

Abstract
2-Methyl-3-buten-2-ol (MBO) is an important biogenic volatile organic compound (BVOC) emitted by pine trees and a potential precursor of atmospheric secondary organic aerosol (SOA) in forested regions. In the present study, hydroxyl radical (OH)-initiated oxidation of MBO was examined in smog chambers under varied initial nitric oxide (NO) and aerosol acidity levels. Results indicate measurable SOA from MBO under low-NO conditions. Moreover, increasing aerosol acidity was found to enhance MBO SOA. Chemical characterization of laboratory-generated MBO SOA reveals that an organosulfate species (C5H12O6S, MW 200) formed and was substantially enhanced with elevated aerosol acidity. Ambient fine aerosol (PM2.5) samples collected from the BEARPEX campaign during 2007 and 2009, as well as from the BEACHON-RoMBAS campaign during 2011, were also analyzed. The MBO-derived organosulfate characterized from laboratory-generated aerosol was observed in PM2.5 collected from these campaigns, demonstrating that it is a molecular tracer for MBO-initiated SOA in the atmosphere. Furthermore, mass concentrations of the MBO-derived organosulfate are well correlated with MBO mixing ratio, temperature, and acidity in the field campaigns. Importantly, this compound accounted for an average of 0.25% and as high as 1% of the total organic aerosol mass during BEARPEX 2009. An epoxide intermediate generated under low-NO conditions is tentatively proposed to produce MBO SOA.
1. Introduction
Biogenic volatile organic compounds (BVOCs) are important precursors of secondary organic aerosol (SOA) in the atmosphere.1,2 Recent studies have focused on biogenic SOA formation from isoprene, monoterpenes, and sesquiterpenes owing to their large global emission rates.1,3,4 2-Methyl-3-buten-2-ol (MBO) is an oxygenated BVOC emitted by certain coniferous tree species.5,6 Although the global emission rate of MBO is much lower than that of the other BVOCs,7 it can be highly abundant in certain regions such as pine forests of the western United States.5,8,9 Due to the high volatility of MBO oxidation products, it is generally not considered as a source of SOA. However, recent studies have examined the potential of MBO for producing SOA and concluded that it could marginally contribute to SOA formation under certain atmospheric conditions.10−13
In the troposphere, MBO can react with hydroxyl radicals (kOH = 5.6 × 10–11 molecule–1 cm3 s–1),10 ozone (kO3 = 8.3 × 10–18 molecule–1 cm3 s–1),10 and nitrate radicals (kNO3 = 1.2 × 10–14 molecule–1 cm3 s–1).14 Assuming the average tropospheric concentrations of OH (1.5 × 106 molecules cm–3), O3 (7 × 1011 molecules cm–3), and NO3 (4.8 × 108 molecules cm–3),15 the estimated chemical lifetimes of MBO reacting with these three oxidants are 3.3, 47.8, and 48.2 h, respectively. Studies have reported SOA formation from MBO initiated by these oxidants.10,11,13,16 Chan et al.11 and Jaoui et al.13 investigated OH-initiated oxidation of MBO under different NO conditions. They both found that in the presence of NO, MBO does not form SOA. However, with the absence of NO, slight SOA formation was observed with SOA yields of less than 1%. Jaoui et al.13 observed 2,3-dihydroxyisopentanol (DHIP) as a MBO SOA tracer from both low-NO chamber experiments and atmospheric particulate matter (PM) samples using the GC/MS technique. In addition, MBO oxidation could produce glycolaldehyde in high yield under both high- and low-NO conditions.10,17,18 Glycolaldehyde and its oxidation product, glyoxal, are both water-soluble and could produce SOA on wet aerosols and clouds.19−21
Recent studies have proposed that organosulfates are an important class of SOA species, especially for biogenic SOA.22−28 Isoprene- and monoterpene-derived organosulfates have been confirmed using the liquid chromatography/electrospray ionization mass spectrometry (LC/ESI-MS) technique.22−29 These organosulfate species have been identified as important atmospheric SOA tracers representing biogenic SOA enhanced by anthropogenic emissions of NOx (=NO + NO2) and SO2.2,30−33 Moreover, aerosol acidity was found to enhance SOA formation from some BVOCs (e.g., isoprene, α-pinene, and β-caryophyllene) as well as the organosulfate component.22−28,34−36 With a structure similar to isoprene, MBO may also produce organosulfates, and SOA formation may be affected by aerosol acidity in a similar manner. In the present study, aerosol samples were collected from both MBO photooxidation chamber experiments and field campaigns in locations with abundant MBO emissions. Ultra-performance liquid chromatography/negative electrospray ionization high-resolution quadrupole time-of-flight mass spectrometry (UPLC/(−)ESI-HR-Q-TOFMS) was used to analyze the filter samples and hence quantitatively investigate the organosulfate formation from MBO photooxidation in the atmosphere.
2. Experimental Section
High-NO Chamber Experiments
Two initially high-NO experiments (UNC1 and UNC2 in Table 1) with neutral versus acidified sulfate seed aerosols were conducted on the same day at the University of North Carolina 274-m3 dual outdoor smog chamber facility (Pittsboro, NC) under natural sunlight. The detailed chamber instrumentation has been described elsewhere.37−40 Briefly, the smog chamber is divided by a Teflon film curtain into two individual chambers with identical volumes (∼136 m3). For the neutral seeded experiment, 0.06 M (NH4)2SO4 solution was atomized into one chamber; 0.06 M ((NH4)2SO4 + H2SO4) solution was used instead in the other chamber for the acidic seeded experiment. Despite the different solution concentrations, similar initial seed aerosol mass concentrations were obtained in both experiments by varying the injection time. NO was injected into the chambers from a high-pressure gas cylinder as soon as seed aerosol volume concentrations stabilized. High-purity liquid MBO (>98%, Aldrich) was then heated in a U-tube and flushed into the chamber with a N2 flow. Ozone and NOx were measured by UV photometric and chemiluminescent monitors (Thermo-Environmental 49P and Bendix Model 8101B analyzers, respectively). MBO concentration was measured by GC/FID. Particle size distributions and the volume concentrations were measured using a scanning mobility particle sizer (SMPS) (TSI 3080) coupled with a condensation particle counter (CPC) (TSI 3022A). Aerosol filter samples (PALL Life Sciences, Teflon, 47-mm diameter, 1.0-μm pore size) were collected during the entire photochemical period at a flow rate of ∼17 L min–1 for each experiment. All uncertainties for measurements by instrumentation used on the smog chamber are provided in Table S4 in the Supporting Information.
Table 1. Smog Chamber Experimental Conditions.
| IDa | MBO (ppmC) | H2O2 (ppm) | NO (ppb) | temperature (K) | RH (%) | [H+]airc (nmol/m3) | seed aerosol (ug/m3 sulfate) | average OC (μg C/m3) | C5H12O6Se (ng/m3) |
|---|---|---|---|---|---|---|---|---|---|
| UNC1 | 4 | 200 | 301–314 | <30 | 50 (AS) | 0.95 | |||
| UNC2 | 4 | 200 | 301–314 | <30 | 45 (1/2 AS + 1/2 SA) | 1.88 | |||
| EPA1 | 15 | 9 | <15b | 300 | <5 | 125 | 40 (ASd) | 6.5 | 6.5 |
| EPA2 | 15 | 9 | <15b | 300 | <5 | 289 | 35 (2/3 AS + 1/3 SAd) | 9.6 | 50.4 |
| EPA3 | 15 | 9 | <15b | 300 | <5 | 902 | 35 (1/2 AS + 1/2 SA) | 11.4 | 53.2 |
| EPA4 | 15 | 9 | <15b | 300 | <5 | 1590 | 32 (1/3 AS + 2/3 SA) | 21.9 | 120.3 |
Experiments UNC1 and UNC2 are the high-NO experiments conducted at the UNC dual outdoor smog chamber; experiments EPA1–EPA4 are the low-NO experiments conducted at the EPA smog chamber.
The NO concentrations in EPA1–EPA4 were not measured, but there was no NO added to the chamber during the experiments. While 15 ppb represents a conservative upper limit for a conventional sensitivity NO instrument, chamber levels were undoubted below 1 ppb based on the experimental operation (see text).
[H+]air is used as an indicator of acidic levels.
“AS” represents ammonium sulfate; “SA” represents sulfuric acid.
C5H12O6S is the chemical formula of the organosulfate species discussed below.
Low-NO Chamber Experiments
A dynamic experiment with four stages (EPA1–EPA4 in Table 1) was conducted at the U.S. EPA (Research Triangle Park, NC) 14.5-m3 Teflon chamber operated in a flow mode to produce a steady-state condition. There was no NO added to the chamber during the experiment. The photolysis of hydrogen peroxide (H2O2) was used as the source of OH radicals. The concentration of MBO throughout the four stages was held at a constant 15 ppmC. The concentration of H2O2 was maintained at ∼9 ppm and RH was less than 5% during the entire experiment. The aerosol acidity was changed between stages by adjusting the fraction of (NH4)2SO4 and H2SO4 in the seed aerosol (shown in Table 1). Particle organic carbon concentrations were measured by a semicontinuous elemental carbon–organic carbon (EC-OC) instrument (Sunset Laboratories, Tigard, OR). Aerosol samples were collected at a flow rate of 15 L min–1 on 47-mm Teflon filters for determination of the hydrogen ion concentration ([H+]air) as nmol H+ m–3 using an Oakton 300 pH probe (Vernon Hills, IL) after dissolution in 10 mL of distilled deionized water. Teflon impregnated glass fiber filters (47 mm; Pall Gelman Laboratory, Ann Arbor, MI) were used for other off-line chemical determinations. The detailed gas-phase and aerosol-phase measurements have been described elsewhere.13,35
Field Measurements: The BEARPEX Campaigns
The BEARPEX (Biosphere Effects on Aerosols and Photochemistry Experiment) campaigns were conducted at a ponderosa pine plantation located between Sacramento and Lake Tahoe in the Sierra Nevada Mountains, California.41,42 MBO is one of the dominant biogenic VOCs emitted by the forest at the BEARPEX site.8,43,44 The field data used for this study were collected September 20–25, 2007, and July 25–30, 2009. The two sampling periods were characterized by very different conditions: 2007 was cooler and wetter ,while 2009 was hotter and drier. A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc., hereinafter referred to as AMS) was used during the BEARPEX 2007 campaign to measure nonrefractory PM1 aerosol components, from which the mass concentrations of individual components (sulfate, nitrate, ammonium, chloride, and organics) were resolved.45 The AMS operation at BEARPEX was described by Farmer et al.46 In the BEARPEX 2009 campaign, the PM1 organic mass was measured by FTIR and sulfate, nitrate, and ammonium were measured using a Metrohm ion chromatograph equipped with a Metrosep A Supp 5 column for anions and a Metrosep C 4 column for cations. The average organic mass concentrations were ∼2.3 and ∼3.7 μg m–3 during 2007 and 2009, respectively. Aerosol acidities (strong acidity) were estimated based on charge balance using the measured concentrations of the three inorganic components. Average acidities were estimated by averaging for each filter sampling period. MBO and isoprene were measured by two different in situ instruments: in 2007 using GC/MS operated by the National Oceanic and Atmospheric Administration (NOAA)9,47 and in 2009 using GC/FID operated by Texas A&M University.48,49
PM2.5 samples were collected using high-volume samplers during the two continuous five-day periods in 2007 and 2009, with three filters each day, to provide sufficient time resolution to examine the diurnal variation of aerosol compositions. This filter sampling approach could also separate the influence of local biogenic emissions in the morning from the anthropogenic emissions from the California Central Valley arriving in the afternoon and subsequent nighttime chemistry.
Field Measurements: The BEACHON-RoMBAS Campaign
The BEACHON-RoMBAS (Biohydro-atmosphere interactions of Energy, Aerosols, Carbon, H2O, Organics and Nitrogen – Rocky Mountain Biogenic Aerosol Study) campaign was conducted July–August, 2011 at the Manitou Forest Observatory located in Pike National Forest, Colorado. Site information has been described elsewhere.50,51 The BEACHON-RoMBAS site was chosen due to abundant BVOC emissions, in which the MBO emission is important, but with limited anthropogenic influence.
Measurements of aerosol components were performed using a high-resolution AMS.45 The sum of MBO and isoprene was measured using two proton-transfer-reaction time-of-flight mass spectrometers (PTR-TOF-MS, Ionicon Analytik GmbH, Austria, University of Innsbruck (UIBK) and National Center for Atmospheric Research (NCAR)). Details about the instruments and data evaluation can be found in Jordan et al.,52 Graus et al.,53 Müller et al.,54 and Cappellin et al.55 Ambient air was sampled at a flow rate of ∼9 SLPM through a 40-m-long Teflon (PFA) line (1/4″ OD) mounted at 25.1 m on the canopy tower. Both instruments were sampling from the same line with a sampling period of about 0.1 s (UIBK) and 10 s (NCAR), respectively. The merged data set was averaged to 6 min. The drift tube was operated at 2.3 mbar (both instruments) and a drift voltage of 580 V (UIBK) and 550 V (NCAR) and a drift tube temperature of 60 °C (both instruments). Calibration was performed by dynamically diluting VOC standards at ppmV levels with scrubbed air. During BEACHON-RoMBAS, the average temperature (∼ 17 °C) and the organic aerosol mass (∼ 1.4 μg m–3) were fairly low; therefore, a 72 h-integrated aerosol filter sampling approach with a flow rate of 1 m3 min–1 was performed using a high-volume sampler to collect sufficient aerosol mass on each filter. The average acidity for each filter sampling period was estimated in the same manner as for the BEARPEX 2007.
Filter Sample Extractions and Chemical Analyses
Filters collected from chamber experiments and field campaigns were stored in individual precleaned packets in a −20 °C freezer before extraction. All filters were extracted in 15 mL of high-purity methanol (LC-MS CHROMASOLV-grade, Sigma-Aldrich) by sonication for 45 min. The methanol extracts were then blown dry under a gentle N2 stream at ambient temperature.24 Soot particles and quartz fibers for all filter extracts were removed using procedures outlined in Surratt et al.24 Dried residues from filter extracts were reconstituted with 150 μL of a 50:50 (v/v) solvent mixture of 0.1% acetic acid in methanol (LC-MS ChromaSolv-Grade, Sigma-Aldrich) and 0.1% acetic acid in water (LC-MS ChromaSolv-Grade, Sigma-Aldrich). The resultant mixtures were then shaken and sonicated for 5 min and stored at −20 °C before analyses. The detailed description of the UPLC/(−)ESI-HR-Q-TOFMS technique and operating conditions can be found in Zhang et al.39 Propyl sulfate was selected as the surrogate standard for quantifying the MBO-derived organosulfate because of its similar solubility and retention time.36 The detection limit is ∼0.05 ng for this surrogate standard.
3. Results and Discussion
Effects of NO and Acidity on SOA Yields from MBO Photooxidation
Figure 1 shows the results of the initially high-NO experiments conducted at the UNC dual outdoor smog chambers on the same day. Similar gas-phase conditions were obtained for both experiments (Figure 1a) and the only variation between the two experiments was the different sulfate seed aerosol acidities. Figure 1b represents the particle volume concentrations (wall loss uncorrected) of the two MBO experiments compared to seed aerosol only experiments without MBO. Slight SOA formation (∼ 7 μg m–3) was observed only in the acidic experiment and the SOA decreased rapidly after reaching the maximum, where the rapid decrease is consistent with Chan et al.11 It should be noted that the SOA was not formed until after the NO concentration approached zero, when half of the initial MBO (∼ 2 ppmC) remained and MBO peroxy radicals likely began to react with hydroperoxy radicals (RO2 + HO2) and/or other peroxy radicals (RO2 + RO2).
Figure 1.
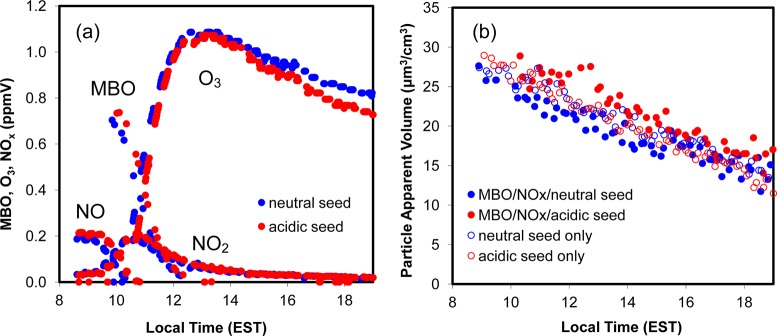
Online measurement results of the high-NO experiments (UNC1 and UNC2). (a) Time profiles of major gas-phase compounds (MBO, NO, NO2, and O3). (b) Wall-loss uncorrected particle apparent volume concentrations (μm3/cm3). Red solid circle represents the acidic seeded MBO experiment; blue solid circle represents the neutral seeded MBO experiments; the hollow circles in red and blue in (b) represent two control experiments with only acidic and neutral seeds, respectively.
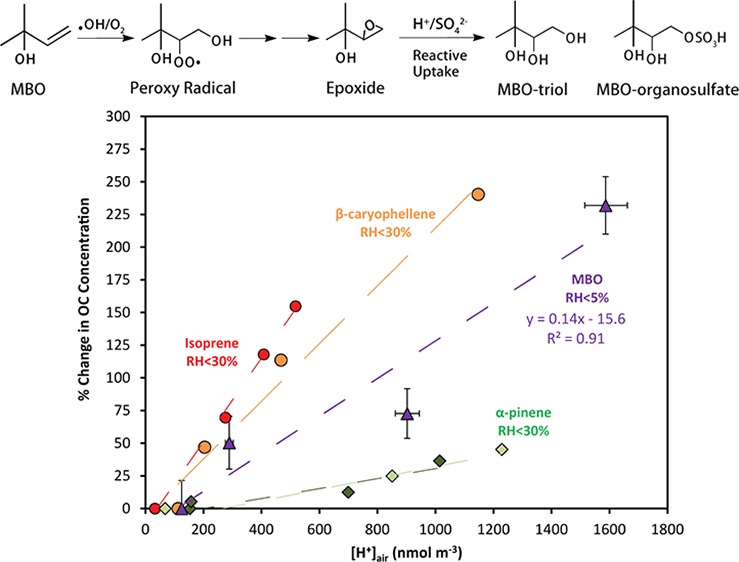
As shown in Table 1, for the low-NO experiment conducted at the EPA smog chamber, measurable SOA was formed in all the four stages with SOA yields up to 1%. In these acidity-varied experiments, [H+]air ranged from 125 to 1587 nmol m–3 and the SOA formation during each stage correlated well with increasing acidity. Figure 2 shows the relationship between the change (%) of organic carbon (OC) concentration compared to the neutral seed case and measured aerosol acidity ([H+]air nmol m–3) for different BVOCs. The data from experiments with isoprene,α-pinene, and β-caryophyllene are reproduced from Surratt et al.23 and Offenberg et al.35 In these prior studies, SOA formation from isoprene, β-caryophyllene, and α-pinene was found to correlate with aerosol acidity as a linear relationship, however, with different slopes. From the present work, the MBO case does also follow the same trend with a slope larger than that of α-pinene and smaller than those of isoprene and β-caryophyllene. It should be noted that the previous studies were performed with RH < 30%, but this study uses RH < 5%, likely causing the aerosols to be more acidic and suggests the acidity effect of MBO may be weaker than this figure indicates compared to the other three BVOCs.
Figure 2.
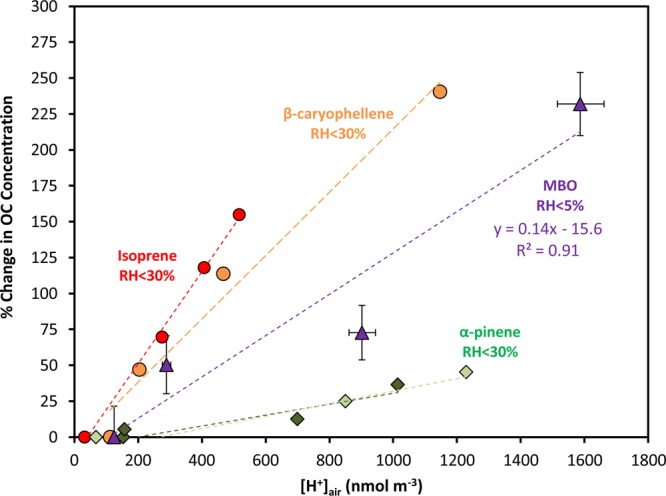
Relationship between the change (%) of organic carbon (OC, in μg C m–3) and the measured seed aerosol acidity ([H+]air nmol m–3) for different BVOCs. The data for isoprene, α-pinene, and β-caryophellene are reproduced from Surratt et al.23 and Offenberg et al.35 The two green shades of α-pinene data are from two individual experiments with different initial conditions, as described by Offenberg et al.35 The MBO data are from the present study. The determination of the error bars is discussed in the Supporting Information.
By combining the two sets of smog chamber experiments, SOA was found to form only when NO was not present; that is, SOA was probably formed from the RO2 + HO2 channel and/or the RO2 + RO2 channel. In addition, the formation of MBO SOA under low-NO conditions was likely enhanced with greater acidity, similar to the observation for the other BVOCs, such as isoprene,23 β-caryophyllene,35 and α-pinene.35
Organosulfate Formation from MBO Photooxidation
Filter samples collected from the chamber experiments discussed above were analyzed using UPLC/(−)ESI-HR-Q-TOFMS. The results suggest an organosulfate species was produced from all experiments. Accurate mass data are consistent with a molecular formula of C5H12O6S (MW 200.0355) for the MBO-derived organosulfate . Figure 3a shows the tandem mass spectra (MS2 spectra) of this organosulfate (m/z 199) from the low-NO experiments, and includes the elemental compositions for each fragment ion determined from the accurate mass measurements. The most abundant fragment is m/z 97 (HSO4–), which indicates the presence of a sulfate ester group in this compound (−OSO3H).22 Since this organosulfate species has the same carbon number (i.e., 5 carbons) as MBO, it maintains the backbone of MBO. Thus, the tentative isomeric structures are proposed in Figure 3a. In a recent study by Jaoui et al.,13 another SOA tracer (DHIP, MW 120), with a structure similar to our proposed organosulfate (i.e., the sulfate ester group is substituted by a hydroxyl group), was observed by GC/MS analyses. For a similar biogenic hydrocarbon, isoprene, previous studies have found that the three major SOA tracers, the 2-methyltetrols (MW 136) and a C5 organosulfate species (MW 216), are derived from the isoprene epoxydiol (IEPOX) isomers.25,36,56 We suggest, by analogy, that both DHIP and the MBO-derived organosulfate found in the present study are formed from an epoxide intermediate, especially since reactive uptake of epoxides has been shown to be kinetically feasible in forming organosulfates and polyols under tropospheric conditions.57 The potential mechanism is discussed further below.
Figure 3.
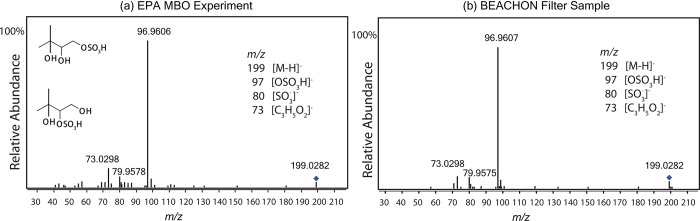
Tandem mass spectra (MS2) of the MBO-derived organosulfate (m/z 199) measured from (a) the EPA low-NO chamber experiment (EPA4, with sampled OC mass concentration ∼21.9 μgC/m3) and (b) the BEACHON aerosol samples. The proposed structural isomers of this organosulfate are shown in (a). The measured mass of the ion is within ±2 mDa of the calculated mass.
The mass concentrations of the MBO-derived organosulfate correlate well with acidity (R2 = 0.87), as shown in Table 1 (and Figure S1). This finding is consistent with previous observations of organosulfates derived from other BVOCs22−24 and suggests that the mechanism of formation of the MBO-derived organosulfate is similar to that of the other organosulfates. Furthermore, increased acidity tends to promote both OC and the MBO-derived organosulfate formation to a similar extent (Figure S1), supporting the same mechanism for the organosulfate and bulk SOA formation. The C5H12O6S organosulfate is also well correlated with OC (R2 = 0.96), indicating the MBO-derived organosulfate can serve as a valid tracer for the OC concentrations.
Field Observations of the MBO-Derived Organosulfate
In addition to the chamber results, the MBO-derived organosulfate was also observed in ambient field samples. In the chemical analyses using UPLC/(−)ESI-HR-Q-TOFMS the same organosulfate species were observed at m/z 199 from BEARPEX 2007, 2009, and BEACHON-RoMBAS 2011 campaigns. The MS2 spectrum and retention time of the m/z 199 organosulfate from the field campaigns are identical to those from MBO chamber experiments (Figure 3), confirming that the m/z 199 organosulfate observed from field campaigns was likely derived from MBO.
Figure 4a shows the averaged MBO mixing ratio and MBO-derived organosulfate mass concentration during each filter-sampling period from BEARPEX 2009. The diurnal variation of the organosulfate mass concentrations is generally consistent with that of MBO mixing ratios. Both the MBO mixing ratio and the MBO-derived organosulfate concentration consistently fell to much lower levels (less than 3 ng/m3) at night. It should be noted that sometimes the MBO mixing ratios in the morning were similar to or even higher than in the afternoon, but the MBO organosulfate level was always higher in the afternoon. This is probably because (1) there will be a certain time delay to form an organosulfate from MBO, (2) the OH and O3 concentrations were highest in the afternoon which likely increased the organosulfate production rate, (3) the factors that enhance organosulfate formation, such as aerosol acidity, were higher in the afternoon when the relative humidity dropped to lower levels and the urban emissions arrived (see Figure 5b). Figure 4b shows the time series of isoprene mixing ratios and the IEPOX-derived organosulfate mass concentrations. The comparison between Figure 4a and 4b suggests that both the isoprene mixing ratio and the IEPOX-derived organosulfate concentration did not follow the diurnal pattern as the MBO did at the BEARPEX site, especially for the IEPOX-derived organosulfates which accumulated to higher concentrations at the end of the week, rather than being dominated by the diurnal trend. At the BEARPEX site, MBO is locally emitted, but isoprene is emitted upwind.42 Thus, the IEPOX-derived organosulfate likely formed upwind with the urban plume before it arrived at the site. Hence, the measured IEPOX-derived organosulfate cannot be simply explained by the trends in local emissions. In some previous studies, the IEPOX-derived organosulfate was found to be the most abundant organosulfate species from ambient aerosols.30,32,33 However, at the BEARPEX site, the MBO organosulfate has a similar abundance and accounts for an average of 0.25% of measured organic PM1, reaching a level of 1% at BEARPEX 2009 (Table S2). The comparison between the MBO-derived and the IEPOX-derived organosulfates further demonstrates that the two C5 organosulfate species arose from different sources and the MBO organosulfate could be a substantial SOA component locally.
Figure 4.

Comparison of diurnal variation of organosulfates and their precursors during the 2009 BEARPEX campaign. (a) Average MBO mixing ratio and the MBO-derived organosulfate; (b) average isoprene mixing ratio and the IEPOX-derived organosulfate. The BVOC mixing ratios are in ppb; the organosulfate mass concentrations are in ng/m3.
Figure 5.
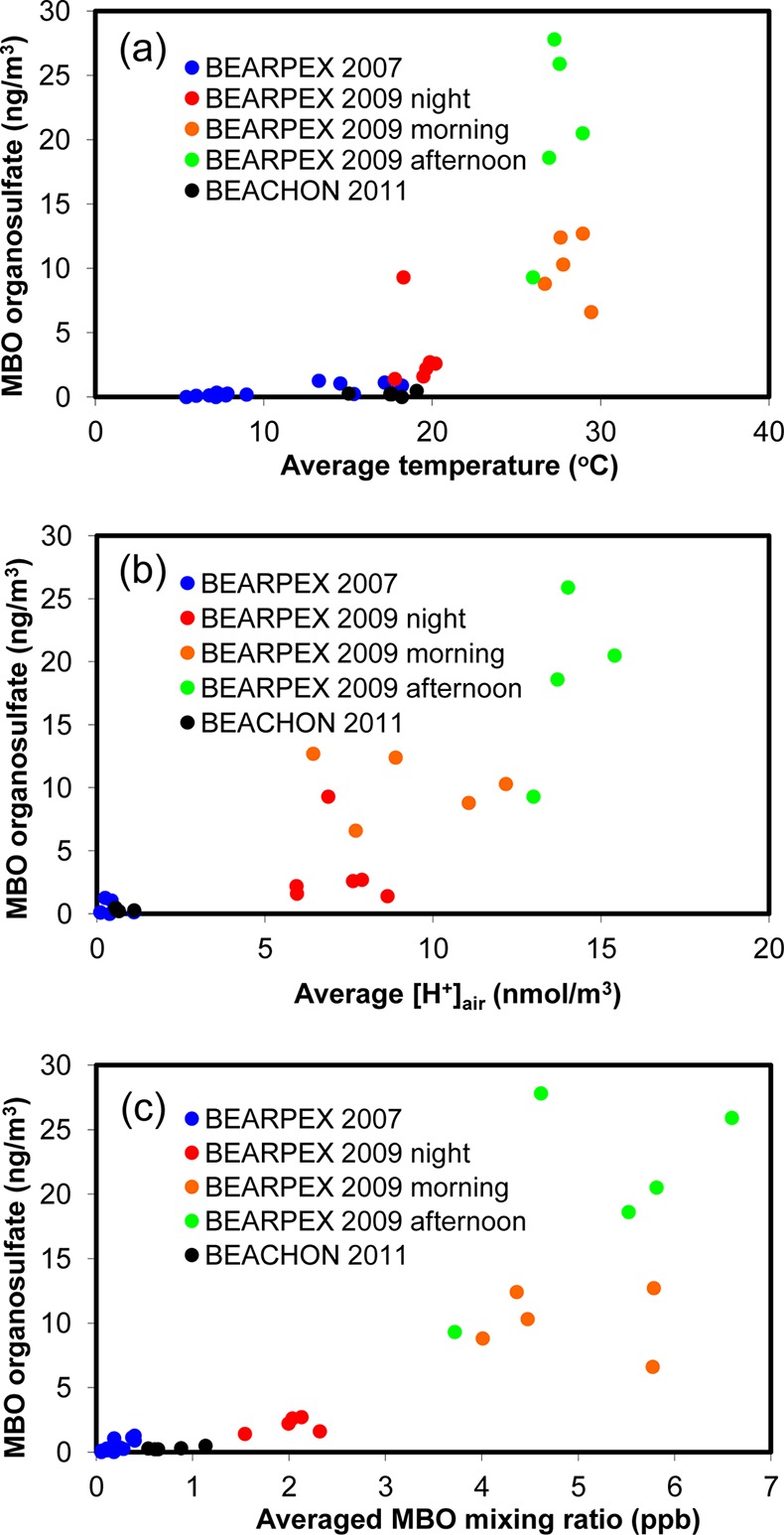
Correlation of the MBO organosulfate mass concentrations to (a) average temperature, (b) average acidity, and (c) average MBO mixing ratio from the BEARPEX and BEACHON-RoMBAS campaigns. The data from different times of day are shown separately for the BEARPEX 2009 results.
Figure 5a–c examines the relationship between the MBO-derived organosulfate concentrations and temperatures, aerosol acidities, and MBO mixing ratios from the BEARPEX and BEACHON-RoMBAS campaigns. Temperatures, aerosol acidities, and MBO mixing ratios were averaged for each filter-sampling period. All the BEARPEX 2009 data are much higher than those of the other two campaigns and have a larger dependence on sampling time during the day. Thus, the correlations analyzed at different times of day (i.e., morning, afternoon, and night) are shown separately for the BEARPEX 2009 data. The temperatures during BEARPEX 2007 and BEACHON-RoMBAS are both lower than that of BEARPEX 2009 (Figure 5a). For BEARPEX 2009, the temperature during the daytime is much higher than that during the nighttime but the temperatures in the morning and afternoon are generally similar. Similar to the temperature conditions, the aerosol acidities during BEARPEX 2007 and BEACHON-RoMBAS are also significantly lower (by an order of magnitude) than those during BEARPEX 2009. Additionally, during BEARPEX 2009 the aerosol acidity is substantially enhanced (by a factor of 3 as shown in Figure 5b) in the afternoon, likely due to the arrival of urban emissions and lower relative humidity. In general, the MBO-derived organosulfate concentration correlates well with the average aerosol acidity (represented as [H+]air in nmol/m3 in Figure 5b, R2 = 0.68). As shown in Figure 5c, the MBO-derived organosulfate concentration correlates well with the average MBO mixing ratio (R2 = 0.75). The MBO mixing ratios increase with increasing temperature as a consequence of the high sensitivity of the MBO emissions to temperature.6
Generally for the BEARPEX 2009 data, higher temperatures and MBO mixing ratios explain the increased formation of organosulfate in the morning compared to the nighttime; the higher acidity partly explains the further enhancement of organosulfate formation in the afternoon compared to the other periods in 2009. The BEACHON-RoMBAS results are similar to the BEARPEX 2007 results in terms of the lower temperatures, aerosol acidities, and MBO mixing ratios. Although the BEACHON-RoMBAS data show slightly higher MBO mixing ratios, the MBO organosulfate formation was not enhanced at the BEACHON-RoMBAS site compared to the BEARPEX 2007 results. One possible explanation might be that under low aerosol acidity, the formation of MBO organosulfate is generally low and not well correlated with MBO mixing ratio. Another possibility is that the sum of MBO and isoprene was measured from BEACHON-RoMBAS and 80% was estimated to be MBO, which might be overestimated. Overall, the field data shown in Figure 5 suggest that MBO is the precursor of this organosulfate species, which is substantially enhanced as aerosol acidity increases.
Potential Mechanisms for Atmospheric MBO SOA Formation
The mechanisms by which MBO-derived organosulfate and other SOA tracers13 form in the atmosphere are still unclear. However, several mechanisms leading to MBO SOA formation have been postulated. Chan et al.11 has proposed that the RO2 + RO2 reaction could produce DHIP, which was observed in the MBO low-NO experiments by Jaoui et al.13 However, the formation of the m/z 199 organosulfate is unlikely via this pathway, since the esterification of an alcohol (i.e., DHIP) with sulfuric acid is kinetically infeasible in the particle phase under either chamber or ambient conditions.58 Although it is possible that aerosol-phase reactions of organic peroxides (ROOR), which are formed from RO2 + RO2 reactions, with acidic sulfate could produce organosulfates, especially for chamber experiments, it unlikely to explain the organosulfate production in the atmosphere because of the dominance of RO2 + HO2 reactions. Consequently, alternative reactions must explain the formation of the m/z 199 organosulfate. As discussed above, the formation pattern of the MBO-derived organosulfate and DHIP is similar to that of the IEPOX-derived organosulfate and the 2-methyltetrols.25,36 Similar to IEPOX chemistry, an epoxide is tentatively proposed as the intermediate (oxidation) product forming the MBO-derived organosulfate as shown in Scheme 1. An MBO-derived epoxide could be produced from ozonolysis, H2O2 oxidation of MBO, or from the further reaction of an MBO hydroxyhydroperoxide. The MBO-derived epoxide can form organosulfates by reaction with sulfuric acid or DHIP by hydrolysis. Epoxidation has been observed from the ozonolysis of olefins.59,60 However, the epoxide yield from olefin ozonolysis tends to be low (<5%)61 and this is likely a minor pathway in the atmosphere. Liu et al.12 have reported MBO epoxide from acid-catalyzed oxidation of MBO with H2O2 (shown in Scheme 1), which is more likely an aqueous-phase process. Therefore, more work is required to establish the formation mechanism of the MBO-derived organosulfate, as well as DHIP, in the atmosphere.
Scheme 1. Proposed Mechanism of MBO Photooxidation Forming SOA (Some Structual Isomers Are Not Shown).

Atmospheric Implications
In the present study, MBO photooxidation is investigated in both smog chamber experiments and field campaigns where MBO emissions are important. SOA formation from MBO was observed under low-NO conditions and after NO was reacted in the high-NO chamber experiments. (While the chamber experiments may have been RO2-rich, sufficient HO2 was present to produce a compound identical to that seen in field samples.) Under both conditions, aerosol acidity was found to promote the formation of MBO SOA. In addition, an organosulfate species (C5H12O6S, MW 200) was substantially enhanced as aerosol acidity increased. From the BEARPEX and BEACHON-RoMBAS campaigns, a species with the same mass spectrometric signature was observed to correlate well with MBO mixing ratio and aerosol acidity. Moreover, the average MBO organosulfate concentration measured from the BEARPEX site is similarly abundant to the IEPOX-derived organosulfate (∼ 15 ng/m3); the latter has been reported to be the most abundant organosulfate tracer in a number of areas.30
The results of this report are significant because MBO is a locally abundant BVOC in certain regions, such as the western United States. Although the SOA yield reported in previous studies is low (<1%), the SOA mass concentration produced could still be appreciable with the high emission rates expected at locations like the BEARPEX site. As discussed above, the formation mechanism of MBO-derived organosulfate is likely similar to that of the IEPOX-derived organosulfate. Lin et al.36 found that the IEPOX-derived organosulfate accounts for ∼5% of total SOA from IEPOX. Assuming the same mass fraction in the MBO case, MBO SOA can account for as high as 0.4 μg m–3, which is 10% of total organic mass on average. However, further investigation is required to provide a more accurate estimate. As a result, the inclusion of SOA formation from MBO in current air quality models may be important,62 especially for regional or local predictions of SOA. The MBO-derived organosulfate has the potential to serve as an SOA tracer for a source apportionment method under further investigation. These new observations of MBO organosulfate formation and the acidity effect are consistent with previous findings of the organosulfate formation from other BVOCs, including isoprene and monoterpenes.24,25,36 Although additional work is warranted, it is likely that the formation mechanisms of these biogenic organosulfate species are similar.
Acknowledgments
This study was supported by an U.S. EPA contract (EP-W-09-023) to the University of North Carolina. The U.S. Environmental Protection Agency through its Office of Research and Development collaborated in the research described here under Contract EP-D-10-070 to Alion Science and Technology. The manuscript is subjected to external peer review and has been not been cleared for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation. The BEARPEX measurements were supported by an NSF grant ATM-0922562 to the University of California, Berkeley, and ATM-0904203 to the University of California, San Diego. The BEACHON-RoMBAS measurements were supported by NSF sponsorship of the National Center for Atmospheric Research (NCAR). UPLC/ESI-HR-Q-TOFMS analyses were conducted in the UNC-CH Biomarker Mass Facility located within the Department of Environmental Sciences and Engineering, which is a part of the UNC-CH Center for Environmental Health and Susceptibility and is supported by NIEHS (Grant 5P20-ES10126). J.D.S. was supported in part by the Electric Power Research Institute (EPRI). Thanks to Ying-Hsuan Lin for helping to operate the UPLC/(−)ESI-HR-Q-TOFMS instrument. The meteorological measurements from BEACHON-RoMBAS were performed by Andrew A. Turnipseed. The IC measurements for BEARPEX 2009 were performed by M. P. Madsen. P.C.J., D.A.D., and J.L.J. were supported by NSF ATM-0919189 and DOE (BER/ASR Program) DE-SC0006035 and DE-SC0006711. GC-FID measurements by Texas A&M were funded by NSF ATM -0934345. Measurements conducted by the University of Innsbruck were supported by the Austrian Science Fund (FWF): L.K. is a recipient of a DOC-fFORTE-fellowship of the Austrian Academy of Sciences at the Institute of Ion Physics and Applied Physics.
Supporting Information Available
Additional information regarding raw data from campaigns (Tables S1–S3), uncertainties of instrumentation and measurement (Table S4), additional EPA experimental data (Figure S1), additional correlation of field data (Figure S2), additional tandem mass spectral data (Figures S3–S4), MBO intercalibration at BEARPEX campaigns (Figures S5–S6), MBO intercalibration at the BEACHON-RoMBAS campaign (Figure S7). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Hallquist M.; Wenger J. C.; Baltensperger U.; Rudich Y.; Simpson D.; Claeys M.; Dommen J.; Donahue N. M.; George C.; Goldstein A. H.; Hamilton J. F.; Herrmann H.; Hoffmann T.; Iinuma Y.; Jang M.; Jenkin M. E.; Jimenez J. L.; Kiendler-Scharr A.; Maenhaut W.; McFiggans G.; Mentel Th. F.; Monod A.; Prévôt A. S.; Seinfeld J. H.; Surratt J. D.; Szmigielski R.; Wildt J. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar]
- Hoyle C. R.; Boy M.; Donahue N. M.; Fry J. L.; Glasius M.; Guenther A.; Hallar A. G.; Huff Hartz K.; Petters M. D.; Petäjä T.; Rosenoern T.; Sullivan A. P. A review of the anthropogenic influence on biogenic secondary organic aerosol. Atmos. Chem. Phys. 2011, 11, 321–343. [Google Scholar]
- Guenther A.; Pierce B.; Lamb B.; Harley P.; Fall R. Natural emission of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ. 2000, 34, 2205–2230. [Google Scholar]
- Guenther A.; Karl T.; Harley P.; Wiedinmyer C.; Palmer P. I.; Geron C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar]
- Goldan P. D.; Kuster W. C.; Fehsenfeld F. C. The observation of a C5 alcohol emission in a North American pine forest. Geophys. Res. Lett. 1993, 20, 1039–1042. [Google Scholar]
- Harley P.; Fridd-Stroud V.; Greenberg J.; Guenther A.; Vasconcellos P. Emission of 2-methyl-3-buten-2-ol by pines: a potentially large natural source of reactive carbon to the atmosphere. J. Geophys. Res. 1998, 103, 25479–25486. [Google Scholar]
- Fu T. M.; Jacob D. J.; Wittrock F.; Burrows J. P.; Vrekoussis M.; Henze D. K. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J. Geophys. Res. 2008, 113, D15303. 10.1029/2007JD009505. [DOI] [Google Scholar]
- Lamanna M. S.; Goldstein A. H. In-situ measurements of C2-C10 VOCs above a Sierra Nevada ponderosa pine plantation. J. Geophys. Res. 1999, 104D1721247–21262. [Google Scholar]
- Bouvier-Brown N. C.; Goldstein A. H.; Worton D. R.; Matross D. M.; Gilman J. B.; Kuster W. C.; Welsh-Bon D.; Warneke C.; de Gouw J. A; Cahill T. M.; Holzinger R. Methyl chavicol: characterization of its biogenic emission rate, abundance, and oxidation products in the atmosphere. Atmos. Chem. Phys. 2009, 9, 2061–2074. [Google Scholar]
- Carrasco N.; Doussin J. F.; O’Connor M.; Wenger J. C.; Picquet-Varrault B.; Durand-Jolibois R.; Carlier P. Simulation chamber studies of the atmospheric oxidation of 2-methyl-3-buten-2-ol: Reaction with hydroxyl radicals and ozone under a variety of conditions. J. Atmos. Chem. 2007, 56, 33–55. [Google Scholar]
- Chan A. W.; Galloway M. M.; Kwan A. J.; Chhabra P. S.; Keutsch F. N.; Wennberg P. O.; Flagan R. C.; Seinfeld J. H. Photooxidation of 2-methyl-3-buten-2-ol (MBO) as a potential source of secondary organic aerosol. Environ. Sci. Technol. 2009, 43, 4647–4652. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Ge M.; Wang W.; Yin S.; Tong S. The uptake of 2-methyl-3-buten-2-ol into aqueous mixed solutions of sulfuric acid and hydrogen peroxide. Phys. Chem. Chem. Phys. 2011, 13, 2069–2075. [DOI] [PubMed] [Google Scholar]
- Jaoui M.; Kleindienst T. E.; Offenberg J. H.; Lewandowski M.; Lonneman W. A. SOA formation from the atmospheric oxidation of 2-methyl-3-buten-2-ol and its implications for PM2.5. Atmos. Chem. Phys. 2012, 12, 2173–2188. [Google Scholar]
- Rudich Y.; Talukdar R. K.; Fox R. W.; Ravishankara A. R. Rate coefficients for the reaction of NO3 with a few olefins and oxygenated olefins. J. Phys. Chem. 1996, 100, 5374–5381. [Google Scholar]
- Seinfeld J. H.; Pandis S. N.. Atmospheric Chemistry and Physics, from Air Pollution to Climate Change, 2nd ed.; John Wiley: New York, 2006; pp 261–265. [Google Scholar]
- Noda J.; Ljungström E. Aerosol formation in connection with NO3 oxidation of unsaturated alcohols. Atmos. Environ. 2002, 36, 521–525. [Google Scholar]
- Fantechi G.; Jensen N. R.; Hjorth J.; Peeters J. Mechanistic studies of the atmospheric oxidation of methyl butenol by OH radicals, ozone, and NO3 radicals. Atmos. Environ. 1998, 32, 3547–3556. [Google Scholar]
- Ferronato C.; Orlando J. J.; Tyndall G. S. Rate and mechanism of the reaction of OH and Cl with 2-methyl-3buten-2-ol. J. Geophys. Res. 1998, 103, 25579–25586. [Google Scholar]
- Liggio J.; Li S. M.; Mclaren R. Reactive uptake of glyoxal by particulate matter. J. Geophys. Res. 2005, 110, D10304. 10.1029/2004JD005113. [DOI] [Google Scholar]
- Volkamer R.; Martini F. S.; Molina L. T.; Salcedo D.; Jimenez J. L.; Molina M. J. A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L19807. 10.1029/2007GL030752. [DOI] [Google Scholar]
- Ervens B.; Turpin B. J.; Weber R. J. Secondary organic aerosol formation in cloud droplets and aqueous particle (aqSOA): a review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102. [Google Scholar]
- Surratt J. D.; Kroll J. H.; Kleindienst T. E.; Edney E. O.; Claeys M.; Sorooshian A.; Ng N. L.; Lewandowski M.; Jaoui M.; Flagan R. C.; Seinfeld J. H. Evidence for organosulfates in secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 517–527. [DOI] [PubMed] [Google Scholar]
- Surratt J. D.; Lewandowski M.; Offenberg J. H.; Jaoui M.; Kleindienst T. E.; Edney E. O.; Seinfeld J. H. Effect of acidity on secondary organic aerosol formation from isoprene. Environ. Sci. Technol. 2007, 41, 5363–5369. [DOI] [PubMed] [Google Scholar]
- Surratt J. D.; Gómez-González Y.; Chan A. W. H.; Vermeylen R.; Shahgholi M.; Kleindienst T. E.; Edney E. O.; Offenberg J. H.; Lewandowski M.; Jaoui M.; Maenhaut W.; Claeys M.; Flagan R. C.; Seinfeld J. H. Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A. 2008, 112, 8345–8378. [DOI] [PubMed] [Google Scholar]
- Surratt J. D.; Chan A. W. H.; Eddingsaas N. C.; Chan M.; Loza C. L.; Kwan A. J.; Hersey S. P.; Flagan R. C.; Wennberg P. O.; Seinfeld J. H. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iinuma Y.; Müller C.; Böge O.; Gnauk T.; Herrmann H. The formation of organic sulfate esters in the limonene ozonolysis secondary organic aerosol (SOA) under acidic conditions. Atmos. Environ. 2007, 41, 5571–5583. [Google Scholar]
- Iinuma Y.; Müller C.; Berndt T.; Böge O.; Claeys M.; Herrmann H. Evidence for the existence of organosulfates from β-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 6678–6683. [DOI] [PubMed] [Google Scholar]
- Iinuma Y.; Böge O.; Kahnt A.; Herrmann H. Laboratory chamber studies on the formation of organosulfates from reactive uptake of monoterpene oxides. Phys. Chem. Chem. Phys. 2009, 36, 7985–7997. [DOI] [PubMed] [Google Scholar]
- Stone E. A.; Yang L.; Yu L. E.; Rupakheti M. Characterization of organosulfates in atmospheric aerosols at four Asian locations. Atmos. Environ. 2012, 47, 323–329. [Google Scholar]
- Froyd K. D.; Murphy S. M.; Murphy D. M.; de Gouw J. A.; Eddingsaas N. C.; Wennberg P. O. Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darer A. I.; Cole-Filipiak N. C.; O’Connor A. E.; Elrod M. J. Formation and stability of atmospherically relevant isoprene-derived organosulfates and organonitrates. Environ. Sci. Technol. 2011, 45, 1895–1902. [DOI] [PubMed] [Google Scholar]
- Hatch L. E.; Creamean J. M.; Ault A. P.; Surratt J. D.; Chan M. N.; Seinfeld J. H.; Edgerton E. S.; Su Y.; Prather K. A. Measurements of isoprene-derived organosulfates in ambient aerosols by Aerosol Time-of-Flight Mass Spectrometry. Part 1: Single particle atmospheric observations in Atlanta. Environ. Sci. Technol. 2011, 45, 5105–5111. [DOI] [PubMed] [Google Scholar]
- Hatch L. E.; Creamean J. M.; Ault A. P.; Surratt J. D.; Chan M. N.; Seinfeld J. H.; Edgerton E. S.; Su Y.; Prather K. A. Measurements of isoprene-derived organosulfates in ambient aerosols by Aerosol Time-of-Flight Mass Spectrometry. Part 2: Temporal variability and formation mechanisms. Environ. Sci. Technol. 2011, 45, 8648–8655. [DOI] [PubMed] [Google Scholar]
- Jang M.; Czoschke N. M.; Lee S.; Kamens R. M. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 2002, 298, 814–817. [DOI] [PubMed] [Google Scholar]
- Offenberg J. H.; Lewandowski M.; Edney E. O.; Kleindienst T. E.; Jaoui M. Influence of aerosol acidity on the formation of secondary organic aerosol from biogenic precursor hydrocarbons. Environ. Sci. Technol. 2009, 43, 7742–7747. [DOI] [PubMed] [Google Scholar]
- Lin Y. −H.; Zhang Z.; Docherty K. S.; Zhang H.; Budisulistiorini S. H.; Rubitschun C. L.; Shaw S. L.; Knipping E. M.; Edgerton E. S.; Kleindienst T. E.; Gold A.; Surratt J. D. Isoprene eposydiols as precursors to secondary organic aerosol formation: Acid-catalyzed reactive uptake studies with authentic compounds. Environ. Sci. Technol. 2012, 46, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Jang M.; Kamens R. M. SOA formation from the photooxidation of α-pinene in the presence of freshly emitted diesel soot exhaust. Atmos. Environ. 2004, 38, 2597–2605. [Google Scholar]
- Leungsakul S.; Jeffries H. E.; Kamens R. M. A kinetic mechanism for predicting secondary aerosol formation from the reactions of d-limonene in the presence of oxides of nitrogen and natural sunlight. Atmos. Environ. 2005, 39, 7063–7082. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Surratt J. D.; Lin Y.-H.; Bapat J.; Kamens R. M. Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions. Atmos. Chem. Phys. 2011, 11, 6411–6424. [Google Scholar]
- Zhang H.; Lin Y.-H.; Zhang Z.; Zhang X.; Shaw S. L.; Knipping E. M.; Weber R. J.; Gold A.; Kamens R. M.; Surratt J. D. Secondary organic aerosol formation from methacrolein photooxidation: Roles of NOx level, relative humidity, and aerosol acidity. Environ. Chem. 2012, 9, 247–262. [Google Scholar]
- Goldstein A. H.; Hultman N. E.; Fracheboud J. M.; Bauer M. R.; Panek J. A.; Xu M.; Qi Y.; Guenther A. B.; Baugh W. Effects of climate variability on the carbon dioxide, water, and sensible heat fluxes above a ponderosa pine plantation in the Sierra Nevada (CA). Agric. Forest Meteorol. 2000, 101, 113–129. [Google Scholar]
- Worton D. R.; Goldstein A. H.; Farmer D. K.; Docherty K. S.; Jimenez J. L.; Gilman J. B.; Kuster W. C.; de Gouw J.; Williams B. J.; Kreisberg N. M.; Hering S. V.; Bench G.; McKay M.; Kristensen K.; Glasius M.; Surratt J. D.; Seinfeld J. H. Origins and composition of fine atmospheric carbonaceous aerosol in the Sierra Nevada Mountains, California. Atmos. Chem. Phys. 2011, 11, 10219–10241. [Google Scholar]
- Schade G. W.; Goldstein A. H.; Gray D. W.; Lerdau M. T. Canopy and leaf level 2-methyl-3-butene-2-ol fluxes from a ponderosa pine plantation. Atmos. Environ. 2000, 34, 3535–3544. [Google Scholar]
- Schade G. W.; Goldstein A. H. Fluxes of oxygenated volatile organic compounds from a ponderosa pine plantation. J. Geophys. Res. 2001, 106D33111–3123. [Google Scholar]
- DeCarlo P. F.; Kimmel J. R.; Trimborn A.; Northway M. J.; Jayne J. T.; Aiken A. C.; Gonin M.; Fuhrer T.; Docherty K.; Worsnop D. R.; Jimenez J. L. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2011, 78, 8281–8289. [DOI] [PubMed] [Google Scholar]
- Farmer D. K.; Kimmel J. R.; Phillips G.; Docherty K. S.; Worsnop D. R.; Sueper D.; Nemitz E.; Jimenez J. L. Eddy covariance maeasurements with high-resolution time-of-flight aerosol mass spectrometry: a new approach to chemically resolved aerosol fluxes. Atmos. Meas. Tech. 2011, 4, 1275–1289. [Google Scholar]
- Gilman J. B.; Burkhart J. F.; Lerner B. M.; Williams E. J.; Kuster W. C.; Goldan P. D.; Murphy P. C.; Warneke C.; Fowler C.; Montzka S. A.; Miller B. R.; Miller L.; Oltmans S. J.; Ryerson T. B.; Cooper O. R.; Stohl A.; de Gouw J. A. Ozone variability and halogen oxidation within the Arctic and sub-Arctic springtime boundary layer. Atmos. Chem. Phys. 2010, 10, 10223–10236. [Google Scholar]
- Park C.; Schade G. W.; Boedeker I. Flux measurements of volatile organic compounds by the relaxed eddy accumulation method combined with a GC-FID system in urban Houston, Texas. Atmos. Environ. 2010, 44, 2605–2614. [Google Scholar]
- Park C.; Schade G. W.; Boedeker I. Characteristics of the flux of isoprene and its oxidation products in an urban area. J. Geophys. Res. 2011, 116, D21303. 10.1029/2011JD015856. [DOI] [Google Scholar]
- Kim S.; Karl T.; Guenther A.; Tyndall G.; Orlando J.; Harley P.; Rasmussen R.; Apel E. Emission and ambient distributions of Biogenic Volatile Organic Compounds (BVOC) in a ponderosa pine ecosystem: interpretation of PTR-MS mass spectra. Atmos. Chem. Phys. 2010, 10, 1759–1771. [Google Scholar]
- Levin E. J. T.; Prenni A. J.; Petters M. D.; Kreidenweis S. M.; Sullivan R. C.; Atwood S. A.; Ortega J.; DeMott P. J.; Smith J. N. An annual cycle of size-resolved aerosol hygroscopicity at a forested site in Colorado. J. Geophys. Res. 2012, 117, D06201. 10.1029/2011JD016854. [DOI] [Google Scholar]
- Jordan A.; Haidacher S.; Hanel G.; Hartungen E.; Mark L.; Seehauser H.; Schottkowsky R.; Sulzer P.; Mark T. D. A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS). Int. J. Mass Spectrom. 2009, 286, 122–128 10.1016/j.ijms.2009.07.005. [DOI] [Google Scholar]
- Graus M.; Müller M.; Hansel A. High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time. J. Am. Soc. Mass. Spectrom. 2010, 21, 1037–1044 10.1016/j.jasms.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Müller M.; Graus M.; Ruuskanen T. M.; Schnitzhofer R.; Bamberger I.; Kaser L.; Titzmann T.; Hortnagl L.; Wohlfahrt G.; Karl T.; Hansel A. First eddy covariance flux measurements by PTR-TOF. Atmos. Meas. Tech. 2010, 3, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellin L.; Biasioli F.; Granitto P. M.; Schuhfried E.; Soukoulis C.; Costa F.; Märk T. D.; Gasperi F. On data analysis in PTR-TOF-MS: From raw spectra to data mining. Sensor. Actuat. B-Chem. 2011, 155, 183–190. [Google Scholar]
- Paulot F.; Crounse J. D.; Kjaergaard H. G.; Kürten A.; Clair J. M. S.; Seinfeld J. H.; Wennberg P. O. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science 2009, 325, 730–733. [DOI] [PubMed] [Google Scholar]
- Eddingsaas N. C.; VanderVelde D. G.; Wennberg P. O. Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols. J. Phys. Chem. A 2010, 114, 8106–8113. [DOI] [PubMed] [Google Scholar]
- Minerath E. C.; Casale M. T.; Elrod M. J. Kinetics feasibility study of alcohol sulfate esterification reactions in tropospheric aerosols. Environ. Sci. Technol. 2008, 42, 4410–4415. [DOI] [PubMed] [Google Scholar]
- Berndt T.; Böge O. Catalyst-free gas-phase epoxidation of alkenes. Chem. Lett. 2005, 34, 584–585. [Google Scholar]
- Berndt T.; Bräsel S. Epoxidation of a series of C2-C6 olefins in the gas phase. Chem. Eng. Technol. 2009, 32, 1189–1194. [Google Scholar]
- Atkinson R. Gas-phase tropospheric chemistry of organic compounds: a review. Atmos. Environ. 1990, 24A, 1–41. [Google Scholar]
- Steiner A. L.; Tonse S.; Cohen R. C.; Goldstein A. H.; Harley R. A. Biogenic 2-methyl-3-buten-2-ol increases regional ozone and HOx sources. Geophys. Res. Lett. 2007, 34, L15806. 10.1029/2007GL030802. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.