

Abstract

Cross-conjugated trienamines are introduced as a new concept in asymmetric organocatalysis. These intermediates are applied in highly enantioselective Diels–Alder and addition reactions, providing functionalized bicyclo[2.2.2]octane compounds and γ′-addition products, respectively. The nature of the transformations and the intermediates involved are investigated by computational calculations and NMR analysis.
Covalent activation of carbonyl compounds, facilitated by chiral amines, plays a dominant role in asymmetric catalysis.1 The past decade has witnessed the development of α-functionalization by enamine catalysis,2 β-functionalization via iminium ion catalysis3 and γ-functionalization by dienamine catalysis.4 Recently, trienamine catalysis has emerged as a new mode of activation involving chiral amine catalyzed, asymmetric Diels–Alder reactions of electron-poor dienophiles with 2,4-dienals5 or 2,4-dienones.6
Trienamine catalysis has focused on the promotion of asymmetric functionalization of the β,ε-carbon centers in the trienamine intermediate, leading to functionalized cyclohexene products (Figure 1a). As recently highlighted by Melchiorre et al., cross-conjugated trienamines have until now been perceived as nonproductive reaction intermediates.7 However, we envisioned that a new development in organocatalysis, in which cross-conjugated trienamine intermediates are involved, might be possible. This cross-conjugated trienamine intermediate could then hopefully undergo either a [4 + 2]-cycloaddition reaction, forming γ′,δ-functionalized products, or a γ′-selective addition reaction (Figure 1b).
Figure 1.
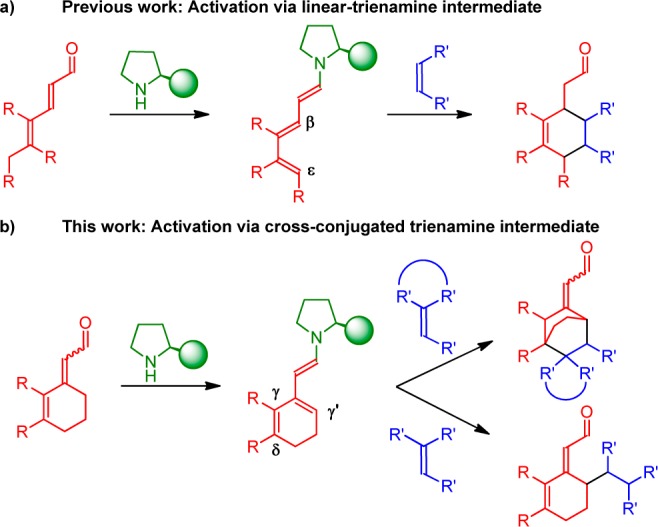
Linear and cross-conjugated trienamines in organocatalysis.
Herein, we present the first asymmetric organocatalyzed Diels–Alder reaction via cross-trienamine intermediates, which provides the enantioselective functionalization at the γ′,δ-carbon centers of cyclic dienals. Furthermore, highly γ′-selective additions have been developed using more polarized olefins. The methodologies presented represent novel reactivity and selectivity concepts in organocatalysis. In contrast to previous work on cross-conjugated dienamines,8 where the enone substrates employed often require specific substitution patterns to ensure exclusive formation of desired products,9 the present approach provides exclusive cross-conjugated reactivity, despite being able to form alternative conjugated intermediates. The cross-conjugated trienamine Diels–Alder reaction shows the conversion of cyclic 2,4-dienals into functionalized bicyclo[2.2.2]octanes or octenes with four stereocenters. These polycyclic compounds have a widespread presence in nature;10 however, their synthesis is not trivial and the development of effective methods, which exhibit high selectivity, still represents a challenge.11
In the present study, we focus first on 3-olefinic oxindoles and 5-olefinic azlactones as dienophiles. The [4 + 2]-cycloaddition of the trienamine intermediate with these dienophiles provides spirocyclic oxindoles and azlactones. Substituted oxindoles are found in a number of natural products exhibiting biological activity12 and have attracted a lot of attention.13 The products obtained from the reaction with the 5-olefinic azlactones are highly functionalized amino acid precursors.14
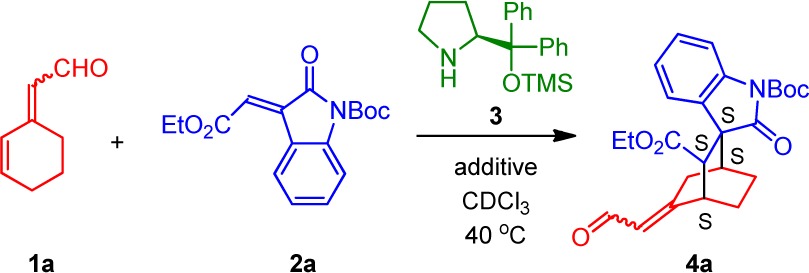
Initial studies revealed that 3-olefinic oxindoles were indeed suitable dienophiles for the [4 + 2]-cycloaddition. Reaction of 2,4-dienal 1a (1:1 E/Z mixture) with the 3-olefinic oxindole 2a, 20 mol % diphenylprolinol silyl ether catalyst 3, and 20 mol % o-fluorobenzoic acid (o-FBA) in CDCl3 at 40 °C gave full conversion to 4a within 20 h (Table 1, entry 1).
Table 1. Optimization of the Cross-Conjugated Trienamine Asymmetric Diels–Alder Reaction of the Cyclic 2,4-Dienal 1a and 3-Olefinic Oxindole 2aa.
| ent | 3 (mol %) | add (mol %) | convb (%) | drb | eec (%) |
|---|---|---|---|---|---|
| 1 | 20 | o-FBA (20) | >95 | 8:1 | 97 |
| 2 | 10 | o-FBA (10) | 44 | 8:1 | ndd |
| 3 | 20 | none | 53 | 8:1 | ndd |
| 4 | 20 | HOAc (20) | >95 | 8:1 | 70 |
| 5 | 20 | BA (20) | >95 | 8:1 | 80 |
| 6 | 20 | o-NBA (20) | >95 | 8:1 | 97 |
Determined by 1H NMR of the crude mixture of 4a.
Determined by chiral stationary phase HPLC.
nd: not determined.
The fact that product 4a results from a cross-conjugated trienamine intermediate is surprising given that the formation of the linear-trieneamine intermediate is not suppressed. To understand why the cycloaddition occurs via the cross-conjugated trienamine, a combination of computational and NMR studies were performed. Calculations (MPW1K/6-31+G(d,p)) on a simplified model system of the two trienamine intermediates A (linear-trienamine intermediate) and B (cross-trienamine intermediate) show that A is favored by 4.2 kcal/mol relative to B (Figure 2).15 This is consistent with the hypothesis that deprotonation at the ε-position is favored over deprotonation at the γ′-position because it provides a fully conjugated π-system. Based on this energy difference, it should be expected that the linear-trienamine intermediate is present in higher concentrations than the cross-trienamine. This was confirmed by 1H NMR studies, which showed that only the linear trienamine could be detected (see Supporting Information (SI) for NMR studies).
Figure 2.

Calculated reaction pathways for the organocatalytic [4 + 2]-cycloaddition of the linear (top) and cross-conjugated (bottom) trienamines. Energies are reported in kcal/mol and are relative to the energy of A+C.
To provide insight as to why the Diels–Alder reaction occurs via the higher energy trienamine intermediate B, the [4 + 2]-cycloaddition of the reaction was modeled computationally.16 In this study, reactions of both A (linear-trienamine intermediate) and B (cross-trienamine intermediate) were examined.17,18 In both cases, the [4 + 2]-cycloaddition was found to be a concerted, highly asynchronous process.19 The transition state structures for the reaction of A+C are both lower in energy than the transition state structures for the reaction of B+C. Both products (D1 and D2) resulting from cycloaddition of A+C are higher in energy than the products (E1 and E2) produced by the cycloaddition of B+C. Comparison of the two thermodynamic products of A+C and B+C (D1 and E1, respectively) shows E1 to be 8.8 kcal/mol lower in energy than D1. It should be noted that the thermodynamic product E1 corresponds to the observed product (4a). The increased thermodynamic stability of the products of the [4 + 2]-cycloaddition through the cross-trienamine B results from the formation of a conjugated π-system, which is not formed in the cycloaddition of A with C.
The data suggest that if the reaction was under kinetic control, the products resulting from the [4 + 2]-cycloaddition of the linear-trienamine intermediate should be the major products. However, the only observed product is that resulting from the cycloaddition with the cross-trienamine intermediate. This suggests that the reaction is thermodynamically driven.
Motivated by the initial synthetic and computational results, a screening of reaction conditions was performed. Decreasing the catalyst and additive loadings to 10 mol % each lowered the conversion (Table 1, entry 2). Acid additives were shown to be necessary for high conversion (entry 3). By employing HOAc as an additive, full conversion and similar diastereoselectivities as for o-FBA were obtained; however, the enantioselectivity dropped significantly (entry 4). Changing the additive to benzoic acid (BA) also resulted in a lower enantioselectivity (entry 5), whereas o-nitrobenzoic acid (o-NBA) provided similar conversion and selectivities as o-FBA (entry 6). The enantioselectivity seems to correlate with the acidity of the additive.
The absolute configuration of compound 4a was established to be (S,S,S,S) by X-ray analysis, based on a chloro-analogue of 4a.20
Next, the effect of substituents on dienal 1 was investigated (Table 2). A 1:1 E/Z mixture of 2,4-dienal 1b, which contains a methyl substituent in the δ-position, gave similar, excellent selectivities as those obtained with 1a (entries 1,2). The γ-substituted 2,4-dienals provided products 7, which contain an endocyclic double bond, after reaction with Wittig reagent 5 (entries 3–6). The lack of tautomerization in these cases is probably due to stabilization of the endocyclic double bond by the electron-donating γ-substituent. Alkyl substituents in the γ-position furnished their respective products in excellent enantioselectivities (entries 3,4). Excellent results were also obtained when methoxy and tert-butyldimethyl silyl ether (OTBDMS) substituents were used (entries 5,6).
Table 2. Scope of the Cross-Conjugated Trienamine Asymmetric Diels–Alder Reaction of Substituted 2,4-Dienals 1a–f with the 3-Olefinic Oxindole Substrate 2aa.

| ent | R1, R2 | yieldb(%) | drc | eed (%) |
|---|---|---|---|---|
| 1e | H, H; 1a | 6a (53) | 8:1 | 97 |
| 2f | H, Me; 1b | 6b (48) | 8:1 | 92 |
| 3 | Me, H; 1c | 7a (51) | 9:1 | 98 |
| 4 | Bn, H; 1d | 7b (53) | 8:1 | 99 |
| 5 | OMe, H; 1e | 7c (52) | 11:1 | 99 |
| 6 | OTBDMS, H; 1f | 7d (62) | >20:1 | 99 |
Reactions were performed on a 0.1 mmol scale; aldehyde 4 was functionalized by in situ Wittig olefination, giving 6 (see SI).
Yields refer to the diastereomerically pure products 6 and 7 isolated by FC. The value for 6 is a combined yield of E/Z isomers.
Determined by 1H NMR of the crude mixture before addition of 5.
Determined by chiral stationary phase HPLC and UPC2.
Reaction was performed at 40 °C.
20 mol % BA was used instead of o-FBA.
To test the generality of the reaction, structural variations were made on the 3-olefinic oxindole 2 (Table 3). Substituents of different electronic natures on the aromatic ring could be tolerated in the reaction and provided their respective products 7e–i in good yields and selectivities (entries 1–5). Utilizing an N-unprotected oxindole provided comparable good results for compound 7j (entry 6). Finally, variations at the olefinic part of the oxindole were examined. A benzoyl substituent gave product 7k in a good yield and excellent diastereo- and enantioselectivity (entry 7), whereas the nitrile-substituted oxindole furnished 7l in a good yield and excellent enantioselectivity, albeit with a lower diastereoselectivity (entry 8).
Table 3. Scope of the Cross-Conjugated Trienamine Promoted, Asymmetric Diels–Alder Reaction of 2,4-Dienal 1f with 3-Olefinic Oxindoles 2b–ia.

| ent | R1, R2 | yieldb (%) | drc | eed (%) |
|---|---|---|---|---|
| 1 | CO2Et, 5-Cl; 2b | 7e (75) | >20:1 | 99 |
| 2 | CO2Et, 5-F; 2c | 7f (54) | >20:1 | 99 |
| 3 | CO2Et, 5-NO2; 2d | 7g (61) | >20:1 | 99 |
| 4e | CO2Et, 5,7-Me,Me; 2e | 7h (66) | >20:1 | 99 |
| 5 | CO2Et, 5-OMe; 2f | 7i (49) | >20:1 | 99 |
| 6f | CO2Et, H; 2g | 7j (65) | >20:1 | 99 |
| 7 | COPh, H; 2h | 7k (62) | >20:1 | 99 |
| 8 | CN, H; 2i | 7l (57) | 7:1 | 99 |
See SI.
Yields refer to the diastereomerically pure products 7 isolated by FC.
Determined by 1H NMR analysis of the crude mixture of the aldehyde.
Determined by chiral stationary phase HPLC.
Compound 7h was prepared at 40 °C for the Diels–Alder reaction.
N-Unprotected oxindole was used.
The absolute configuration of 7 was established by X-ray analysis of a derivatized product of the aldehyde precursor from product 7e.20 Configurations of the other products of type 7 were assigned by analogy.
Finally, the cross-conjugated trienamine concept has been extended to other dienophiles. β-Aryl-substituted olefinic azlactones 8a–d were shown to undergo Diels–Alder reactions yielding products 9a–d in excellent enantioselectivities (Table 4). Good yields were obtained with azlactones carrying electron-withdrawing substituents on the aryl moiety (entries 1,2); however, by employment of electron-rich substituents the yield dropped (entry 4).
Table 4. Scope of the Cross-Conjugated Trienamine Asymmetric Diels–Alder Reaction of 2,4-Dienal 1f with β-Aryl-Substituted Olefinic Azlactones 8a.

| ent | Ar | yieldb (%) | drc | ee (%) |
|---|---|---|---|---|
| 1 | 4-(CO2Me)-C6H4; 8a | 9a (58) | 5:1 | 99d |
| 2 | 3-Cl-C6H4; 8b | 9b (54) | 4:1 | 98e |
| 3 | Ph; 8c | 9c (40) | 4:1 | 98e |
| 4 | 2-thiophenyl; 8d | 9d (34) | 3:1 | 92e |
See SI.
Yields refer to the diastereomerically pure products 9 isolated by FC.
Determined by 1H NMR of the crude mixture of the aldehyde.
Determined by chiral stationary phase UPC2.
Determined by chiral stationary phase HPLC.
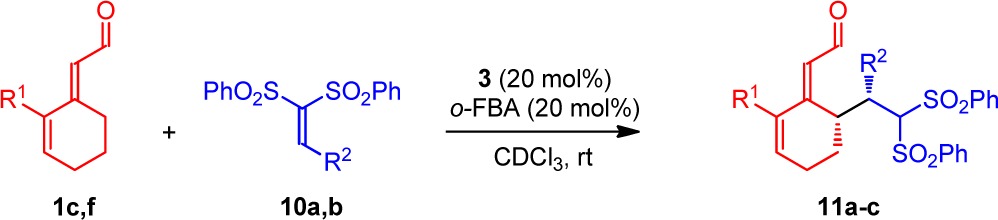
The cross-trienamine reaction concept is not limited to Diels–Alder reactions, as employing vinyl bis-sulfones 10 gave a selective γ′-addition (Table 5).21 Reaction of 1,1-bis(phenylsulfonyl)ethylene 10a with 1f provided product 11a in good yield and high enantioselectivity (entry 1). β-Phenyl vinyl bis-sulfone 10b reacted with 1f and 1c (entries 2,3), giving a good yield of the major diastereomer. The enantioselectivity was excellent, and the configuration was established by X-ray analysis of 11b.20
Table 5. Scope of the Cross-Conjugated Trienamine Asymmetric γ′-Addition of 2,4-Dienals 1 with Vinyl Bis-sulfones 10a.
| ent | R1 | R2 | yieldb (%) | drc | eed (%) |
|---|---|---|---|---|---|
| 1 | OTBDMS | H | 11a (73) | – | 93 |
| 2 | OTBDMS | Ph | 11b (41) | 2:1 | 99 |
| 3 | Me | Ph | 11c (48) | 3:1 | 99 |
See SI.
Yields refer to the diastereomerically pure products 11 isolated by FC.
Determined by 1H NMR of the crude mixture.
Determined by chiral stationary phase UPC2.
In summary, we have developed a new concept in organocatalysis, cross-trienamines. These intermediates are demonstrated to undergo highly enantioselective Diels–Alder reactions with 3-olefinic oxindoles and 5-olefinic azlactones, providing an attractive path to important bicyclic structures. Furthermore, we have also shown that cross-trienamines can react in a highly enantioselective γ′-addition with vinyl bis-sulfones. Additionally, calculations and NMR studies were carried out to provide insight into the cross-trienamine intermediates.
Acknowledgments
This work was financially supported by the Aarhus University, OChem School, FNU and the Carlsberg Foundation. Dr. Jacob Overgaard is gratefully acknowledged for performing X-ray analysis. Thanks are expressed to Dr. Hao Jiang for rewarding discussions. M.S. thanks the Austrian Science Fund (FWF) for a postdoctoral Erwin Schrödinger fellowship (J 3053-N19).
Supporting Information Available
Experimental procedures, analytical data, computational details, and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- For recent reviews on organocatalysis:; a MacMillan D. W. C. Nature 2008, 455, 304. [DOI] [PubMed] [Google Scholar]; b Melchiorre P. Angew. Chem., Int. Ed. 2009, 48, 1360. [DOI] [PubMed] [Google Scholar]; c Bertelsen S.; Jørgensen K. A. Chem. Soc. Rev. 2009, 38, 2178. [DOI] [PubMed] [Google Scholar]; d Grondal C.; Jeanty M.; Enders D. Nat. Chem. 2010, 2, 167. [DOI] [PubMed] [Google Scholar]; e Cheong P. H. Y.; Legault C. Y.; Um J. M.; Celebi-Olecm N.; Houk K. N. Chem. Rev. 2011, 111, 5042. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Jensen K. L.; Dickmeiss G.; Jiang H.; Albrecht L.; Jørgensen K. A. Acc. Chem. Res. 2012, 45, 248. [DOI] [PubMed] [Google Scholar]
- For a review on enamine catalysis:Mukherjee S.; Yang J. W.; Hoffmann S.; List B. Chem. Rev. 2007, 107, 5471. [DOI] [PubMed] [Google Scholar]
- For a review on iminium ion catalysis:Erkkilä A.; Majander I.; Pihko P. M. Chem. Rev. 2007, 107, 5416. [DOI] [PubMed] [Google Scholar]
- For a review on dienamine catalysis:; a Ramachary D. B.; Reddy Y. V. Eur. J. Org. Chem. 2012, 865. [Google Scholar]
- a Jia Z.-J.; Jiang H.; Li J.-L.; Gschwend B.; Li Q.-Z.; Yin X.; Grouleff J.; Chen Y.-C.; Jørgensen K. A. J. Am. Chem. Soc. 2011, 133, 5053. [DOI] [PubMed] [Google Scholar]; b Jiang H.; Gschwend B.; Albrecht L.; Hansen S. G.; Jørgensen K. A. Chem.—Eur. J. 2011, 17, 9032. [DOI] [PubMed] [Google Scholar]; c Jia Z.-J.; Zhou Q.; Zhou Q.-Q.; Chen P.-Q.; Chen Y.-C. Angew. Chem., Int. Ed. 2011, 50, 8638. [DOI] [PubMed] [Google Scholar]; d Liu Y.; Nappi M.; Arceo E.; Vera S.; Melchiorre P. J. Am. Chem. Soc. 2011, 133, 15212. [DOI] [PubMed] [Google Scholar]; e Liu Y.; Nappi M.; Escucero-Adán E. C.; Melchiorre P. Org. Lett. 2012, 14, 1310. [DOI] [PubMed] [Google Scholar]
- Xiong X.-F.; Zhou Q.; Gu J.; Dong L.; Liu T.-Y.; Chen Y.-C. Angew. Chem., Int. Ed. 2012, 51, 4401. [DOI] [PubMed] [Google Scholar]
- Arceo E; Melchiorre P. Angew. Chem., Int. Ed. 2012, 51, 5290. [DOI] [PubMed] [Google Scholar]
- Examples of organocatalytic reactions of enones via cross-conjugated dienamines:; a Thayumanavan T.; Buchiramachary D.; Kandasamy S.; Tanaka F.; Barbas C. F. III. Tetrahedron Lett. 2002, 67, 301. [Google Scholar]; b Ramachary D. B.; Chowdari N. S.; Barbas C. F. III Angew. Chem., Int. Ed. 2003, 42, 4233. [DOI] [PubMed] [Google Scholar]; c Aznar F.; García B.; Cabal M.-P. Adv. Synth. Catal. 2006, 348, 2443. [Google Scholar]; d Wu L.-Y.; Bencivenni G.; Mancinelle M.; Mazzanti A.; Bartoli G.; Melchiorre P. Angew. Chem., Int. Ed. 2009, 48, 7196. [DOI] [PubMed] [Google Scholar]
- Examples in which cross-dienamine reactivity is observed without enforced cross-conjugated dienamine formation:; a Yamamoto Y.; Momiyama N.; Yamamoto H. J. Am. Chem. Soc. 2004, 126, 5962. [DOI] [PubMed] [Google Scholar]; b Sundén H.; Ibrahem I.; Ariksson L.; Córdova A. Angew. Chem., Int. Ed. 2005, 44, 4877. [DOI] [PubMed] [Google Scholar]; c Sundén H.; Rios R.; Xu Y.; Eriksson L.; Córdova A. Adv. Synth. Catal. 2007, 349, 2549. [Google Scholar]; d Bencivenni G.; Wu L.-Y.; Mazzanti A.; Giannichi B.; Pesciaioli F.; Song M.-P.; Bartoli G.; Melchiorre P. Angew. Chem., Int. Ed. 2009, 48, 7200. [DOI] [PubMed] [Google Scholar]
- a Huang J. M.; Yokoyama R.; Yang C.-S.; Fukuyama Y. J. Nat. Prod. 2001, 64, 428. [DOI] [PubMed] [Google Scholar]; b Liu W; Gu Q.; Zhu W.; Cui C.; Fan G.; Zhu T.; Liu H.; Fang Y. Tetrahedron Lett. 2005, 46, 4993. [Google Scholar]; c Li D.; Wang F.; Cai S.; Zeng X.; Xiao X.; Gu Q.; Zhu W. J. Antibiot. 2007, 60, 317. [DOI] [PubMed] [Google Scholar]; d Ishiuchi K.; Kubota T.; Hayashi S.; Shibata T.; Kobayashi J. Tetrahedron Lett. 2009, 50, 6534. [Google Scholar]
- a Hong R.; Chen Y.; Deng L. Angew. Chem., Int. Ed. 2005, 44, 3478. [DOI] [PubMed] [Google Scholar]; b Dong S.; Zhu J.; Porco J. A. Jr. J. Am. Chem. Soc. 2008, 130, 2738. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nicolaou K. C.; Toh Q.-Y.; Chen D. Y.-K. J. Am. Chem. Soc. 2008, 130, 11292. [DOI] [PubMed] [Google Scholar]; d Chen L.; Hua Z.; Jin Z. Org. Lett. 2011, 13, 3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bindra J. S.The Alkaloids; Manske R. H. F., Ed.; Academic Press: New York, 1973; Vol. 14, p 84. [Google Scholar]; b Galliford C. V.; Scheidt K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. [DOI] [PubMed] [Google Scholar]; c Peddibhotla S. Curr. Bioact. Compd. 2009, 5, 20. [Google Scholar]; d Trost B. M.; Brennan M. K. Synthesis 2009, 3003. [Google Scholar]
- Recent reviews and selected examples covering organocatalytic, enantioselective formation of spirocyclic oxindoles:; a Rios R. Chem. Soc. Rev. 2012, 41, 1060. [DOI] [PubMed] [Google Scholar]; b Jiang K.; Jia Z.-J.; Chen S.; Wu L.; Chen Y.-C. Chem.—Eur. J. 2010, 16, 2852. [DOI] [PubMed] [Google Scholar]; c Wei Q.; Gong L.-Z. Org. Lett. 2010, 12, 1008. [DOI] [PubMed] [Google Scholar]; d Tan B.; Candeias N. R.; Barbas C. F. J. Am. Chem. Soc. 2011, 133, 4672. [DOI] [PubMed] [Google Scholar]
- For example, see:Alba A.-N. R.; Rios R. Chem.—Asian J. 2011, 6, 720. [DOI] [PubMed] [Google Scholar]
- Calculations were performed with GAUSSIAN09. Geometries were optimized with the MPW1K/6-31+G(d,p) level of theory and basis set (Lynch B. J.; Patton L. F.; Harris M.; Truhlar D. G. J. Phys. Chem. A 2000, 104, 4811.) in the gas phase. Reported values are zero-point corrected electronic energies. See SI for full details and complete references on theoretical methods. [Google Scholar]
- For structures and energies see SI.
- Reactions leading to all four diastereomers for reactions of both A and B with C were examined; however, only the energies for the reactions leading to the kinetic and thermodynamic products are presented in the main text. For further information on the reactions leading to the other diastereomers, see SI.
- The [4 + 2]-cycloaddition was also examined with wB97xd/pc-1 and B3LYP/6-31+G(d,p). See SI for details.
- IRC calculations show a distinct shoulder on the path to the products. However, no intermediate could be located.
- See SI for details.
- The selective addition to the γ′-position can be explained by the larger orbital coefficient at this position, as compared to the ε-position, in the HOMO of the trienamine intermediate. See SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.