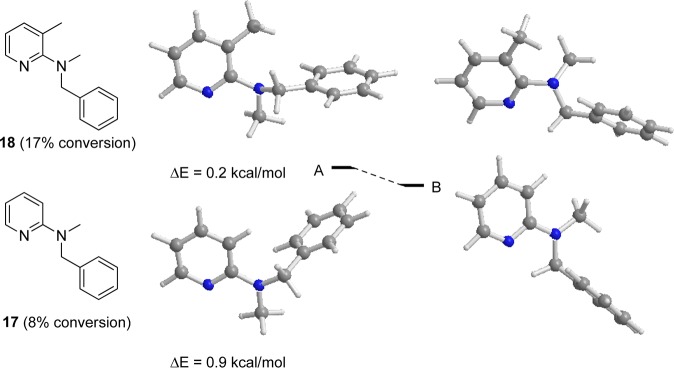
Energy profile (PBE1PBE) for interconversion of stable 17 and 18 rotamers A and B via rotation around the C–N bond. Energy values (in kilocalories per mole) are referred to the more stable rotamer A.
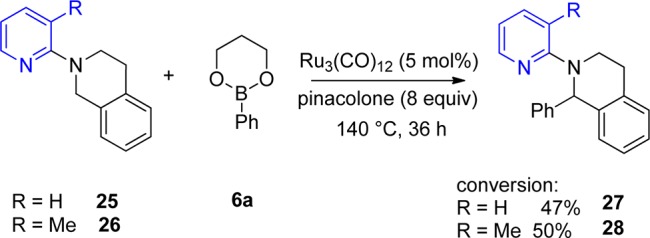
On cyclic tetrahydroisoquinoline substrates 25 and 26, which have of course a locked conformation in which the CH2 group has to point toward the pyridine nitrogen, the reaction also worked but with moderate conversion (Scheme 7). However, this example shows that the 3-methyl group functions only to favor the conformation that can be arylated. Since 27 and 28 are formed in the same yield essentially, influences of the 3-methyl group other than steric ones can be excluded.
Scheme 7. Direct Arylation of N-Substituted Tetrahydroisoquinolines 25 and 26.
After having established the substrate scope of the transformation, we set out to investigate the mechanism of this arylation process. For similar reactions, two different mechanisms were proposed by the groups of Sames8 and Maes.9
So far, all reactions were carried out under an argon atmosphere with pinacolone as solvent (Table 5, entry 1). Sames and co-workers8 argued that the ketone was important to form an intermediate metal–alkoxy complex that can then undergo transmetalation. Maes and co-workers,9 on the other hand, reported that an alcohol as solvent is important to trap a formed alkyl–borate species upon concomitant formation of hydrogen. Hence, we investigated the effect of different solvents and atmospheres to check if one of these two mechanisms is operable in our case.
Table 5. Screening of Conditionsa.
| entry | atmosphere | solvent | convb (%) | yieldc,d (%) |
|---|---|---|---|---|
| 1 | argon | pinacolone | 86 | 69 [64] |
| 2 | argon | acetone/dioxane (1:1) | 82 | 62 |
| 3 | argon | cyclohexanone | 83 | 61 |
| 4 | argon | acetophenone | 80 | 61 |
| 5 | air | pinacolone | 91 | 73 [61] |
| 6 | CO | pinacolone | 65 | 43 |
| 7 | H2 | pinacolone | 97 | 93 [91] |
| 8 | argone | 3-ethyl-3-pentanol | 96 | 45 |
| 9 | argonf | pinacolone | 99 | 59 [53] |
Reaction conditions: 4a (0.5 mmol), 6a (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion based on GC analysis with respect to 4a (dodecane as internal standard).
Yield determined by GC analysis with respect to 4a (dodecane as internal standard).
Number in brackets is yield of 5a.
The reaction was performed in a vial with septum and argon balloon.
Microwave reaction: 170 °C for 2.5 h.
The nature of the ketone seems to be irrelevant, since all investigated ketones gave similar conversions and yield (Table 5, entries 1–4). In the case of acetone, a 1:1 mixture with dioxane had to be used in order to reach the required temperature. In terms of operational simplicity, we tested whether the reaction could also be carried out in air, that is, if the catalyst is stable under air at such elevated temperatures. Gratifyingly, we found that the conversion remained in a similar range under air and also the yield was within experimental error (Table 5, entry 5). We also carried out the reaction under atmospheres of CO and H2. As expected, CO as strong coordinating ligand led to a decreased catalyst activity (Table 5, entry 6). The group of Maes9 suggested that the Ru(II) hydride species obtained after C–H insertion undergoes transmetalation with the boronic ester directly leading to a pinacolborane species. Since this pinacolborane could poison the catalyst, they suggest trapping it with an alcohol to give a boronate and H2. Indeed, they were able to detect H2 via Raman spectroscopy, supporting their mechanistic suggestion. We hypothesized that an atmosphere of H2 should decrease the conversion and yield if the reaction proceeds via this mechanism, since trapping of the borane species might be hampered. Interestingly, the reaction worked even better under hydrogen, suggesting that either no hydrogen is produced in the current transformation or it has no detrimental influence on the arylation (Table 5, entry 7). Regarding the solvent, pinacolone can be replaced by 3-ethyl-3-pentanol, albeit the yield was lower (Table 5, entry 8). Hence, it can be excluded that in the case of pinacolone as ketone, a reduction to the corresponding alcohol is the initial step of the reaction. If that were the case, the yield with 3-ethyl-3-pentanol as solvent should be significantly higher, in the range of the yields obtained with ketones as solvents. The improved result under H2 atmosphere cannot be explained at this point. It could be possible that under H2 atmosphere a more reactive catalytic species is formed, but evidence for that is lacking.
To test whether pinacolborane (HBpin) indeed hampers the reaction by poisoning the catalyst, we performed the reaction in the presence of 1 equiv of HBpin. Maes and co-workers9 speculated that such a pinacolborane species could poison the catalyst eventually by oxidative addition, and the reaction should actually be shut down almost completely when such a great excess of HBpin is present. We observed that the conversion indeed decreased to 75% in comparison to 95% without HBpin. However, even with a large excess of HBpin with respect to the catalyst, the conversion is still high. This result indicates that HBpin eventually produced in the reaction is not poisoning the catalyst, and the decrease of conversion in the control experiment could be explained by other reasons (e.g., dilution, change of the internal temperature, etc.).
It seems that the mechanism described by the Maes group9 is either not operable in this case or at least not the preferred one for this particular transformation. Seemingly, the arylation proceeds rather via the mechanism suggested by Sames and co-workers8 in our case. Notably, the reaction is also feasible under microwave irradiation (Table 5, entry 9), which shortens the reaction time significantly (2.5 h instead of 24 h) with only a slight decrease in yield (59% microwave vs 64% conventional heating).
Since an H2 atmosphere was found to improve the yield in our standard reaction, we investigated whether this can be generalized to other boronic esters. As already shown in Table 5, the yield for reaction of 4a with boronic ester 6a improved from 64% to 91% under H2 atmosphere (Table 6, entries 1 and 2). Also, in the case of 4-methylphenylboronic ester, the yield improved significantly from 62% to 83% (Table 6, entries 3 and 4). However, other boronic esters, such as 4-methoxy- or 4-trifluoromethyl-substituted ones (Table 6, entries 5–8), performed worse. Due to this discrepancy, reaction under argon atmosphere is the more reliable choice.
Table 6. Ru(0)-Catalyzed Arylation of 4a under Argon and Hydrogena.
| entry | Ar | atmosphere | 5 | convb (%) | yieldc (%) |
|---|---|---|---|---|---|
| 1 | Ph | argon | 5a | 86 | 64 |
| 2 | Ph | H2 | 5a | 97 | 91 |
| 3 | 4-Me-Ph | argon | 5f | 88 | 62 |
| 4 | 4-Me-Ph | H2 | 5f | 98 | 83 |
| 5 | 4-OMe-Ph | argon | 5h | 50 | 39 |
| 6 | 4-OMe-Ph | H2 | 5h | 23 | ni |
| 7 | 4-CF3–Ph | argon | 5k | 61 | 41 |
| 8 | 4-CF3–Ph | H2 | 5k | 65 | 35 |
Reaction conditions: 4a (0.5 mmol), 6a (0.75 mmol), Ru3(CO)12 (5 mol %), and pinacolone (0.5 mL).
Conversion based on GC analysis with respect to 4a (dodecane as internal standard).
ni = not isolated.
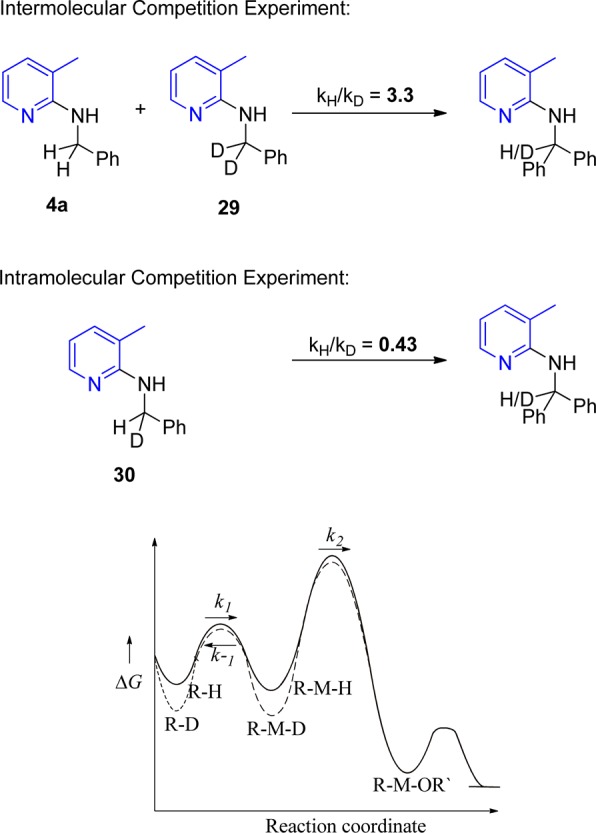
Kinetic isotope effect (KIE) studies were carried out for inter- and intramolecular experiments to gain additional mechanistic insight. The KIE was found to be kH/kD = 3.3 for the intermolecular experiment with starting material 4a and its deuterated counterpart 29. However, this primary isotope effect does not provide evidence whether C–H bond cleavage occurs during the rate-determining step of the reaction.16 Interestingly, the intramolecular KIE for substrate 30 was found to be kH/kD = 0.43, which can be explained by a reversible C–H activation step (also termed inverse equilibrium isotope effect).17 Naturally, the C–H bond is preferentially broken over the C–D bond. However, the R–Ru–D intermediate is more stable as compared to the R–Ru–H species. Hence, the reverse reaction for R–Ru–D is slower, and this intermediate can be accumulated (k–1H > k–1D). Obviously the subsequent step in the catalytic cycle is significantly slower (i.e., rate-determining), and due to the much higher concentration of R–Ru–D in the reaction mixture, this species is converted more often, resulting in the observed inverse equilibrium isotope effect. These results demonstrate that the C–H activation step is not the rate-determining step; otherwise, a reversed KIE would not be observed. The energy diagram in Scheme 8 shows qualitatively the energy profile of this reaction up to the proposed Ru–alkoxy complex (no detailed DFT calculations were carried out since there is not known enough about the nature of the intermediate Ru complexes).
Scheme 8. Competitive Deuterium-Labeling Experiment.
Mechanistic Proposal
In agreement with the work of Kakiuchi et al.,7 we propose the mechanism shown in Scheme 9. In the first step, the Ru(0) complex is coordinated to the pyridine nitrogen before C–H insertion to 31 takes place. The ketone reacts with this intermediate complex via hydride transfer and forms a metal–alkoxy (Ru–OR) species 33, which facilitates transmetalation with Ar–B(OR)2. Therefore, the ketone works as hydrogen and boron scavenger simultaneously. We could detect the formed alcohol species by GC–mass spectrometry (MS). The reductive elimination delivers finally product 5 and regenerates the catalyst.
Scheme 9. Proposed Mechanism.

Potentially, imine formation from the amine substrate and subsequent sp2 arylation via nucleophilic attack of the boronic acid ester could also be possible. However, we excluded this possibility for several reasons. First of all, the reaction also works when NH is substituted by a CH2 group, where imine formation is not possible. Also, when the reaction mixtures were analyzed, no imine was detected in any case (GC-MS analysis) from either the substrate or the product. This is in contrast to our previously reported Ru(II)-catalyzed arylation protocol,12 where we detected the arylated imine in most cases. Additionally, the reaction also proceeds in H2 atmosphere, where imine formation is certainly disfavored. Also, the tetrahydroisoquinoline substrates 25 and 26 give the product. In these cases, no imine but only an iminium cation could be formed. Hence, we excluded the possibility of intermediate imine formation and subsequent arylation.
Very recently, a reductive deprotection protocol for the pyridine directing group was reported.9 In this example the pyridine ring was first reduced (Pd/C/H2) to the corresponding cyclic imine before it was cleaved by hydrazine and acetic acid as reported previously.8 Due to the initial reduction step, it was expected that this protocol would not be suitable for our systems since rather “benzylic deprotection” will occur, leading to diphenylmethane as well as 3-methylpyridine-2-amine. Indeed, the benzylic product 5a delivered only diphenylmethane as expected. Therefore, we adopted a strategy recently reported by Studer and co-workers18 (Scheme 10). Initially the amino group of 5a is Boc-protected before N-methylation of the pyridyl group. A final hydrolysis of the pyridinium salt delivers the Boc-protected diphenylmethanamine in high yield (84% overall). The Boc-protected compounds can be used in further reactions or deprotected by well-established protocols.19
Scheme 10. Cleavage of the Directing Group.

Conclusion
In conclusion, benzylic amines were readily arylated with various arenes via cyclometalation with Ru3(CO)12. 3-Substituted pyridine emerged as a useful directing group for this transformation. Various bulky groups (CH3, CF3, Ph) were applied and usually led to the desired products in good yield. Only chlorine was inefficient, being apparently not bulky enough. The free NH group seems to be crucial for this reaction but could also be replaced by CH2 without decrease in yield. The reaction seems to be sensitive toward the electronic and steric nature of the substituents on the aryl donor and the benzylic amine. This operationally simple reaction can be performed under air and showed even better activity under H2 atmosphere in selected examples. Finally, removal of the directing group was successfully demonstrated. The performed mechanistic investigations support the mechanism proposed by Kakiuchi et al.,7 which is also supported by the group of Sames.8
Experimental Section
General Methods
All reactions were carried out under argon, unless otherwise mentioned. Argon was purified by passage through Drierite. Unless otherwise noted, chemicals were purchased from commercial suppliers and used without further purification. Microwave reactions were performed on a Biotage Initiator 60 microwave unit (max pressure 20 bar, IR temperature sensor). High-resolution mass spectrometry (HRMS) for literature-unknown compounds was performed by liquid chromatography in combination with hybrid ion trap and high-resolution time-of-flight mass spectrometry (LC-IT-TOF-MS) in only positive-ion detection mode with the recording of standard (MS) and tandem (MS/MS) spectra. NMR spectra were recorded in CDCl3 with tetramethylsilane (TMS) as internal standard, and chemical shifts are reported in parts per million (ppm). GC-MS runs were performed on a standard capillary column BGB 5 (30 m × 0.32 mm i.d.).
General Procedure I for Preparation of Benzylic Amines
2-Chloro-3-substituted pyridine (1 equiv), amine (1.2 equiv), K2CO3 (3.5 equiv), Pd(OAc)2 (2 mol %), and 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP; 2 mol %) were placed in an oven-dried 6 mL vial with a septum screw cap and a magnetic stirring bar. The vial was evacuated and flushed with argon (three times). After addition of dry toluene to the reaction mixture, the vial was closed with a fully covered solid Teflon-lined cap. The reaction vial was then heated in a reaction block at 130 °C for 16 h. After cooling to room temperature (rt), the solid material was removed by filtration and washed with 10 mL of CH2Cl2. The combined organic layers were evaporated, and the resulting crude product was purified by flash column chromatography [petroleum ether/ethyl acetate (PE/EtOAc) = 10:1].
General Procedure II for C–H Activation Reaction
Pyridine derivative (0.5 mmol, 1 equiv), arylboronic acid ester (0.75 mmol, 1.5 equiv), and Ru3(CO)12 (0.025 mmol, 5 mol %) were placed in an oven-dried 6 mL-vial with a septum screw cap and a magnetic stirring bar. The vial was evacuated and flushed with argon (three times). After addition of 0.5 mL of dry pinacolone to the reaction mixture, the vial was closed with a fully covered solid Teflon-lined cap. The reaction vial was then heated in a reaction block at 140 °C for 24–36 h. After cooling to rt, 2 mL of EtOAc and 2 mL of water were added to the reaction mixture, which was stirred for 5 min at rt. The reaction mixture was extracted with EtOAc (three times). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by flash column chromatography (PE/EtOAc = 49:1) and dried under high vacuum.
General Procedure III for Preparation of Tertiary Amines
2-Bromo-3-substituted pyridine (1 equiv), amine (1.4 equiv), NaOtBu (2 equiv), tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba)3; 2 mol %], and DPPP [1,3-bis(diphenylphosphino)propane, 2 mol %] were placed in an oven-dried 6 mL vial with a septum screw cap and a magnetic stirring bar. The vial was evacuated and flushed with argon (three times). After addition of dry toluene to the reaction mixture, the vial was closed with a fully covered solid Teflon-lined cap. The reaction vial was then heated in a reaction block at 75 °C for 16 h. After cooling to rt, the solid material was removed by filtration and washed with 10 mL of CH2Cl2. The combined organic layers were evaporated, and the resulting crude product was purified by flash column chromatography (PE/EtOAc = 15:1/10:1).
General Procedure IV for Preparation of Amides
A 3 M solution of CH3MgCl in tetrahydrofuran (THF; 1.2 equiv) was added dropwise to a solution of N-benzylpyridin-2-amine (1 equiv) in dry THF (5 mL) at rt, and the mixture was stirred for 10 min at that temperature. The acyl chloride (3 equiv) was dissolved in 2 mL of THF and then added slowly to the solution. The stirring was continued at rt for 1 h (or full conversion, monitored by thin-layer chromatography, TLC). Then the reaction was quenched with H2O, and the resulting solution was extracted with Et2O (3 × 5 mL). The combined organic layers were washed with NaHCO3 (2×) and brine (2×), dried over Na2SO4, filtered, and concentrated in vacuo.
N-Benzyl-3-methylpyridin-2-amine (4a)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), benzylamine (128 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Analytical data are in accordance with the literature.20 Colorless solid (182 mg, 92% yield); mp 48–49 °C; 1H NMR (CDCl3, 200 MHz) δ 2.09 (s, 3H), 4.36 (s, 1H), 4.70 (d, J = 5.3 Hz, 2H), 6.57 (dd, J = 7.1, 5.1 Hz, 1H), 7.23–7.43 (m, 6H), 8.06 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 45.9, 113.0, 116.6, 127.3, 128.0, 128.7, 136.9, 140.1, 145.6, 156.8.
N-Benzyl-3-chloropyridin-2-amine (4b)
Reaction of 2,3-dichloropyridine (148 mg, 1 mmol, 1 equiv), benzylamine (128 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Analytical data are in accordance with the literature.21 Yellow oil (200 mg, 91% yield); 1H NMR (CDCl3, 200 MHz) δ 4.68 (d, J = 5.6 Hz, 2H), 5.26 (s, 1H), 6.54 (dd, J = 7.6, 4.9 Hz, 1H), 7.23–7.39 (m, 5H), 7.45 (dd, J = 7.6, 1.6 Hz, 1H) 8.04 (dd, J = 4.9, 1.6 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 45.6, 113.2, 115.4, 127.4, 127.8, 128.7, 136.2, 139.4, 146.2, 154.0.
N-Benzyl-3-(trifluoromethyl)pyridin-2-amine (4c)
Reaction of 2-chloro-3-(trifluoromethyl)pyridine (182 mg, 1 mmol, 1 equiv), benzylamine (128 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless oil (238 mg, 95% yield); 1H NMR (CDCl3, 200 MHz) δ 4.66 (d, J = 5.3 Hz, 2H), 5.11 (s, 1H), 6.56 (dd, J = 7.5, 5.0 Hz, 1H), 7.16–7.28 (m, 5H), 7.59 (dd, J = 7.6, 0.8 Hz, 1H), 8.20 (d, J = 4.4 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 45.4, 108.7 (q, J = 31.3 Hz), 118.8, 124.6 (q, J = 271.5 Hz) 127.4, 127.6, 128.8, 135.1 (q, J = 5.1 Hz), 139.2, 151.9, 154.4. HRMS calcd for C13H11F3N2+: [M + H]+ 253.0947. Found: [M + H]+ 253.0955.
N-Benzyl-3-phenylpyridin-2-amine (4d)
Reaction of N-benzyl-3-chloropyridin-2-amine 4c from the above protocol (219 mg, 1 mmol, 1 equiv), phenylboronic acid (366 mg, 3 mmol, 3 equiv), K2CO3 (276 mg, 2 mmol, 2 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (DCPTPB; 10 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless solid (255 mg, 98% yield); mp 58–60 °C; 1H NMR (CDCl3, 200 M + Hz) δ 4.64 (d, J = 5.6 Hz, 2H), 4.88 (s, 1H), 6.66 (dd, J = 7.2, 5.1 Hz, 1H), 7.18–7.42 (m, 11H), 8.14 (dd, J = 4.9, 1.5 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 45.6, 113.1, 122.4, 127.1, 127.5, 127.9, 128.6, 129.0, 129.3, 137.2, 138.0, 140.0, 147.2, 155.5. HRMS calcd for C18H16N2+: [M + H]+ 261.1386. Found: [M + H]+ 261.1390.
N-(4-Isopropoxybenzyl)-3-methylpyridin-2-amine (4e)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), (4-isopropoxyphenyl)methanamine (198 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless oil (185 mg, 72% yield); 1H NMR (CDCl3, 200 MHz) δ 1.33 (d, J = 6.0 Hz, 6H), 2.06 (s, 3H), 4.27 (s, 1H), 4.51 (sep, J = 6.0 Hz, 1H), 4.59 (d, J = 5.2 Hz, 2H), 6.54 (dd, J = 7.1, 5.1 Hz, 1H), 6.86 (d, J = 8.6 Hz, 2H), 7.20–7.32 (m, 3H), 8.05 (dd, J = 5.1, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 21.2, 45.5, 70.0, 112.9, 116.0, 116.6, 129.3, 131.9, 136.9, 145.6, 156.8, 157.3. HRMS calcd for C16H20N2O+: [M + H]+ 257.1648. Found: [M + H]+ 257.1642.
N-(4-Methoxybenzyl)-3-methylpyridin-2-amine (4f)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), 4-methoxybenzylamine (164 mg, 1.2 mmol, 1.2 equiv), K2CO3 (414 mg, 3 mmol, 3 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Yellow oil (183 mg, 80% yield); 1H NMR (CDCl3, 200 MHz) δ 2.07 (s, 3H), 3.80 (s, 3H), 4.30 (s, 1H), 4.61 (d, J = 5.2 Hz, 2H), 6.55 (dd, J = 7.1, 5.1 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H), 7.21–7.34 (m, 3H), 8.06 (dd, J = 4.9, 1.0 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 45.4, 55.4, 112.9, 114.1, 116.6, 129.3, 132.1, 136.9, 145.5, 156.8, 158.9. HRMS calcd for C14H16N2O+: [M + H]+ 229.1335. Found: [M + H]+ 229.1338.
3-Methyl-N-(4-methylbenzyl)pyridin-2-amine (4g)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), 4-methylbenzylamine (145 mg, 1.2 mmol, 1.2 equiv), K2CO3 (414 mg, 3 mmol, 3 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless solid (188 mg, 88% yield); mp 46–47 °C; 1H NMR (CDCl3, 200 MHz) δ 2.01 (s, 3H), 2.30 (s, 3H), 4.26 (s, 1H), 4.59 (d, J = 5.2 Hz, 2H), 6.50 (dd, J = 7.1, 5.1 Hz, 1H), 7.08–7.26 (m, 5H), 8.00 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 21.2, 45.8, 112.9, 116.6, 128.0, 129.4, 136.8, 136.9, 137.0, 145.5, 156.8. HRMS calcd for C14H16N2+: [M + H]+ 213.1386. Found: [M + H]+ 213.1380.
N-(4-Fluorobenzyl)-3-methylpyridin-2-amine (4h)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), (4-fluorophenyl)methanamine (150 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless oil (158 mg, 73% yield); 1H NMR (CDCl3, 200 MHz) δ 2.08 (s, 3H), 4.36 (s, 1H), 4.65 (d, J = 5.4 Hz, 2H), 6.56 (dd, J = 7.1, 5.1 Hz, 1H), 6.96–7.05 (m, 2H), 7.21–7.37 (m, 3H), 8.03 (dd, J = 5.0, 1.2 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 45.1, 113.2, 115.5 (d, JCF = 21.3 Hz), 116.6, 129.5 (d, JCF = 8.0 Hz), 135.9 (d, JCF = 3.1 Hz), 137.1, 145.6, 156.6, 162.2 (d, JCF = 244.9 Hz). HRMS calcd for C13H13FN2+: [M + H]+ 217.1136. Found: [M + H]+ 217.1128.
3-Methyl-N-[4-(trifluoromethyl)benzyl]pyridin-2-amine (4i)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), [4-(trifluoromethyl)phenyl]methanamine (210 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless solid (195 mg, 73% yield); mp 54–55 °C; 1H NMR (CDCl3, 200 MHz) δ 2.14 (s, 3H), 4.50 (s, 1H), 4.78 (d, J = 5.7 Hz, 2H), 6.58 (dd, J = 7.1, 5.1 Hz, 1H), 7.25–7.29 (m, 1H), 7.53 (d, J = 9.7 Hz, 4H), 8.03 (dd, J = 5.0, 1.2 Hz, 1H).; 13C NMR (CDCl3, 50 MHz) δ 17.1, 45.2, 113.5, 116.7, 124.4 (q, JCF = 271.9 Hz), 125.6 (q, JCF = 3.9 Hz), 127.9, 129.4 (q, JCF = 32.3 Hz), 137.2, 144.6, 145.6, 156.4. HRMS calcd for C14H13F3N2+: [M + H]+ 267.1104. Found: [M + H]+ 267.1099.
Methyl 4-{[(3-Methylpyridin-2-yl)amino]methyl}benzoate (4j)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), methyl 4-(aminomethyl)benzoate (198 mg, 1.2 mmol, 1.2 equiv), K2CO3 (414 mg, 3 mmol, 3 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless solid (223 mg, 87% yield); mp 122–123 °C; 1H NMR (CDCl3, 200 MHz) δ 2.12 (s, 3H), 3.90 (s, 3H), 4.48 (s, 1H), 4.77 (d, J = 5.7 Hz, 2H), 6.56 (dd, J = 7.1, 5.1 Hz, 1H), 7.23–7.27 (m, 1H), 7.43 (d, J = 8.2 Hz, 2H), 7.97–8.02 (m, 3H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 45.3, 52.2, 113.3, 116.6, 127.5, 129.0, 130.0, 137.1, 145.6, 145.8, 156.5, 167.1. HRMS calcd for C15H16N2O2+: [M + H]+ 257.1285. Found: [M + H]+ 257.1296.
N-(4-Methylbenzyl)-3-(trifluoromethyl)pyridin-2-amine (4k)
Reaction of 2-chloro-3-(trifluoromethyl)pyridine (182 mg, 1 mmol, 1 equiv), methylbenzylamine (145 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless oil (260 mg, 98% yield); 1H NMR (CDCl3, 200 MHz) δ 2.33 (s, 3H), 4.67 (d, J = 5.2 Hz, 2H), 5.13 (s, 1H), 6.62 (dd, J = 7.5, 5.0 Hz, 1H), 7.12–7.26 (m, 4H), 7.65 (dd, J = 7.6, 0.8 Hz, 1H), 8.27 (d, J = 4.4 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 45.2, 108.6 (q, J = 31.3 Hz), 111.6, 124.6 (q, J = 271.5 Hz) 127.6, 129.4, 135.1 (q, J = 5.1 Hz), 136.1, 137.0, 151.8, 154.5. HRMS calcd for C14H13F3N2+: [M + H]+ 267.1104. Found: [M + H]+ 267.1093.
3-Chloro-N-(4-methylbenzyl)pyridin-2-amine (4l)
Reaction of 2,3-dichloropyridine (148 mg, 1 mmol, 1 equiv), 4-methylbenzylamine (145 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless oil (201 mg, 86% yield); 1H NMR (CDCl3, 200 MHz) δ 2.35 (s, 3H), 4.65 (d, J = 5.4 Hz, 2H), 5.23 (s, 1H), 6.54 (dd, J = 7.6, 4.9 Hz, 1H), 7.14–7.30 (m, 4H), 7.45 (dd, J = 7.6, 1.6 Hz, 1H) 8.04 (dd, J = 4.9, 1.6 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 45.4, 113.1, 115.4, 127.8, 129.4, 136.1, 136.3, 137.0, 146.2, 154.0. HRMS calcd for C13H13ClN2+: [M + H]+ 233.0840. Found: [M + H]+ 233.0849.
N-(4-Methylbenzyl)-3-phenylpyridin-2-amine (4m)
Reaction of 3-chloro-N-(4-methylbenzyl)pyridin-2-amine 4l from the above protocol (233 mg, 1 mmol, 1 equiv), phenylboronic acid (366 mg, 3 mmol, 3 equiv), K2CO3 (276 mg, 2 mmol, 2 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (DCPTPB; 10 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Colorless solid (230 mg, 84% yield); mp 68–69 °C; 1H NMR (CDCl3, 200 MHz) δ 2.33 (s, 3H), 4.62 (d, J = 5.4 Hz, 2H), 4.87 (s, 1H), 6.69 (dd, J = 7.2, 5.1 Hz, 1H), 7.10–7.45 (m, 10H), 8.18 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 45.5, 113.0, 122.4, 127.6, 127.9, 129.0, 129.3, 136.7, 136.9, 137.2, 138.1, 147.2, 155.5 (one phenyl carbon is overlapping). HRMS calcd for C19H18N2+: [M + H]+ 275.1543. Found: [M + H]+ 275.1556.
N-Benzhydryl-3-methylpyridin-2-amine (5a)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Analytical data are in accordance with the literature.4b Colorless solid (88 mg, 64% yield); mp 91–93 °C; 1H NMR (CDCl3, 200 MHz) δ 2.07 (s, 3H), 4.60 (d, J = 6.8 Hz, 1H), 6.42–6.48 (m, 2H), 7.12–7.29 (m, 11H), 7.89 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 58.6, 113.2, 116.4, 127.1, 127.7, 128.6, 137.0, 143.6, 145.7, 155.8.
N-Benzhydryl-3-(trifluoromethyl)pyridin-2-amine (5c)
Reaction of N-benzyl-3-(trifluoromethyl)pyridin-2-amine 4c (126 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (128 mg, 78% yield); 1H NMR (CDCl3, 200 MHz) δ 5.45 (d, J = 6.7 Hz, 1H), 6.54–6.63 (m, 2H), 7.19–7.32 (m, 10H), 7.66 (d, J = 7.3 Hz, 1H), 8.19 (d, J = 4.6, 1H); 13C NMR (CDCl3, 50 MHz) δ 58.5, 108.8 (q, J = 31.2 Hz), 112.1, 124.6 (q, J = 271.3 Hz) 127.4, 127.6, 128.8, 135.1 (q, J = 5.1 Hz), 142.7, 151.9, 153.6. HRMS calcd for C19H15F3N2+: [M + H]+ 329.1260. Found: [M + H]+ 329.1271.
N-Benzhydryl-3-phenylpyridin-2-amine (5d)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless solid (151 mg, 90% yield); mp 90–92 °C; 1H NMR (CDCl3, 200 MHz) δ 5.18 (d, J = 7.4 Hz, 1H), 6.51 (d, J = 7.5 Hz, 1H), 6.64 (dd, J = 7.2, 5.0 Hz, 1H), 7.14–7.44 (m, 16H), 8.08 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 58.5, 113.5, 122.4, 127.1, 127.5, 128.0, 128.6, 128.9, 129.4, 137.4, 138.1, 143.5, 147.4, 154.6. HRMS calcd for C24H20N2+: [M + H]+ 337.1699. Found: [M + H]+ 337.1713.
3-Methyl-N-[phenyl(m-tolyl)methyl]pyridin-2-amine (5g)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-(m-tolyl)-1,3,2-dioxaborinane (132 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (88 mg, 61% yield); 1H NMR (CDCl3, 200 MHz) δ 2.11 (s, 3H), 2.29 (s, 3H), 4.63 (d, J = 7.2 Hz, 1H), 6.46–6.52 (m, 2H), 7.01–7.34 (m, 10H), 7.95 (dd, J = 5.0, 1.2 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 21.6, 58.5, 113.1, 116.4, 124.7, 127.0, 127.6, 127.9, 128.5, 128.6, 129.3, 137.0, 138.2, 143.6, 143.7, 145.8, 155.8. HRMS calcd for C20H20N2+: [M + H]+ 289.1699. Found: [M + H]+ 289.1679.
N-[(3-Chlorophenyl)(phenyl)methyl]-3-methylpyridin-2-amine (5h)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-(2-chlorophenyl)-1,3,2-dioxaborinane (147 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (58 mg, 38% yield); 1H NMR (CDCl3, 200 MHz) δ 2.13 (s, 3H), 4.59 (d, J = 6.6 Hz, 1H), 6.46–6.57 (m, 2H), 7.19–7.32 (m, 10H), 7.95 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 58.3, 113.6, 116.6, 125.8, 127.3, 127.5, 127.6, 127.8, 128.9, 129.8, 134.5, 137.2, 143.0, 145.7, 155.6. HRMS calcd for C19H17ClN2+: [M + H]+ 309.1153. Found: [M + H]+ 309.1138.
3-Methyl-N-[phenyl(p-tolyl)methyl]pyridin-2-amine (5i)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-(p-tolyl)-1,3,2-dioxaborinane (132 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (89 mg, 62% yield); mp 103–105 °C; 1H NMR (CDCl3, 200 MHz) δ 2.13 (s, 3H), 2.32 (s, 3H), 4.64 (d, J = 6.8 Hz, 1H), 6.46–6.54 (m, 2H), 7.09–7.32 (m, 10H), 7.96 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 21.2, 58.3, 113.1, 116.4, 127.0, 127.6, 127.7, 128.6, 129.3, 136.8, 137.0, 140.7, 143.7, 145.8, 155.8. HRMS calcd for C20H20N2+: [M + H]+ 289.1699. Found: [M + H]+ 289.1699.
N-{[4-(tert-Butyl)phenyl](phenyl)methyl}-3-methylpyridin-2-amine (5j)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-[4-(tert-butyl)phenyl]-1,3,2-dioxaborinane (164 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (106 mg, 64% yield); mp 120–122 °C; 1H NMR (CDCl3, 200 MHz) δ 1.29 (s, 9H), 2.13 (s, 3H), 4.66 (d, J = 6.8 Hz, 1H), 6.48–6.53 (m, 2H), 7.20–7.34 (m, 10H), 7.96 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.3, 31.5, 34.6, 58.2, 113.1, 116.4, 125.6, 127.0, 127.4, 127.6, 128.5, 137.0, 140.6, 143.7, 145.8, 150.0, 155.9. HRMS calcd for C23H26N2+: [M + H]+ 331.2169. Found: [M + H]+ 331.2178.
N-[(4-Methoxyphenyl)(phenyl)methyl]-3-methylpyridin-2-amine (5k)
Reaction of N-benzyl-3-methylpyridin-2-amine (99 mg, 0.5 mmol, 1 equiv), 2-(4-methoxyphenyl)-1,3,2-dioxaborinane (144 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (59 mg, 39% yield); mp 59–61 °C; 1H NMR (CDCl3, 200 MHz) δ 2.14 (s, 3H), 3.79 (s, 3H), 4.63 (d, J = 6.5 Hz, 1H), 6.46–6.55 (m, 2H), 6.82–6.89 (m, 2H), 7.22–7.36 (m, 8H), 7.97 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 55.4, 58.0, 113.2, 114.0, 116.4, 127.0, 127.6, 128.6, 128.9, 135.8, 137.0, 143.8, 145.8, 155.8, 158.7. HRMS calcd for C20H20N2O+: [M + H]+ 305.1648. Found: [M + H]+ 305.1655.
N-[(4-Fluorophenyl)(phenyl)methyl]-3-methylpyridin-2-amine (5l)
Reaction of N-benzyl-3-methylpyridin-2-amine (99 mg, 0.5 mmol, 1 equiv), 2-(4-fluorophenyl)-1,3,2-dioxaborinane (135 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (97 mg, 66% yield); mp 101–103 °C; 1H NMR (CDCl3, 200 MHz) δ 2.07 (s, 3H), 4.55 (d, J = 6.7 Hz, 1H), 6.42–6.50 (m, 2H), 6.86–6.97 (m, 2H), 7.16–7.26 (m, 8H), 7.90 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 58.0, 113.4, 115.4 (d, J = 21.3 Hz), 116.5, 127.3, 127.7, 128.7, 129.2 (d, J = 8.1 Hz), 137.2, 139.3 (d, J = 3.1 Hz), 143.4, 145.7, 155.6, 161.9 (d, J = 245.0 Hz). HRMS calcd for C19H17N2F+: [M + H]+ 293.1449. Found: [M + H]+ 293.1448.
N-[(4-Chlorophenyl)(phenyl)methyl]-3-methylpyridin-2-amine (5m)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-(4-chlorophenyl)-1,3,2-dioxaborinane (147 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (51 mg, 33% yield); 1H NMR (CDCl3, 200 MHz) δ 2.12 (s, 3H), 4.59 (d, J = 6.6 Hz, 1H), 6.45–6.55 (m, 2H), 7.21–7.32 (m, 10H), 7.94 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 58.2, 113.5, 116.5, 127.5, 127.8, 128.7, 128.8, 129.0, 132.7, 137.1, 142.1, 143.2, 145.7, 155.6. HRMS calcd for C19H17ClN2+: [M + H]+ 309.1153. Found: [M + H]+ 309.1138.
3-Methyl-N-{phenyl[4-(trifluoromethyl)phenyl]methyl}pyridin-2-amine (5n)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (99 mg, 0.5 mmol, 1 equiv), 2-[4-(trifluoromethyl)phenyl]-1,3,2-dioxaborinane (173 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (70 mg, 41% yield); mp 56–58 °C; 1H NMR (CDCl3, 200 MHz) δ 2.17 (s, 3H), 4.65 (d, J = 6.3 Hz, 1H), 6.54–6.60 (m, 2H), 7.26–7.60 (m, 10H), 7.97 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 58.6, 113.7, 116.6, 124.4 (q, J = 272.7 Hz), 125.5 (q, J = 3.8 Hz), 127.7, 127.8, 127.9, 128.9, 129.2 (q, J = 32.3 Hz), 137.2, 142.9, 145.7, 147.6, 155.5. HRMS calcd for C20H17F3N2+: [M + H]+ 343.1417. Found: [M + H]+ 343.1433.
N-[Phenyl(p-tolyl)methyl]-3-(trifluoromethyl)pyridin-2-amine (5t)
Reaction of N-benzyl-3-(trifluoromethyl)pyridin-2-amine 4c (126 mg, 0.5 mmol, 1 equiv), 2-(p-tolyl)-1,3,2-dioxaborinane (132 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (132 mg, 77% yield); 1H NMR (CDCl3, 200 MHz) δ 2.32 (s, 3H), 5.44 (d, J = 6.2 Hz, 1H), 6.51 (d, J = 7.1 Hz, 1H), 6.60 (dd, J = 7.6, 5.0 Hz, 1H), 7.10–7.36 (m, 9H), 7.66 (d, J = 7.5 Hz, 1H), 8.19 (d, J = 4.7, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 58.3, 108.7 (q, J = 31.3 Hz), 112.0, 124.6 (q, J = 271.6 Hz) 127.3, 127.4, 127.5, 128.7, 129.4, 135.0 (q, J = 5.1 Hz), 137.0, 139.8, 142.9, 152.0, 153.6. HRMS calcd for C20H17F3N2+: [M + H]+ 343.1417. Found: [M + H]+ 343.1429.
N-{[4-(tert-Butyl)phenyl](phenyl)methyl}-3-(trifluoromethyl)pyridin-2-amine (5u)
Reaction of N-benzyl-3-(trifluoromethyl)pyridin-2-amine 4c (126 mg, 0.5 mmol, 1 equiv), 2-[4-(tert-butyl)phenyl]-1,3,2-dioxaborinane (164 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (134 mg, 70% yield); 1H NMR (CDCl3, 200 MHz) δ 1.29 (s, 9H), 5.47 (d, J = 6.6 Hz, 1H), 6.53–6.64 (m, 2H), 7.12–7.35 (m, 9H), 7.66 (d, J = 7.6 Hz, 1H), 8.20 (d, J = 4.8, 1H); 13C NMR (CDCl3, 50 MHz) δ 31.5, 34.6, 58.1, 108.7 (q, J = 31.3 Hz), 112.0, 124.7 (q, J = 271.7 Hz) 125.7, 127.3, 127.5, 128.7, 129.4, 135.1 (q, J = 5.1 Hz), 139.6, 142.9, 150.2, 152.0, 153.7. HRMS calcd for C23H23F3N2+: [M + H]+ 385.1886. Found: [M + H]+ 385.1910.
N-[(4-Methoxyphenyl)(phenyl)methyl]-3-(trifluoromethyl)pyridin-2-amine (5v)
Reaction of N-benzyl-3-(trifluoromethyl)pyridin-2-amine 4c (126 mg, 0.5 mmol, 1 equiv), 2-(4-methoxyphenyl)-1,3,2-dioxaborinane (144 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (109 mg, 61% yield); 1H NMR (CDCl3, 200 MHz) δ 3.79 (s, 3H), 5.41 (d, J = 6.5 Hz, 1H), 6.51 (d, J = 7.0 Hz, 1H), 6.62 (dd, J = 7.6, 5.0 Hz, 1H), 6.82–6.90 (m, 2H), 7.18–7.32 (m, 7H), 7.67 (d, J = 6.6 Hz, 1H), 8.21 (d, J = 4.5, 1H); 13C NMR (CDCl3, 50 MHz) δ 55.3, 57.9, 108.7 (q, J = 31.3 Hz), 112.0, 114.1, 124.6 (q, J = 271.5 Hz) 127.3, 127.4, 128.7, 128.8, 134.9, 135.0 (q, J = 5.5 Hz), 142.9, 152.0, 153.6, 158.9. HRMS calcd for C20H17F3N2O+: [M + H]+ 359.1366. Found: [M + H]+ 359.1386.
N-[(4-Fluorophenyl)(phenyl)methyl]-3-(trifluoromethyl)pyridin-2-amine (5w)
Reaction of N-benzyl-3-(trifluoromethyl)pyridin-2-amine 4c (126 mg, 0.5 mmol, 1 equiv), 2-(4-fluorophenyl)-1,3,2-dioxaborinane (135 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (88 mg, 51% yield); 1H NMR (CDCl3, 200 MHz) δ 5.39 (d, J = 6.1 Hz, 1H), 6.53 (d, J = 6.8 Hz, 1H), 6.63 (dd, J = 7.6, 5.0 Hz, 1H), 7.00–7.04 (m, 2H), 7.23–7.33 (m, 7H), 7.67 (d, J = 7.6 Hz, 1H), 8.20 (d, J = 4.8, 1H); 13C NMR (CDCl3, 50 MHz) δ 57.9, 108.9 (q, J = 31.3 Hz), 112.3, 115.6 (d, J = 21.2 Hz), 124.6 (q, J = 271.6 Hz) 127.5, 127.6, 128.9, 129.1 (d, J = 8.1 Hz), 135.1 (q, J = 5.1 Hz), 138.5 (d, J = 3.2 Hz), 142.5, 151.9, 153.5, 162.1 (d, J = 245.5 Hz). HRMS calcd for C19H14F4N2+: [M + H]+ 347.1166. Found: [M + H]+ 347.1178.
3-Phenyl-N-[phenyl(p-tolyl)methyl]pyridin-2-amine (5x)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 2-(p-tolyl)-1,3,2-dioxaborinane (132 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless oil (149 mg, 85% yield); 1H NMR (CDCl3, 200 MHz) δ 2.29 (s, 3H), 5.17 (d, J = 7.4 Hz, 1H), 6.47 (d, J = 7.5 Hz, 1H), 6.63 (dd, J = 7.2, 5.0 Hz, 1H), 7.04–7.44 (m, 15H), 8.08 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 58.4, 113.3, 122.3, 126.9, 127.4, 127.5, 127.9, 128.5, 129.0, 129.3, 129.4, 136.6, 137.3, 138.1, 140.5, 143.6, 147.4, 154.6. HRMS calcd for C25H22N2+: [M + H]+ 351.1856. Found: [M + H]+ 351.1873.
N-{[4-(tert-Butyl)phenyl](phenyl)methyl}-3-phenylpyridin-2-amine (5y)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv.), 2-[4-(tert-butyl)phenyl]-1,3,2-dioxaborinane (164 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless solid (189 mg, 96% yield); mp 74–76 °C; 1H NMR (CDCl3, 200 MHz) δ 1.27 (s, 9H), 5.20 (d, J = 7.5 Hz, 1H), 6.50 (d, J = 7.6 Hz, 1H), 6.62 (dd, J = 7.2, 5.0 Hz, 1H), 7.12–7.44 (m, 15H), 8.08 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 31.5, 34.5, 58.2, 113.2, 122.3, 125.5, 126.9, 127.2, 127.5, 127.9, 128.5, 129.0, 129.4, 137.3, 138.1, 140.4, 143.7, 147.4, 149.8, 154.6. (one phenyl carbon is overlapping). HRMS calcd for C28H28N2+: [M + H]+ 393.2325. Found: [M + H]+ 393.2349.
N-[(4-Fluorophenyl)(phenyl)methyl]-3-phenylpyridin-2-amine (5z)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 2-(4-fluorophenyl)-1,3,2-dioxaborinane (135 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless solid (128 mg, 72% yield); mp 79–81 °C; 1H NMR (CDCl3, 200 MHz) δ 5.12 (d, J = 7.3 Hz, 1H), 6.48 (d, J = 7.3 Hz, 1H), 6.65 (dd, J = 7.3, 5.0 Hz, 1H), 6.90–6.99 (m, 2H), 7.14–7.44 (m, 13H), 8.08 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 58.1, 113.5, 115.3 (d, J = 21.3 Hz), 122.4, 127.2, 127.5, 128.1, 128.7, 128.9, 129.1 (d, J = 8.0 Hz), 129.4, 137.4, 138.0, 139.2 (d, J = 3.2 Hz), 143.2, 147.3, 154.4, 161.9 (d, J = 245.1 Hz). HRMS calcd for C24H19N2F+: [M + H]+ 355.1605. Found: [M + H]+ 355.1621.
N-[(4-Chlorophenyl)(phenyl)methyl]-3-phenylpyridin-2-amine (5aa)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 2-(4-chlorophenyl)-1,3,2-dioxaborinane (147 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless oil (118 mg, 64% yield); 1H NMR (CDCl3, 200 MHz) δ 5.12 (d, J = 7.2 Hz, 1H), 6.47 (d, J = 7.3 Hz, 1H), 6.64 (dd, J = 7.2, 5.0 Hz, 1H), 7.15–7.42 (m, 15H), 8.07 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 58.2, 113.6, 122.4, 127.3, 127.5, 128.0, 128.6, 128.7, 128.9, 129.4, 132.7, 137.4, 137.9, 142.0, 142.9, 147.3, 154.3 (one phenyl carbon is overlapping). HRMS calcd for C24H19N2Cl+: [M + H]+ 371.1310. Found: [M + H]+ 371.1294.
3-Phenyl-N-{phenyl[4-(trifluoromethyl)phenyl]methyl}pyridin-2-amine (5ab)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 2-[4-(trifluoromethyl)phenyl]-1,3,2-dioxaborinane (173 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless oil (62 mg, 31% yield); 1H NMR (CDCl3, 200 MHz) δ 5.16 (d, J = 7.1 Hz, 1H), 6.53 (d, J = 7.1 Hz, 1H), 6.67 (dd, J = 7.3, 5.0 Hz, 1H), 7.18–7.55 (m, 15H), 8.07 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 58.6, 113.8, 122.5, 124.4 (q, JCF = 272.3 Hz), 125.5 (q, JCF = 3.8 Hz), 127.5, 127.6, 127.7, 128.0, 128.8, 129.3, 137.4, 137.8, 142.5, 147.2, 147.5, 154.2 (two phenyl carbons are overlapping). HRMS calcd for C25H19N2F3+: [M + H]+ 405.1573. Found: [M + H]+ 405.1568.
1-(4-{Phenyl[(3-phenylpyridin-2-yl)amino]methyl}phenyl)ethanone (5ac)
Reaction of N-benzyl-3-phenylpyridin-2-amine 4d (130 mg, 0.5 mmol, 1 equiv), 1-[4-(1,3,2-dioxaborinan-2-yl)phenyl]ethanone (153 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II for 36 h. Colorless oil (98 mg, 52% yield); 1H NMR (CDCl3, 200 MHz) δ 2.53 (s, 3H), 5.18 (d, J = 7.1 Hz, 1H), 6.52 (d, J = 7.1 Hz, 1H), 6.66 (dd, J = 7.2, 5.0 Hz, 1H), 7.19–7.44 (m, 13H), 7.87 (d, J = 8.2 Hz, 2H), 8.06 (dd, J = 5.0, 1.7 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 26.7, 58.7, 113.7, 122.4, 127.4, 127.5, 127.6, 128.0, 128.7, 128.8, 128.9, 129.4, 135.9, 137.4, 137.8, 142.6, 147.3, 149.0, 154.3, 197.8. HRMS calcd for C26H22N2O+: [M + H]+ 379.1805. Found: [M + H]+ 379.1799.
N-[(4-Isopropoxyphenyl)(phenyl)methyl]-3-methylpyridin-2-amine (5af)
Reaction of N-(4-isopropoxybenzyl)-3-methylpyridin-2-amine 4e (128 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (42 mg, 25% yield); 1H NMR (CDCl3, 200 MHz) δ 1.30 (d, J = 6.0 Hz, 6H), 2.11 (s, 3H), 4.49 (sep, J = 6.0 Hz, 1H), 4.61 (d, J = 7.0 Hz, 1H), 6.44–6.52 (m, 2H), 6.77–6.85 (m, 2H), 7.17–7.34 (m, 8H), 7.95 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 22.2, 58.0, 69.9, 113.1, 115.8, 116.4, 126.9, 127.5, 128.5, 128.9, 135.6, 137.0, 143.8, 145.8, 155.8, 157.0. HRMS calcd for C22H24N2O+: [M + H]+ 333.1961. Found: [M + H]+ 333.1963.
Methyl 4-{[(3-Methylpyridin-2-yl)amino](phenyl)methyl}benzoate (5ag)
Reaction of methyl 4-{[(3-methylpyridin-2-yl)amino]methyl}benzoate 4j (128 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (43 mg, 26% yield); 1H NMR (CDCl3, 200 MHz) δ 2.13 (s, 3H), 3.86 (s, 3H), 4.65 (d, J = 6.6 Hz, 1H), 6.49–6.55 (m, 2H), 7.21–7.33 (m, 6H), 7.41 (d, J = 8.2 Hz), 7.92–7.99 (m, 3H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 52.1, 58.7, 113.5, 116.5, 127.4, 127.5, 127.9, 128.8, 129.9, 137.1, 142.9, 145.7, 148.8, 155.5, 167.1. HRMS calcd for C21H20N2O2+: [M + H]+ 333.1598. Found: [M + H]+ 333.1587.
N-[(4-Chlorophenyl)(p-tolyl)methyl]-3-methylpyridin-2-amine (5ah)
Reaction of 3-methyl-N-(4-methylbenzyl)pyridin-2-amine 4g (106 mg, 0.5 mmol, 1 equiv), 2-(4-chlorophenyl)-1,3,2-dioxaborinane (147 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (81 mg, 50% yield); 1H NMR (CDCl3, 200 MHz) δ 2.03 (s, 3H), 2.23 (s, 3H), 4.49 (d, J = 6.6 Hz, 1H), 6.34 (d, J = 6.7 Hz, 1H), 6.43 (dd, J = 7.1, 5.1 Hz, 1H), 7.01–7.16 (m, 9H), 7.86 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 21.2, 57.9, 113.4, 116.5, 127.7, 128.6, 128.9, 129.5, 132.6, 137.1, 137.2, 140.2, 142.2, 145.7, 155.6. HRMS calcd for C20H19N2Cl+: [M + H]+ 323.1310. Found: [M + H]+ 323.1317.
3-Methyl-N-{p-tolyl[4-(trifluoromethyl)phenyl]methyl}pyridin-2-amine (5ai)
Reaction of 3-methyl-N-(4-methylbenzyl)pyridin-2-amine 4g (106 mg, 0.5 mmol, 1 equiv), 2-[4-(trifluoromethyl)phenyl]-1,3,2-dioxaborinane (173 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (58 mg, 33% yield); 1H NMR (CDCl3, 200 MHz) δ 2.16 (s, 3H), 2.35 (s, 3H), 4.63 (d, J = 6.3 Hz, 1H), 6.49–6.59 (m, 2H), 7.14–7.29 (m, 5H), 7.52 (q, J = 9.7 Hz, 4H), 7.97 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.2, 21.2, 58.4, 113.6, 116.6, 124.4 (q, JCF = 272.0 Hz), 125.4 (q, JCF = 3.8 Hz), 127.7, 127.8, 129.1 (q, JCF = 32.3 Hz), 129.7, 137.2, 137.4, 140.0, 145.7, 147.8, 155.6. HRMS calcd for C21H19N2F3+: [M + H]+ 357.1573. Found: [M + H]+ 357.1587.
N-(Di-p-tolylmethyl)-3-phenylpyridin-2-amine (5aj)
Reaction of N-(4-methylbenzyl)-3-phenylpyridin-2-amine 4m (137 mg, 0.5 mmol, 1 equiv), 2-(p-tolyl)-1,3,2-dioxaborinane (132 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (133 mg, 73% yield); mp 127–129 °C; 1H NMR (CDCl3, 200 MHz) δ 2.19 (s, 6H), 5.08 (d, J = 7.4 Hz, 1H), 6.35 (d, J = 7.5 Hz, 1H), 6.52 (dd, J = 7.2, 5.0 Hz, 1H), 6.95–7.34 (m, 14H), 7.99 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 58.1, 113.2, 122.3, 127.4, 127.9, 128.9, 129.2, 129.3, 136.5, 137.2, 138.1, 140.7, 147.4, 154.6. HRMS calcd for C26H24N2+: [M + H]+ 365.2012. Found: [M + H]+ 365.2043.
N-{[4-(tert-Butyl)phenyl](p-tolyl)methyl}-3-phenylpyridin-2-amine (5ak)
Reaction of N-(4-methylbenzyl)-3-phenylpyridin-2-amine 4m (137 mg, 0.5 mmol, 1 equiv), 2-[4-(tert-butyl)phenyl]-1,3,2-dioxaborinane (164 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (137 mg, 67% yield); mp 97–99 °C; 1H NMR (CDCl3, 200 MHz) δ 1.18, (s, 9H), 2.20 (s, 3H), 5.11 (d, J = 7.5 Hz, 1H), 6.38 (d, J = 7.6 Hz, 1H), 6.53 (dd, J = 7.2, 5.0 Hz, 1H), 6.96–7.36 (m, 14H), 8.00 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 31.5, 34.5, 57.9, 113.1, 122.3, 125.4, 127.1, 127.4, 127.9, 129.0, 129.2, 129.3, 136.5, 137.3, 138.2, 140.6, 140.8, 147.4, 149.7, 154.7. HRMS calcd for C29H30N2+: [M + H]+ 407.2482. Found: [M + H]+ 407.2515.
N-[(4-Fluorophenyl)(p-tolyl)methyl]-3-phenylpyridin-2-amine (5al)
Reaction of N-(4-methylbenzyl)-3-phenylpyridin-2-amine 4m (137 mg, 0.5 mmol, 1 equiv), 2-(4-fluorophenyl)-1,3,2-dioxaborinane (135 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless solid (110 mg, 60% yield); mp 100–102 °C; 1H NMR (CDCl3, 200 MHz) δ 2.29 (s, 3H), 5.11 (d, J = 7.2 Hz, 1H), 6.43 (d, J = 7.3 Hz, 1H), 6.64 (dd, J = 7.3, 5.0 Hz, 1H), 6.90–7.45 (m, 14H), 8.07 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 57.8, 113.4, 115.3 (d, JCF = 21.3 Hz), 122.4, 127.4, 128.0, 128.9, 129.1, 129.4, 136.9, 137.4, 138.0, 139.4 (d, JCF = 3.1 Hz), 140.3, 147.4, 154.5, 161.9 (d, JCF = 244.9 Hz) (one carbon is overlapping). HRMS calcd for C25H21N2F+: [M + H]+ 369.1762. Found: [M + H]+ 369.1787.
3-Phenyl-N-{p-tolyl[4-(trifluoromethyl)phenyl]methyl}pyridin-2-amine (5am)
Reaction of N-(4-methylbenzyl)-3-phenylpyridin-2-amine 4m (137 mg, 0.5 mmol, 1 equiv), 2-[4-(trifluoromethyl)phenyl]-1,3,2-dioxaborinane (173 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (69 mg, 33% yield); 1H NMR (CDCl3, 200 MHz) δ 2.21 (s, 3H), 5.06 (d, J = 6.9 Hz, 1H), 6.39 (d, J = 6.9 Hz, 1H), 6.58 (dd, J = 7.3, 5.0 Hz, 1H), 7.00 (s, 4H), 7.14–7.46 (m, 10H), 7.98 (dd, J = 5.0, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 21.2, 53.4, 113.7, 122.5, 125.5 (q, JCF = 3.8 Hz), 127.5, 127.6, 128.1, 128.9, 129.4, 129.6, 137.3, 137.4, 137.9, 139.7, 147.3, 147.9, 154.3 (two carbons are overlapping with other peaks). HRMS calcd for C26H21N2F3+: [M + H]+ 419.1735. Found: [M + H]+ 419.1765.
N-Benzyl-1-methyl-1H-benzo[d]imidazol-2-amine (9)
1-Methyl-2-(methylthio)-1H-benzo[d]imidazole (356 mg, 2 mmol, 1 equiv) was dissolved in 20 mL of dry dichloromethane (DCM) and cooled to 5 °C. Then m-chloroperoxybenzoic acid (m-CPBA; 692 mg, 4 mmol, 2 equiv) was added slowly to the solution, and the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was added to a NaHCO3 solution and the organic phase was separated. The organic layer was washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The mixture of sulfoxide and sulfone product (400 mg) was recrystallized from EtOAc. In an 8 mL vial was placed the mixture (400 mg), benzylamine (856 mg, 8 mmol, 4 equiv), and 6 mL of BF3·Et2O. The vial was capped (with a Teflon septum cap), evacuated, and refilled with argon via a needle through the septum three times. Dry toluene (5 mL) was then added via syringe. The septum cap was quickly replaced with a fully covered solid Teflon-lined cap. The reaction vial was placed in a reaction block at 140 °C and stirred for 16 h. Then the vial was cooled to room temperature and the reaction mixture was concentrated in vacuo. The product was purified by flash column chromatography (PE/EtOAc 4:1). Colorless solid (310 mg, 65% yield); mp 164–166 °C; 1H NMR (CDCl3, 200 MHz) δ 3.46 (s, 3H), 4.44 (s, 1H), 4.72 (d, 2H, J = 5.5 Hz), 7.06–7.18 (m, 3H), 7.30–7.53 (m, 6H). 13C NMR (CDCl3, 50 MHz) δ 28.4, 47.7, 107.2, 116.6, 119.8, 121.4, 127.8, 128.2, 128.8, 135.1, 138.7, 142.2, 154.4.
N-Benzhydryl-1-methyl-1H-benzo[d]imidazol-2-amine (10)
Reaction of N-benzyl-1-methyl-1H-benzo[d]imidazol-2-amine 13 (119 mg, 0.5 mmol, 1 equiv), 5,5-dimethyl-2-phenyl-1,3,2-dioxaborinane 2 (143 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Yellow solid (70 mg, 45% yield); mp 189–191 °C;1H NMR (CDCl3, 200 MHz) δ 3.54 (s, 3H), 4.62 (d, J = 6.6 Hz, 1H), 6.49 (d, J = 6.8 Hz, 1H), 7.07–7.10 (m, 3H), 7.29–7.49 (m, 11H); 13C NMR (CDCl3, 50 MHz) δ 28.5, 60.5, 107.2, 117.0, 119.8, 121.3, 127.5, 127.6, 128.8, 135.1, 142.2, 142.4, 153.4. HRMS calcd for C21H19N3+: [M + H]+ 314.1652. Found: [M + H]+ 314.1660.
3-Methyl-2-(phenylethynyl)pyridine
2-Bromo-3-methylpyridine (172 mg, 1 mmol, 1 equiv), phenylacetylene (122 mg, 1.2 mmol, 1.2 equiv), pyrrolidine (142 mg, 2 mmol, 2 equiv), PdCl2 (4 mg, 0.02 mmol, 2 mol %), PPh3 (10 mg, 0.04 mmol, 4 mol %), and 2 mL of degassed water were placed in an oven-dried 6 mL vial with a Teflon cap and a magnetic stirring bar. The reaction vial was then heated in a reaction block at 120 °C for 3 h. After cooling to rt, the reaction mixture was extracted with diethyl ether (4 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (PE/EtOAc = 9:1) to give the pure product as red oil (139 mg, 72% yield). Analytical data are in accordance with the literature.221H NMR (CDCl3, 200 MHz) δ 2.52 (s, 3H), 7.15 (dd, J = 7.7, 4.8 Hz, 1H), 7.33–7.40 (m, 3H), 7.51–7.63 (m, 3H), 8.45 (dd, J = 4.7, 1.0 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 19.6, 87.6, 93.2, 122.6, 122.8, 128.5, 129.0, 132.1, 136.0, 137.1, 143.2, 147.5.
3-Methyl-2-phenethylpyridine (11)
3-Methyl-2-(phenylethynyl)pyridine from the above protocol (193 mg, 1 mmol, 1 equiv), triethylamine (253 mg, 2.5 mmol, 2.5 equiv), 10% palladium on carbon (30 mg), and 30 mL of EtOH were charged in a round-bottomed flask. The reaction mixture was stirred at room temperature under atmospheric H2 pressure for 16 h. The solvent was removed under reduced pressure and the residue was dissolved in 25 mL of Et2O. The solid material was filtered off. The organic layer was washed with saturated NaHCO3 and with brine, dried over Na2SO4, filtered, and concentrated. Analytical data are in accordance with the literature.23 Pale yellow oil (196 mg, 99% yield); 1H NMR (CDCl3, 200 MHz) δ 2.23 (s, 3H), 3.02–3.10 (m, 4H) 7.05 (dd, J = 7.6, 4.8 Hz, 1H), 7.19–7.42 (m, 6H), 8.42 (d, J = 3.7 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 18.8, 35.1, 37.5, 121.4, 126.0, 128.5, 128.6, 131.3, 137.7, 142.1, 146.8, 159.6.
2-(Benzyloxy)-3-methylpyridine (12)
2-Chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), phenylmethanol (140 mg, 1.3 mmol, 1.3 equiv), KOtBu (224 mg, 2 mmol, 2 equiv), and 5 mL of dioxane were charged in a round-bottomed flask. The reaction mixture was refluxed for 24 h. After cooling to rt, 2 mL of H2O was added to the solution and the aqueous phase was extracted with EtOAc (3 × 5 mL). The combined organic layer was washed with saturated NaHCO3 and with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (PE/EtOAc = 19:1) to give the pure product. Analytical data are in accordance with the literature.24 Colorless oil (140 mg, 70% yield); 1H NMR (CDCl3, 200 MHz) δ 2.26 (s, 3H), 5.44 (s, 2H), 6.82 (dd, J = 7.1, 5.1 Hz, 1H), 7.31–7.52 (m, 6H), 8.03 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 16.0, 67.3, 116.9, 121.1, 127.6, 127.7, 128.5, 138.0, 138.7, 144.1, 162.0.
3-Methyl-N-phenethylpyridin-2-amine (13)
Reaction of 2-chloro-3-methylpyridine (128 mg, 1 mmol, 1 equiv), 2-phenylethanamine (145 mg, 1.2 mmol, 1.2 equiv), K2CO3 (483 mg, 3.5 mmol, 3.5 equiv), Pd(OAc)2 (4 mg, 0.02 mmol, 2 mol %), and BINAP (12 mg, 0.02 mmol, 2 mol %) in 2.5 mL of dry toluene was carried out according to general procedure I. Pale yellow oil (183 mg, 86% yield); 1H NMR (CDCl3, 200 MHz) δ 1.98 (s, 3H), 2.98 (t, J = 6.7 Hz, 2H), 3.78 (q, J = 6.4 Hz, 2H), 4.13 (s, 1H), 6.54 (dd, J = 7.1, 5.1 Hz, 1H), 7.20–7.38 (m, 6H), 8.07 (dd, J = 5.0, 1.1 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 16.9, 35.8, 42.8, 112.7, 116.7, 126.4, 128.6, 129.0, 136.8, 139.9, 145.6, 156.8. HRMS calcd for C14H16N2+: [M + H]+ 213.1386. Found: [M + H]+ 213.1395.
2-(2,2-Diphenylethyl)-3-methylpyridine (14)
Reaction of 3-methyl-2-phenethylpyridine 11 (99 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (102 mg, 75% yield); 1H NMR (CDCl3, 200 MHz) δ 2.01 (s, 3H), 3.51 (d, J = 7.8 Hz, 2H), 4.74 (t, J = 7.8 Hz, 1H), 6.97 (dd, J = 7.6, 4.8 Hz, 1H), 7.11–7.29 (m, 11H), 8.38 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 18.8, 40.8, 50.5, 121.2, 126.2, 128.2, 128.3, 131.8, 137.6, 144.7, 146.7, 158.4. HRMS calcd for C20H19N+: [M + H]+ 274.1590. Found: [M + H]+ 274.1601.
N-(1,2-Diphenylethyl)-3-methylpyridin-2-amine (16)
Reaction of 3-methyl-N-phenethylpyridin-2-amine 13 (106 mg, 0.5 mmol, 1 equiv), 2-phenyl-1,3,2-dioxaborinane (122 mg, 0.75 mmol, 1.5 equiv), and Ru3(CO)12 (16 mg, 0.025 mmol, 5 mol %) in 0.5 mL of dry pinacolone was carried out according to general procedure II. Colorless oil (56 mg, 39% yield); 1H NMR (CDCl3, 200 MHz) δ 2.05 (s, 3H), 3.21 (d, J = 6.6 Hz, 2H), 4.48 (d, J = 7.0 Hz, 1H), 5.55 (t, J = 6.8 Hz, 1H), 6.49 (dd, J = 7.1, 5.1 Hz, 1H), 7.06–7.35 (m, 11H), 7.96 (dd, J = 5.0, 1.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.0, 43.7, 55.6, 112.9, 116.7, 126.5, 126.7, 126.9, 128.3, 128.4, 129.6, 136.9, 138.0, 143.5, 145.7, 156.0. HRMS calcd for C20H20N2+: [M + H]+ 289.1699. Found: [M + H]+ 289.1708.
N-Benzyl-N-methylpyridin-2-amine (17)
Reaction of 2-bromopyridine (158 mg, 1 mmol, 1 equiv), N-methyl-1-phenylmethanamine (169 mg, 1.4 mmol, 1.4 equiv), NaOtBu (192 mg, 2 mmol, 2 equiv), Pd2(dba)3 (18 mg, 0.02 mmol, 2 mol %), and DPPP (16 mg, 0.04 mmol, 4 mol %) in 4 mL of dry toluene was carried out according to general procedure III. Analytical data are in accordance with the literature.25 Pale yellow oil (186 mg, 94% yield); 1H NMR (CDCl3, 200 MHz) δ 3.05 (s, 3H), 4.79 (s, 2H), 6.46–6.57 (m, 2H), 7.19–7.45 (m, 6H), 8.16–8.20 (m, 1H); 13C NMR (CDCl3, 50 MHz) δ 36.2, 53.3, 105.8, 111.9, 127.0, 127.1, 128.6, 137.4, 138.8, 148.1, 159.0.
N-Benzyl-N,3-dimethylpyridin-2-amine (18)
Reaction of 2-bromo-3-methylpyridine (172 mg, 1 mmol, 1 equiv), N-methyl-1-phenylmethanamine (169 mg, 1.4 mmol, 1.4 equiv), NaOtBu (192 mg, 2 mmol, 2 equiv), Pd2(dba)3 (18 mg, 0.02 mmol, 2 mol %), and DPPP (16 mg, 0.04 mmol, 4 mol %) in 4 mL of dry toluene was carried out according to general procedure III. Colorless oil (186 mg, 88% yield); 1H NMR (CDCl3, 200 MHz) δ 2.32 (s, 3H), 2.75 (s, 3H), 4.33 (s, 2H), 6.82 (dd, J = 7.3, 4.9 Hz, 1H), 7.24–7.40 (m, 6H), 8.16 (dd, J = 4.8, 1.5 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 19.0, 39.1, 57.8, 117.4, 124.5, 126.9, 128.0, 128.4, 139.3, 139.5, 145.2, 162.6. HRMS calcd for C14H16N2+: [M + H]+ 213.1386. Found: [M + H]+ 213.1385.
N-Benzyl-N-(pyridin-2-yl)acetamide (19)
Reaction of N-benzylpyridin-2-amine 1a (184 mg, 1 mmol, 1 equiv), acetyl chloride (237 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. Analytical data are in accordance with the literature.26 Yellow oil (213 mg, 94% yield); 1H NMR (CDCl3, 200 MHz) δ 2.07 (s, 3H), 5.10 (s, 2H), 7.08–7.30 (m, 7H), 7.61–7.70 (m, 1H), 8.49 (d, 3J = 4.3 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 23.2, 51.1, 121.7, 122.1, 127.2, 127.8, 128.4, 137.6, 138.1, 149.2, 155.2, 170.5.
N-Benzyl-N-(3-methylpyridin-2-yl)acetamide (20)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (198 mg, 1 mmol, 1 equiv), acetyl chloride (237 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. The resulting crude product was purified by flash column chromatography (PE/EtOAc = 1:1). Colorless oil (227 mg, 95% yield); 1H NMR (CDCl3, 200 MHz) δ 1.79 (s, 3H), 1.87 (s, 3H), 4.73 (s, 1H), 5.14 (s, 1H), 7.19 (s, 6H), 7.52 (d, 3J = 7.6 Hz, 1H), 8.40 (d, 3J = 4.6 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.0, 22.2, 51.1, 123.6, 127.5, 128.3, 129.3, 131.6, 136.9, 140.2, 147.6, 154.0, 169.9. HRMS calcd for C15H16N2O+: [M + H]+ 241.1335. Found: [M + H]+ 241.1335.
N-Benzyl-N-(pyridin-2-yl)benzamide (21)
Reaction of N-benzylpyridin-2-amine 1a (184 mg, 1 mmol, 1 equiv), benzoyl chloride (420 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. Colorless solid (224 mg, 78% yield); mp 113–115 °C; 1H NMR (CDCl3, 200 MHz) δ 5.35 (s, 2H), 6.59 (d, J = 8.0 Hz, 1H), 6,97 (dd, J = 7.4, 4.9 Hz, 1H), 7.14–7.38 (m, 11H), 8.43 (dd, J = 4.9, 1.9 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 51.7, 121.1, 122.8, 127.3, 128.1, 128.3, 128.4, 128.8, 130.3, 136.0, 137.3, 137.8, 148.8, 155.9, 170.8. HRMS calcd for C19H16N2O+: [M + H]+ 289.1335. Found: [M + H]+ 289.1327.
N-Benzyl-N-(3-methylpyridin-2-yl)benzamide (22)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (198 mg, 1 mmol, 1 equiv), benzoyl chloride (420 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. The resulting crude product was purified by flash column chromatography (PE/EtOAc = 1:1). Colorless solid (251 mg, 83% yield); mp 114–116 °C; 1H NMR (CDCl3, 200 MHz) δ 1.41 (s, 3H), 4.82 (d, J = 13.8 Hz, 1H), 5.50 (d, J = 13.8 Hz, 1H), 6.93–7.29 (m, 12H), 8.36 (dd, J = 4.6, 1.7 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.1, 52.4, 122.8, 127.5, 127.6, 128.3, 128.8, 129.4, 130.2, 131.4, 136.1, 137.0, 140.0, 147.1, 154.4, 169.7. HRMS calcd for C20H18N2O+: [M + H]+ 303.1492. Found: [M + H]+ 303.1492.
N-Benzyl-N-(pyridin-2-yl)pivalamide (23)
Reaction of N-benzylpyridin-2-amine 1a (184 mg, 1 mmol, 1 equiv), pivaloyl chloride (363 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. Colorless solid (211 mg, 79% yield); mp 69–71 °C; 1H NMR (CDCl3, 200 MHz) δ 1.06 (s, 9H), 4.95 (s, 2H), 6.92 (dd, J = 7.9, 0.8 Hz, 1H), 7.17–7.24 (m, 6H), 7.57–7.65 (m, 1H), 8.49–8.52 (m, 1H); 13C NMR (CDCl3, 50 MHz) δ 29.3, 41.4, 54.6, 122.8, 123.5, 127.2, 128.4, 137.9, 138.0, 149.2, 156.5, 178.9. HRMS calcd for C17H20N2O+: [M + H]+ 269.1648. Found: [M + H]+ 269.1645.
N-Benzyl-N-(3-methylpyridin-2-yl)pivalamide (24)
Reaction of N-benzyl-3-methylpyridin-2-amine 4a (198 mg, 1 mmol, 1 equiv), pivaloyl chloride (363 mg, 3 mmol, 3 equiv), and MeMgCl (3 M in THF, 0.4 mL, 1.2 mmol, 1.2 equiv) in 7 mL of THF was carried out according to general procedure IV. The resulting crude product was purified by flash column chromatography (PE/EtOAc = 6:1). Colorless solid (221 mg, 78% yield); mp 67–69 °C; 1H NMR (CDCl3, 200 MHz) δ 1.03 (s, 9H), 2.11 (s, 3H), 4.61 (s, 1H), 4.97 (s, 1H), 7.14–7.31 (m, 6H), 7.54 (d, 3J = 7.4 Hz, 1H), 8.33 (dd, J = 4.7, 1.7 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.8, 28.7, 41.2, 54.1, 123.6, 127.2, 128.3, 128.7, 131.7, 137.5, 140.0, 146.6, 155.4, 178.6. HRMS calcd for C18H22N2O+: [M + H]+ 283.1805. Found: [M + H]+ 283.1807.
2-(Pyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline (25)
Reaction of 2-bromopyridine (158 mg, 1 mmol, 1 equiv), 1,2,3,4-tetrahydroisoquinoline (186 mg, 1.4 mmol, 1.4 equiv), NaOtBu (192 mg, 2 mmol, 2 equiv), Pd2(dba)3 (18 mg, 0.02 mmol, 2 mol %), and DPPP (16 mg, 0.04 mmol, 4 mol %) in 4 mL of dry toluene was carried out according to general procedure III. Analytical data are in accordance with the literature.27 Colorless solid (199 mg, 95% yield); mp 47–49 °C; 1H NMR (CDCl3, 200 MHz) δ 2.98 (t, J = 5.9 Hz, 2H), 3.86 (t, J = 5.9 Hz, 2H), 4.72 (s, 2H), 6.59–6.71 (m, 2H), 7.21 (q, J = 3.1 Hz, 4H), 7.46–7.55 (m, 1H), 8.25 (dd, J = 4.9, 1.2 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 29.1, 42.6, 47.2, 106.7, 112.6, 126.3, 126.5, 126.7, 128.5, 134.5, 135.5, 137.5, 148.1, 158.8.
2-(3-Methylpyridin-2-yl)-1,2,3,4-tetrahydroisoquinoline (26)
Reaction of 2-bromo-3-methylpyridine (172 mg, 1 mmol, 1 equiv), 1,2,3,4-tetrahydroisoquinoline (186 mg, 1.4 mmol, 1.4 equiv), NaOtBu (192 mg, 2 mmol, 2 equiv), Pd2(dba)3 (18 mg, 0.02 mmol, 2 mol %), and DPPP (16 mg, 0.04 mmol, 4 mol %) in 4 mL of dry toluene was carried out according to general procedure III. Yellow oil (203 mg, 91% yield); 1H NMR (CDCl3, 200 MHz) δ 2.35 (s, 3H), 3.07 (t, J = 5.8 Hz, 2H), 3.41 (t, J = 5.8 Hz, 2H), 4.45 (s, 2H), 6.88 (dd, J = 7.3, 4.9 Hz, 1H), 7.19 (s, 4H), 7.42–7.46 (m, 1H), 8.22 (dd, J = 4.8, 1.8 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 18.5, 29.9, 48.5, 51.6, 117.8, 124.9, 125.9, 126.2, 126.9, 128.9, 134.6, 135.4, 139.4, 145.3, 161.9. HRMS calcd for C15H16N2+: [M + H]+ 225.1386. Found: [M + H]+ 225.1378.
tert-Butyl Benzhydryl(3-methylpyridin-2-yl)carbamate (36)
A 3 M solution of CH3MgCl in THF (1.2 mL, 3.6 mmol, 1.2 equiv) was added dropwise to a solution of N-benzhydryl-3-methylpyridin-2-amine 5a (822 mg, 3 mmol, 1 equiv) in dry THF (20 mL) at rt, and the mixture was stirred for 10 min at that temperature. Di-tert-butyl dicarbonate (1.96 g, 9 mmol, 3 equiv) was dissolved in 10 mL of THF and then added slowly to the solution. The stirring was continued at rt for 1 h. Then the reaction was quenched with H2O, and the resulting solution was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The resulting crude product was purified by flash column chromatography (PE/EtOAc = 19:1) to give 36 as a colorless solid (1.03 g, 92% yield); mp 114–115 °C; 1H NMR (CDCl3, 200 MHz) δ 1.27 (s, 9H), 2.03 (s, 3H), 6.56 (s, 1H), 6.92–7.30 (m, 10 H), 7.69 (s; 2H), 8.29 (d, J = 4.4 Hz, 1H); 13C NMR (CDCl3, 50 MHz) δ 17.7, 28.1, 67.0, 80.7, 122.3, 126.9, 127.8, 129.9, 132.2, 138.8, 139.6, 146.0, 153.2, 153.9. HRMS calcd for C24H26N2O2+: [M + H]+ 375.2067. Found: [M + H]+ 375.2057.
tert-Butyl Benzhydrylcarbamate (37)
Methyl trifluoromethanesulfonate (45 mg, 0.275 mmol, 1.1 equiv) was added dropwise to a solution of 36 (94 mg, 0.25 mmol, 1 equiv) in CH2Cl2 (6 mL) at 0 °C, and the resulting solution was stirred for 12 h at that temperature. Then the solvent was removed under vacuum, and the residue was dissolved in MeOH (3 mL). A 2 M aqueous NaOH solution (1.5 mL) was added, and stirring was continued at 50 °C for 6 h. The solvents were removed, and the resulting residue was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was recrystallized from MeOH/water to give the desired product as a colorless solid (64 mg, 91% yield). Analytical data are in accordance with the literature:28 mp 121–122 °C; 1H NMR (CDCl3, 200 MHz) δ 1.43 (s, 9H), 5.17 (s, 1H), 5.90 (s, 1H), 7.22–7.36 (m, 10 H); 13C NMR (CDCl3, 50 MHz) δ 28.3, 58.4, 79.8, 127.2, 127.3, 128.6, 142.1, 155.0.
Acknowledgments
We acknowledge the Austrian Science Foundation (FWF, Project P21202-N17) for financial support of this work.
Supporting Information Available
One table listing catalyst screening, additional text describing DFT calculations, and NMR spectra of all prepared compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected papers on C–H activation, see the following: ; a Murai S.; Kakiuchi F.; Sekine S.; Tanaka Y.; Kamatani A.; Sonoda M.; Chatani N. Nature 1993, 366, 529–531. [Google Scholar]; b Arndtsen B. A.; Bergman R. G.; Mobley T. A.; Peterson T. H. Acc. Chem. Res. 1995, 28, 154–162. [Google Scholar]; c Handbook of C–H Transformations: Applications in Organic Synthesis, Vol. 2; Dyker G., Ed.; Wiley–VCH: Weinheim, Germany, 2005. [Google Scholar]; d Sames D.; Godula K. Science 2006, 312, 67–72. [DOI] [PubMed] [Google Scholar]; e Campeau L.-C.; Schipper D. J.; Fagnou K. J. Am. Chem. Soc. 2008, 130, 3266–3267. [DOI] [PubMed] [Google Scholar]; f Fagnou K.; Liegault B.; Lapointe D.; Caron L.; Vlassova A. J. Org. Chem. 2009, 74, 1826–1834. [DOI] [PubMed] [Google Scholar]; g Huestis M.; Fagnou K. Org. Lett. 2009, 11, 1357–1360. [DOI] [PubMed] [Google Scholar]; h Ackermann L.; Vicente R.; Kapdi A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. [DOI] [PubMed] [Google Scholar]; i Schnürch M.; Dastbaravardeh N.; Ghobrial M.; Mrozek B.; Mihovilovic M. D. Curr. Org. Chem 2011, 15, 2694–2730. [Google Scholar]; j Yeung C. S.; Dong V. M. Chem. Rev. 2011, 111, 1215–1292. [DOI] [PubMed] [Google Scholar]
- For selected papers on sp2 C–H bond functionalization, see the following: ; a Kakiuchi F.; Chatani N. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar]; b Chen X.; Li J.-J.; Hao X.-S.; Goodhue C. E.; Yu J.-Q. J. Am. Chem. Soc. 2006, 128, 78–79. [DOI] [PubMed] [Google Scholar]; c Fairlamb I. J. S. Annu. Rep. Prog. Chem., Sect. B: Org. Chem 2007, 103, 68–89. [Google Scholar]; d McGlacken G. P.; Bateman L. M. Chem. Soc. Rev. 2009, 38, 2447–2464. [DOI] [PubMed] [Google Scholar]
- For selected papers on sp3 C–H bond functionalzation, see the following: ; a Shabashov D.; Daugulis O. Org. Lett. 2005, 7, 3657–3659. [DOI] [PubMed] [Google Scholar]; b Murahashi S.-I.; Komiya N.; Terai H. Angew. Chem., Int. Ed. 2005, 44, 6931–6933. [DOI] [PubMed] [Google Scholar]; c Chen X.; Goodhue C. E.; Yu J.-Q. J. Am. Chem. Soc. 2006, 128, 12634–12635. [DOI] [PubMed] [Google Scholar]; d Campos K. R. Chem. Soc. Rev. 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]; e Tsuchikama K.; Kasagawa M.; Endo K.; Shibata T. Org. Lett. 2009, 11, 1821–1823. [DOI] [PubMed] [Google Scholar]; f Chaumontet M.; Piccardi R.; Baudoin O. Angew. Chem., Int. Ed. 2009, 48, 179–182. [DOI] [PubMed] [Google Scholar]; g Rousseaux S.; Gorelsky S. I.; Chung B. K. W.; Fagnou K. J. Am. Chem. Soc. 2010, 132, 10692–10705. [DOI] [PubMed] [Google Scholar]; h Jazzar R.; Hitce J.; Renaudat A.; Sofack-Kreutzer J.; Baudoin O. Chem.—Eur. J. 2010, 16, 2654–2672. [DOI] [PubMed] [Google Scholar]; i Bellina F.; Rossi R. Chem. Rev. 2010, 110, 1082–1146. [DOI] [PubMed] [Google Scholar]; j Sundararaju B.; Tang Z.; Achard M.; Sharma G. V. M; Toupet L.; Bruneau C. Adv. Synth. Catal. 2010, 352, 3141–3146. [Google Scholar]; k Ghobrial M.; Harhammer K.; Mihovilovic M. D.; Schnürch M. Chem. Commun. 2010, 46, 8836–8838. [DOI] [PubMed] [Google Scholar]; l Sundararaju B.; Achard M.; Sharma G. V. M; Bruneau C. J. Am. Chem. Soc. 2011, 133, 10340–10343. [DOI] [PubMed] [Google Scholar]; m Pan S.; Endo K.; Shibata T. Org. Lett. 2011, 13, 4692–4695. [DOI] [PubMed] [Google Scholar]; n Baudoin O. Chem. Soc. Rev. 2011, 40, 4902–4911. [DOI] [PubMed] [Google Scholar]; o Wasa M.; Engle K. M.; Lin D. W.; Yoo E. J.; Yu J.-Q. J. Am. Chem. Soc. 2011, 133, 19598–19601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected papers on cyclometalation C–H bond activation, see the following: ; a Ishiyama T.; Hartwig J. J. Am. Chem. Soc. 2000, 122, 12043–12044. [Google Scholar]; b Park Y. J.; Jo E.-A.; Jun C.-H. Chem. Commun. 2005, 1185–1187. [DOI] [PubMed] [Google Scholar]; c Kalyani D.; Sanford M. S. In Directed Metallation; Topics in Organometallic Chemistry, Vol. 24; Chatani N., Ed.; Springer: Heidelberg, Germany, 2007; p 85. [Google Scholar]; d Ackermann L. In Directed Metallation; Topics in Organometallic Chemistry, Vol. 24; Chatani N., Ed.; Springer: Heidelberg, Germany, 2007; p 222. [Google Scholar]; e Kim M.; Kwak J.; Chang S. Angew. Chem., Int. Ed. 2009, 48, 8935–8939. [DOI] [PubMed] [Google Scholar]
- For selected papers on chelation assisted sp3 C–H bond functionalization, see ; Kalyani D.; Deprez N. R.; Desai L. V.; Sanford M. S. J. Am. Chem. Soc. 2005, 127, 7330–7331. [DOI] [PubMed] [Google Scholar]
- Jun C.-H. Chem. Commun. 1998, 1405–1406. [Google Scholar]
- Kakiuchi F.; Matsuura Y.; Kan S.; Chatani N. J. Am. Chem. Soc. 2005, 127, 5936–5945. [DOI] [PubMed] [Google Scholar]
- Pastine S. J.; Gribkov D. V.; Sames D. J. Am. Chem. Soc. 2006, 128, 14220–14221. [DOI] [PubMed] [Google Scholar]
- Prokopcova H.; Bergman S. D.; Aelvoet K.; Smout V.; Herrebout W.; Van der Veken B.; Meerpoel L.; Maes B. U. W. Chem.—Eur. J. 2010, 16, 13063–13067. [DOI] [PubMed] [Google Scholar]
- Dastbaravardeh N.; Schnürch M.; Mihovilovic M. D. Org. Lett. 2012, 14, 1930–1933. [DOI] [PubMed] [Google Scholar]
- Park Y. J.; Park J.-W.; Jun C.-H. Acc. Chem. Res. 2008, 41, 222–234. [DOI] [PubMed] [Google Scholar]
- Dastbaravardeh N.; Schnürch M.; Mihovilovic M. D. Org. Lett. 2012, 14, 3792–3795. [DOI] [PubMed] [Google Scholar]
- Koley M.; Schnürch M.; Mihovilovic M. D. Tetrahedron 2011, 67, 4169–4178. [Google Scholar]
- Gribkov D. V.; Pastine S. J.; Schnürch M.; Sames D. J. Am. Chem. Soc. 2007, 129, 11750–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr R. G.; Yang W.. Density-functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [Google Scholar]
- Simmons E. M.; Hartwig J. F. Ange. Chem., Int. Ed. 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- Jones W. D. Acc. Chem. Res. 2003, 36, 140–146. [DOI] [PubMed] [Google Scholar]
- Jana K. J.; Grimme S.; Studer A. Chem.—Eur. J. 2009, 15, 9078–9084. [DOI] [PubMed] [Google Scholar]
- For N-Boc deprotection, see the following: ; a Norma J. T.; Simon W. M.; Frost H. N.; Ewing M. Tetrahedron Lett. 2004, 45, 905–906. [Google Scholar]; b Srinivasan N.; Yurek-George A.; Ganesan A. Mol. Div. 2005, 9, 291–293. [DOI] [PubMed] [Google Scholar]
- Murai S.; Chatani N.; Asaumi T.; Yorimitsu S.; Ikeda T.; Kakiuchi F. J. Am. Chem. Soc. 2001, 123, 10935–10941. [DOI] [PubMed] [Google Scholar]
- Koley M.; Wimmer L.; Schnürch M.; Mihovilovic M. D. Eur. J. Org. Chem. 2011, 1972–1979. [Google Scholar]
- Beveridge R. E.; A. B. A. Synthesis 2010, 6, 1000–1008. [Google Scholar]
- Reimann E.; Schwaetzer I.; Zymalkowski F. Justus Liebigs Ann. Chem 1975, 1070–1080. [DOI] [PubMed] [Google Scholar]
- Lanni E. L.; Bosscher M. A.; Ooms B. D.; Shandro C. A.; Ellsworth B. A.; Anderson C. E. J. Org. Chem. 2008, 73, 6425–6428. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Gao T.-T.; Shao L.-X. Tetrahedron 2011, 67, 5150–5155. [Google Scholar]
- Manley P. J.; Bilodeau M. T. Org. Lett. 2002, 4, 3127–3129. [DOI] [PubMed] [Google Scholar]
- Toma G.; Fujita K.-i.; Yamaguchi R. Eur. J. Org. Chem. 2009, 4586–4588. [Google Scholar]
- Maddani M. R.; Moorthy S. K.; Prabhu K. R. Tetrahedron 2010, 66, 329–333. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.