

Abstract
MicroRNAs (miRNAs), the newly discovered regulators of gene expression, act by promoting degradation of mRNA and/or by inhibiting protein expression. Dysregulation of miRNA expression has been noted in an expanding number of diseases; and in some instances, manipulating miRNA expression holds promise as a new form of therapy. Herpetic stromal keratitis (HSK) is an important vision-impairing lesion and currently any role that miRNA dysregulation plays during its pathogenesis is only just beginning to be investigated. In this review, we discuss the likely participation of specific miRNAs during HSK and discuss the prospect of modulating their expression as a means of therapy.
Introduction
The discovery of a novel mechanism of gene regulation by microRNAs (miRNAs) in nematodes by Ambros and colleagues1 stimulated an intense search for similar regulatory mechanisms in mammalian cells. The search has yielded abundantly with at least 800 different miRNA species identified, with many of them dysregulated in metabolic pathways that result in neoplasia, autoimmunity, and an expanding number of other human diseases. In several instances, either restoring miRNA levels or blocking their activity represents a valuable means of therapy. Currently, our understanding of miRNA involvement during the pathogenesis of stromal keratitis caused by herpes simplex virus (HSV) is very much in its infancy. This topic is discussed along with a speculative assessment of the prospects of manipulating selected species of miRNAs as a therapeutic maneuver that could reach the clinic.
What Are miRNAs?
miRNAs are genome-encoded, small, single-stranded RNAs that posttranscriptionally downregulate gene expression either by degradation or translation repression of the target mRNA. All canonical miRNA biogenesis processes begin in the nucleus where stem loops in the pri-miRNA are recognized by the enzyme Drosha and its cofactor DGCR8, which cause nuclear cleavage to result in 70 nt precursors called pre-miRNA (Fig. 1). The pre-miRNA is exported to the cytoplasm with the help of exportin 5 and further processed with the help of another enzyme, Dicer. The Dicer also helps in the loading of the miRNA single strand into the miRNA-induced silencing complex (miRISC). The miRNA guides the miRISC to the target mRNA and the degree of complementarity between the miRNA and the target mRNA determines the fate of the mRNA. If the two have high complementary sequences, the target mRNA is degraded by miRISC consisting of Argonaut proteins. If there is partial complimentary sequences between the miRNA and target mRNA, this would result in translational repression by several miRISC complexes bound to different sites on the target mRNA. Accordingly, miRNAs usually act to fine-tune gene expression rather than to act as on/off switches. It is important to note that not all cellular miRNAs use the canonical pathways. There are few other mechanisms used by cells to generate functional miRNAs which have been reviewed in detail by others2 and these are not discussed herein.
Figure 1. .
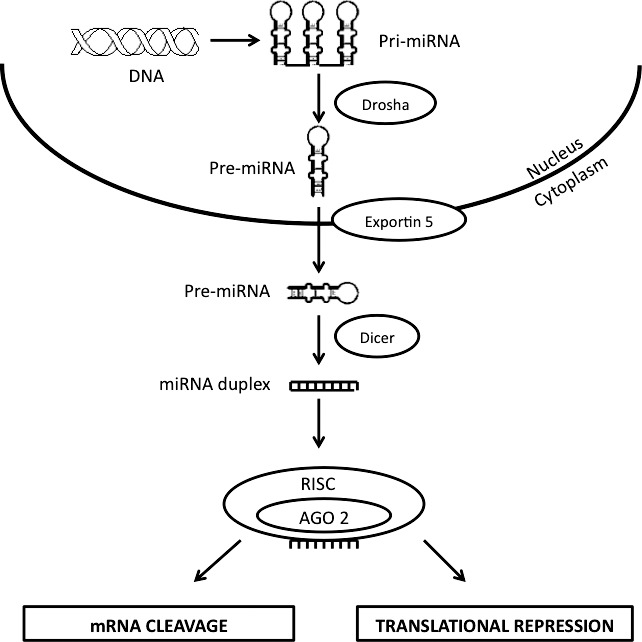
miRNA biogenesis. The generation of miRNAs involves nuclear and cytoplasmic processing steps. In the nucleus, pri-miRNAs, transcribed by RNA polymerase from DNA, are processed by Drosha into pre-miRNAs that are then transported to cytoplasm via exportin 5. Once in the cytoplasm, these pre-miRNAs are recognized and cleaved by Dicer into 20 to 22 bp duplexes that enter RISC. In RISC, the passenger strand is cleaved while the guide strand directs RISC machinery to target mRNAs resulting in mRNA degradation or inhibition of protein translation.
Most miRNA genes are expressed under control of their own promoters and regulatory sequences and act alone in gene regulation. However, others are arranged in clusters and are expressed and function together (e.g., the miR-19-92 cluster). The impact of miRNA expression during human and animal diseases has become known from a variety of experimental approaches. These have included measuring the consequences of gene knockout or overexpression, as can be done in experimental rodent studies. Alternatively, miRNA expression can be reduced in vivo by antagomir nanoparticle approaches, or be enhanced either by administrating miRNA in nanoparticles, or by causing their biosynthesis using vectors encoding miRNAs.
Herpetic Stromal Keratitis
Infection with HSV generally leads to lesions on surface structures that include the cornea of the eye. The infection can be acute, involving largely the epithelium, and resolve quite quickly, especially if treated with antivirals. Some cases, however, result in chronic lesions that involve the corneal stroma and these impair vision and can even result in blindness. HSK lesions in humans are commonly the consequence of one or more episodes of HSV reactivation from latency.3 The tissue damage that occurs in HSK is thought to result in large part from the host inflammatory reaction and residual scarring set off by the infection. The strongest case that tissue damage in HSK represents an immunopathological response to infection comes from animal studies, particularly the mouse.4 Such studies have identified several essential steps in lesion pathogenesis, some or all of which could be subject to regulation by yet to be identified miRNA species. Several miRNAs are expressed by healthy mouse corneas under physiological conditions.5 Among these, miR-132, miR-182, and miR-1 are expressed at low levels; miR-146b shows intermediate expression; while miR-21, miR-23b, and miR-24 are expressed at high levels in corneas. However, their contribution in modulating critical events in corneal immunopathology during HSK largely remains unknown. The details of HSK pathogenesis have received some recent reviews6,7 and herein we mention only a few essential points. Firstly, the inciting HSV infection in humans usually derives from a breakdown of latent infection in the trigeminal ganglia, which is caused by exposure to a variety of stressful stimuli. Accordingly, understanding how to sustain latency and prevent reactivation is an important issue, and miRNAs could influence these events.
Early steps following HSV infection largely represent the response of innate defenses and these may be sufficient to control the virus before significant inflammatory lesions occur. The overt HSK lesions appear to be inflammatory reactions orchestrated mainly by T cells, particularly CD4 T cells of the Th-1 subset.8 However, the principal tissue damage and resultant corneal scarring is mainly attributed to inflammatory products derived from nonlymphoid inflammatory cells such as neutrophils and macrophages.9,10 Finally, the onset and extent of HSK lesions is influenced by the neovascularization that occurs in the normally transparent avascular cornea.11 Understanding how to limit and resolve corneal neovascularization (CV) is an important therapeutic objective. As we discuss, the CV process is subject to control by one or more species of miRNAs, with a recent publication implicating the relevance of miR-132.12
miRNAs and Latency
The hallmark of all herpes virus infections is their ability to persist indefinitely in the body in a state called latency. In the case of HSV, latency is maintained in neuronal tissue, which with ocular infection means the trigeminal ganglion. During latency, the virus produces no proteins, so the agent may be invisible to the adaptive immune system. Unfortunately, latency terminates in occasional neurons, which is probably a continual process.13 Productive replication occurs in such neurons and the virus disseminates to peripheral sites where it can cause recurrent lesions. It is conceivable that the regulatory effects of miRNAs could influence the initiation, maintenance, and breakdown of latency. All of these points here have yet to be firmly established.
With regard to the establishment of latency, HSV itself encodes a number of miRNAs and these could influence the extent to which either latent or productive infection of neurons occurs.14 For example, the transcript expressed uniquely during latency, LAT, serves as a primary miRNA precursor that encodes four distinct miRNAs in HSV-infected cells. One of these miRNAs, mir-H2-3p, can influence the expression of the viral immediate early gene ICP0.14 Another miRNA, miR-H6, which lies upstream of the LAT promoter, modulates the expression of ICP4.14 These observations came from in vitro transfection assays; but if—as is likely—the same effect occurs in vivo, the miRNA regulation could be highly relevant since both ICP4 and ICP0 proteins are involved in the initial phases of productive replication following primary and recurrent infections.15,16 Conceivably, virus encoded miRNAs could influence the amount of ICP4 and ICP0 expression in a latently infected neuron and this might determine if the productive cycle is initiated. However, viral-encoded miRNA regulation of HSV latency in vivo remains an unresolved and debatable issue17 that merits more investigation.
Perhaps more amenable to therapeutic manipulation to influence latency will be to identify any host-encoded miRNAs that are responsible for maintaining latency and that may lose this function in response to circumstances that cause reactivation. Such circumstances include changes in hormone levels caused by stress or other events, changes in immune status, and exposure to UV irradiation being among the most investigated. The herpetic recurrences that follow stressful experiences could be mediated by changes in glucocorticoid levels, and many studies have documented that glucocorticoids can influence miRNA expression.18 For example, glucocorticoids can cause upregulation of the miRNA clusters miR-15/16 or miR-233,19 but as yet, none have been related to latency. It would be of interest to determine if any changes of these miRNA clusters precede the onset of viral reactivation. Additionally, glucocorticoids may also influence gene expression changes by histone modifications, an event also implicated in the maintenance of latency.20 The glucocorticoid binding to cytoplasmic glucocorticoid receptors triggers events that lead to induction of some anti-inflammatory genes. These effects occur in part via recruitment of histone modifying transcriptional coactivators and other cofactors to glucocorticoid responsive elements in the responsive genes.21 Moreover, glucocorticoid signaling can recruit histone deacetylases to the NFkB complex and inhibit inflammatory gene expression.22 These epigenetic events influenced by glucocorticoids could be caused by miRNA expression changes. For example, miR-449 and miR-1 are involved in regulating the activity of histone acetylation.23,24 It remains to be seen whether glucocorticoids may promote virus reactivation via changing the levels of histone modification associated miRNAs (Fig. 2).
Figure 2. .
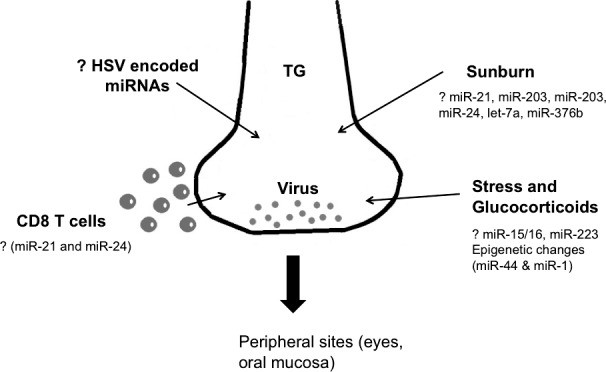
Speculative role of miRNAs in HSV-1 reactivation from latency. After primary infection, HSV-1 becomes latent in trigeminal ganglia. Certain conditions such as sunburn, stress, impaired T cell responses, and immunosuppression, can reactivate the virus to come to the peripheral sites (eyes, oral mucosa) to induce lesions. miRNAs may regulate all of these above-mentioned stimuli. Sunburn can trigger HSV reactivation and it involves changes in expression levels of miRNAs (miR-21, miR-203). Likewise, stress induced glucocorticoids induce changes in miRNAs (miR-15/16 and miR-223) and also lead to epigenetic modifications, a process which involves miRNA change (miR-44 and miR-1) and is implicated in maintenance of latency. CD8 T cells patrol trigeminal ganglia and inhibit HSV reactivation, but functionality of CD8 T cells can also be modulated by miRNA activity (miR-21 and miR-24). The role of HSV encoded miRNAs in HSV reactivation remains unknown so far.
Another event that often causes the onset of recurrent herpes lesions is exposure to excessive sunlight, such as sunburn. Exposure to UV light can also trigger reactivation of HSV in a keratitis model in mice.25 How UV irradiation or sunburn induces herpes virus reactivation remains unclear, but recent reports indicate a potential role for miRNAs in this process. For instance, the knockdown of Ago2 and Dicer, essential components of the miRNA processing pathway, reduces survival of HELA cells exposed to UV radiation.26 This could mean that miRNAs play a critical role in UV-induced cell cycle events and apoptosis. Perhaps of more relevance, dysregulation in miR-21, miR-203, miR-205, miR-24, let-7a, and miR-376b is observed in murine epidermal tissue and human keratinocytes upon UV exposure.27 At least two of these, miR-21 and miR-24,28,29 can influence CD8 T cell functionality which, as mentioned subsequently, may participate in the maintenance of latency30,31 (Fig. 2).
Another well-studied situation that may influence the stability and breakdown of latency is immune status. For example, immunosuppressed patients are more likely to suffer herpes recrudescence, with severe lesions occurring in those with AIDS.32 As discussed subsequently, miRNAs are known to influence the functional capacity of both innate and adaptive immune components. The role of innate immune events in the control of latency has received minimal attention, but there is strong evidence that the functional competence of viral specific CD8 T cell responses in the trigeminal ganglion influences the stability of latency.30,31 Currently, we do not know if miRNA dysregulation impacts on the immune control of latency, but this topic is under active investigation by many groups that include our own.
miRNAs and Angiogenesis
The cornea's function depends on its transparency since it must minimally interfere with the passage of light to the retina. This may be one reason the normal cornea lacks a blood vasculature since vessels diffract light. However, a significant consequence of HSV ocular infection is that pathological angiogenesis occurs, a process driven and facilitated by many factors.6 A major angiogenic factor involved in CV is VEGF; and counteracting VEGF and events set off when it binds to its specific receptors represents a valuable means of therapy.33 Pathological angiogenesis is also a major problem in neoplastic diseases and several species of miRNAs show changes that may account for the neovascularization. The miRNA that showed the largest change of expression during angiogenesis was miR-132.34 We have also shown that miR-132 is also upregulated in animals with CV following HSV infection.12
In the HSV system, the upregulation of miR-132 may be in part the consequence of stimulation by an as-yet-unidentified mechanism by IL-17, a cytokine produced early in HSK pathogenesis by gamma delta cells of the innate immune system.35 The upregulated miR-132 appears to act to augment VEGF signaling via an inhibitory effect on a negative regulator of VEGF function, Ras-GAP.34 Of interest, inhibiting the expression of miR-132 using antagomirs in the form of nanoparticles was an effective means of diminishing CV, as well as reducing the severity of HSK (Fig. 3; Table). Additionally, miR-132 blockade results in potent reduction in pathological angiogenesis in retinal models (Westenskow PD, et al. IOVS 2012;53:ARVO E-Abstract 4120). Of note, this modality seems to work better than VEGF trap approaches and resulted in fewer off-target effects and lower compensatory increases in angiogenic molecules, which is often the case with VEGF inhibition approaches.36 It is also to be noted that miR-132 function depends on context. Thus, miR-132 promotes IFN production in Kaposi sarcoma herpes virus–infected lymphatic endothelial cells37 and regulates dendritic maturation in newborn neurons.38 However, it exerts an anti-inflammatory effect at the neural immune interface.39 Although we realize that miR-132 has an angiogenic function during HSK, it is also possible that it interacts with other miRNAs in its modulation of tissue damage.
Figure 3. .
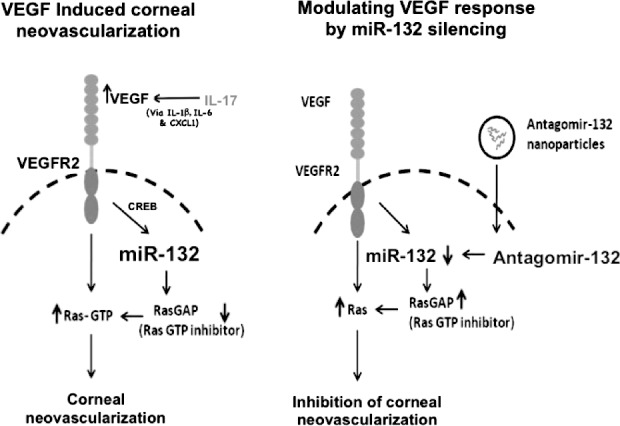
Illustration of antagomir-132–mediated inhibition of CV. Left panel describes outcome (CV) in HSV-infected untreated mice: HSV infection leads to upregulation of IL-17 in corneas. IL-17 (along with IL-6 and virus infected epithelial cells) increases VEGF (probably via increasing IL-1b, IL-6, and Cxcl1) levels in the eyes. VEGF thus acting through VEGFR2 receptors on the blood vessel endothelial cells upregulates miR-132 expression via CREB transcription factor. MiR-132 removes RasGAP (intrinsic inhibitor of Ras) leading to activation of Ras and CV. Right panel describes modulation of CV by miR-132 silencing: Administration of antagomir-132 nanoparticles leads to deposition of antagomir-132 cargo in blood vessel endothelial cells resulting in silencing of miR-132. This leads to higher levels of RasGAP, which thereby inhibits angiogenic Ras activity resulting in inhibition of CV. Reprinted with permission from Mulik S, Xu J, Reddy PB, et al. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181:525–534. Copyright 2012 The American Society for Investigative Pathology.
Table. .
Speculation of the Role of miRNAs, Phase They Should Be Administered, Their Blockade/Overexpression And Reduction in HSK Lesion Severity (Mild +, Moderate ++, Severe +++)
|
Steps |
miRNAs |
Action |
Preclinical (0–6 d p.i.)/Clinical Phase (7–12 d p.i.) Administration |
Reduction in HSK Lesions/CV (overexpression or blockade) |
| Corneal neovascularization | ||||
| VEGF signaling | miR-132 | Promotes VEGF signaling | Clinical phase | Blockade (++) |
| MMP-9 | miR-885-5p, miR-491-5p, miR-212 | Negatively regulates MMP-9 activity | Preclinical phase | Overexpression (+) |
| Inflammation | ||||
| IL-17–induced inflammation | miR-23b | Negatively regulates NFkB and IL-17 activity | Preclinical phase | Overexpression (++) |
| T cell–mediated damage | miR-155 | Generation of Th1 and Th17 cells | Clinical phase | Blockade (++) |
| miR-29 | IFN-γ production by Th1 cells | Preclinical phase | Blockade (+) | |
| miR-326 and miR-309 | Generation of Th17 cells | Preclinical phase | Blockade (+) | |
| miR-17 and miR-20a | Inhibits T cell activation | Preclinical phase | Blockade (+) | |
| miR-182 | T cell proliferation | Preclinical phase | Blockade (+) | |
| Resolution | ||||
| T cell responses | miR-146a | Resolution of T cell–mediated damage | Clinical phase | Overexpression (+++) |
| Necessary for Tregs to control Th1 responses | ||||
| Regulates NFkB and proinflammatory cytokines | ||||
| IL-10 production | miR-21 and miR-208a | Induces IL-10 production | Clinical phase | Overexpression (++) |
| Neutrophil recruitment | miR-146b | Lowers IL-8 and RANTES | Clinical phase | Overexpression (+) |
| Lipid mediator switch | miR-292-2 | Lowers leukotrienes and turns on SPM | Clinical phase | Overexpression (++) |
| Tregs | miR-10a | Stabilizes tregs | Clinical phase | Overexpression (+) |
In addition to VEGF, matrix metalloproteinase-9 (MMP-9) and other metalloproteinases play pivotal roles in promoting pathological blood vessel development during HSK.40 Recent reports indicate that the regulation of MMP-9 expression involves modulating effect by miRNAs. Accordingly, miR-885-5p, miR-491-5p, miR-212, and miR-132 negatively regulate the levels of MMP-9 expression in mammary glands and in cancer models41,42 (Table). How miR-132 promotes angiogenesis during HSK simultaneously by degrading angiogenic MMP-9 needs further investigation.12,43 However, miR-132 blockade–induced reduction in CV and expression of miR-132 in blood vessel endothelial cells while MMP-9 in neutrophils make MMP-9 an unlikely target of miR-132 during HSK.
Almost certainly there are additional miRNAs that influence the many stages of CV. For example, it is known that the activity of VEGF in the cornea is limited by being bound to a soluble form of one of its receptors.44 HSV infection disrupts the balance between VEGF and its soluble receptor,43 a process that likely involves expression changes in miRNAs. Additional events that could be regulated by miRNAs include disruption of blood vessel endothelial junctions, pericyte detachment, invasion of endothelial cells toward the center of the cornea, as well as their proliferative effects. It is also likely that miRNAs fine-tune the activity of apoptotic/antiapoptotic molecules in pathological blood vessel endothelial cells. Accordingly, the provision of appropriate miRNAs may induce apoptosis of endothelial cells and this may result in regression of established blood vasculature during HSK. Furthermore, pathological blood vessels—which occur in corneas during HSK—are leaky, and whether miRNAs influence leakiness of such vessels by modulating the activity of Src kinases or by regulating the functionality of proteins involved in maintenance of endothelial junction integrity requires exploration.
miRNAs and Inflammation
Several types of innate immune sensors are triggered when HSV infects the cornea and many molecules with diverse function are rapidly produced. These include proinflammatory cytokines, chemokines, interferons and angiogenic factors such as VEGF. Among the best-studied innate sensors are the toll-like receptors with TLR2, TLR3, and TLR9: all involved as HSV sensors.45,46 Additional intracellular sensors may also recognize HSV and these include the recently described sensor STING that recognizes HSV-induced membrane fusion and inflammasomes that lead to production of the proinflammatory cytokine IL-1b.47–49 The innate recognition phase sets the scene for both infection control, mediated by interferons and innate effectors such as granulocytes, macrophages, and NK cells, as well as establishing conditions that result in tissue damage orchestrated by adaptive components of immunity. Most persons with HSV keratitis resolve infection with little or no stromal disease. Those less fortunate develop HSK with this outcome most likely to occur after one or more recurrences from latency. Why individuals differ in their response pattern to HSV infection remains poorly understood, but differential regulation of innate and adaptive immunity by one or more species of miRNA is a likely possibility that merits investigation.
With regard to the extent of innate immune sensing, evidence accumulates showing that TLR function is subject to control by numerous miRNA species. As lucidly reviewed by O'Neil and colleagues,50 miRNAs may regulate the strength, location, and timing of TLR responses and also appear to control the switch from an early innate immune-induced–proinflammatory response to conditions that represent the resolution phase of the inflammatory process. In fact, at least 10 miRNAs are upregulated when one or more TLRs engage their ligands and others are downregulated. Apparently, the TLRs themselves are not targeted by miRNAs and regulation mainly affects the activity of signaling molecules.50 In this context, TLR signal transducers such as MyD88, IRAK, TAB, TRAF6, and IKK are all targets of one or more species of miRNAs. In addition, transcriptional factors induced by TLR ligation are subject to miRNA regulation and some miRNAs also degrade the products of TLR signaling.50 One such miRNA is let-7, which binds to the mRNA of IL-6 and decreases expression of this cytokine, which is a major participant in HSK pathogenesis and also a stimulant of the angiogenic factor VEGF.51 Additionally, other cytokines such as TNF-α, IL-10, IL-12, and IFN-γ are directly repressed by miRNA action.50 However, what role if any these aforementioned miRNAs play during the innate phase of HSK pathogenesis remains to be seen.
Additional components of innate responses to HSV include granulocytes, macrophages, and NK cells. The function of all of these cell types is influenced by miRNA expression.52–54 For example, miR-223 regulates the proliferation and activation status of neutrophils.55 Furthermore, it's also conceivable that miRNAs are involved in regulating tissue-damaging molecules such as the production of reactive oxygen and nitrogen radicals by neutrophils and the generation of neutrophil extracellular traps. Macrophage function during infection is influenced my several miRNAs that include miR-155, miR-146, miR-132, miR-147, miR-9, let-7e, miR-27b, and miR-125b.56,57 Similarly, NK cell killer function, such as granzyme activity, is influenced by miR-155 and miR-27a*.58,59
Although it is still in its early days, it has become evident that miRNAs derived from innate immune cells—and perhaps exported from such cells in the form of exosomes60—are important regulators of inflammation, and the miRNAs also influence the balance of adaptive immune responses that determine whether immunity or tissue damage ensues. Indeed, the enforced expression of TLR-induced miRNAs might well turn out to be useful negative regulators of the inflammatory process, as well as tools to shape the desired pattern of adaptive immunity to one that has minimal tissue damage.
As mentioned in a previous section, the tissue-damaging events in HSK are orchestrated predominantly by effector CD4 T cells, with the actual damage mainly mediated by neutrophils and macrophages.9,61,62 Multiple miRNAs are known to influence the induction and effector function of T cells, the recruitment and activation of inflammatory cells, as well as the participation of additional cell types such as regulatory T cells involved in lesion resolution. The topic is complex and fast moving. In consequence, we mention only what may be the most pertinent miRNA expression events that impact on the pathogenesis of HSK and that represent promising candidates for therapeutic manipulation to change the outcome of disease.
The miRNA miR-23b, generally implicated in liver regeneration,63,64 is a likely candidate since it also acts as a critical regulator in several autoimmune syndromes in humans and mice whose pathogenesis is similar to HSK.65 Accordingly, miR-23b normally plays an anti-inflammatory role by inhibiting the transcription factor NFkB that in turn regulates the production of numerous proteins involved in tissue damage, as well as the recruitment of proinflammatory cells. The expression of miR-23b is downregulated by a still poorly understood mechanism when the proinflammatory cytokine IL-17 binds to its receptor. As a consequence, NFkB becomes hyperactivated and inflammation progresses. Although HSK is mainly orchestrated by inflammatory Th1 cells, the cytokine IL-17A produced by innate cells does play a prominent role during the onset of HSK and may also participate in neovascularization.35,66 Thus, it is conceivable that miR-23b dysregulation occurs during HSK and this may influence the severity of lesions that ensue. This aspect merits investigation, as does the prospect that administration of additional miR-23b may represent a useful adjunct to therapy (Table).
miR-155 is another miRNA frequently implicated in inflammatory lesions and serves to positively regulate inflammation.67–69 It is also required for normal immune function,70,71 as well as for generation of pathogenic T cells,72 and acts as an oncomir in various cancers.73,74 Animals deficient in miR-155 because of gene knockout (KO) are resistant to the induction of some autoimmune diseases.67,72 We have also observed that miR-155 KO animals develop significantly reduced HSK lesions, although they become highly susceptible to virus-induced encephalitis (S. Mulik, S. Bhela, and B.T. Rouse, unpublished observations, [2012]). The miR-155 may influence tissue damage in several ways that include regulating the production of critical chemokines such as those for neutrophils, which play a prominent role in HSK pathogenesis.68 Our preliminary results in the HSK system indicate that chemokines such as the neutrophil attracting CXCL1, are reduced in miR-155–deficient animals. In addition, as is shown in some autoimmune models, miR-155 deficiency may also result in the increased production of the critical anti-inflammatory mediator IL-10, as well as reduced generation of Th1 and Th17 effectors that orchestrate HSK lesions.72 In some inflammatory and autoimmune settings, loss of miR-155 also results in profound reduction in the inflammatory milieu as well as tissue damage.69 Since counteracting T cell function and inflammation would be useful to limit tissue damage, administering antagomirs to diminish miR-155 expression may be a valuable approach to achieve this objective during HSK, as we are currently evaluating (Table).
There are other miRNAs that we speculate may significantly influence the pathogenesis of HSK. In this regard, miR-29 was recently shown to target Tbet and Eomes transcription factors, both of which promote IFN-γ production by Th1 cells,75 central orchestrators of HSK pathogenesis. Similarly, miR-326 and miR-301a are involved in the generation of Th-17 cells, a subset recently shown to influence the later stages of HSK.35 Inhibition of miR-326/301a resulted in attenuated EAE lesions in mice via effects on Th-17 cells.76,77 miRNAs are also known to be involved in sequential steps of T cell proliferation. For example, miR-142-3p/5p, miR-17, and miR-20a inhibit T cell activation.78,79 On the other hand, IL-2 induced miR-182 degrades Foxo 1 and promotes clonal expansion of activated helper T lymphocytes.80
It is now realized that fully differentiated T cells, once considered a stable population, may take on other activities depending on environmental cues. For example, Tregs can lose their regulatory function and adopt effector phenotypes.81 It appears that changes in miRNA expression may be involved in this cellular functional plasticity.82 For example, in response to retinoic acid and TGFb, Tregs increase miR-10a expression that limits their conversion into follicular helper T cells and Th-17 cell subsets. Whether and how miRNAs influence various stages of T cell activation or T cell flexibility during HSK is largely unknown (Table).
miRNAs in Anti-Inflammation and Resolution
The aim of all research on inflammatory diseases is to discover optimal ways to resolve lesions and understand how they function at a mechanistic level. Under natural circumstances, inflammatory responses are usually resolved either because the inciting stimuli are removed and/or because the balance of host events changes. Such changes could be influenced, at least in part, by dysregulation of one or more species of miRNAs. Several host events can push inflammation toward resolution. These include the expansion of regulatory T cells, the increased activity of several cytokines particularly IL-10 and TGFb, the production of one or more species of host galectins, as well as multiple specialized pro-resolving mediators derived from polyunsaturated fatty acids. How miRNA changes relate to the increased activity of these anti-inflammatory and inflammation resolving events is beginning to be unraveled, at least for some of them. Two such candidates are miR-146a and miR-21, which play prominent roles in protecting tissues from inflammatory damage.57
miR-146a exerts this regulatory effect by policing NF-κB dysregulation. In the context of innate immunity, miR-146a inhibits TLR signaling and NF-κB activation and dampens the production of proinflammatory mediators such as IL-6 and TNF-α.83 A diminished miR-146a response also results in malfunction of the adaptive immune responses. Of note, regulatory T cells express high levels of miR-146a and Treg-specific deletion of miR-146a expression leads to severe autoimmune disease due to uncontrolled Th-1 responses.84 This further corroborates a regulatory role of miR-146a in restraining inflammation and maintaining peripheral tolerance. In addition, miR-146a was implicated in resolving both CD4 and CD8 T cell–mediated immunopathological responses.85 In mice, TCR stimulation–induced expression of miR-146a and T cell–specific miR-146a deficiency resulted in hyperactive acute antigenic responses, chronic inflammatory autoimmune responses as well as T cell–mediated autoimmune disease.85 It will be of interest to see what effects miR-146a overexpression will have on the balance between protective Tregs and eye damaging Th-1 cells during HSK. We speculate that the potent regulatory environment induced by miR-146a may hamper lesion development (Table). Moreover, dampening of proinflammatory milieu by administering additional miR-146a could represent a useful form of therapy.
Another miRNA likely involved in resolution is miR-21. This miRNA halts inflammation in various models partly via its ability to induce IL-10,86,87 a molecule previously shown to suppress HSK lesions.88 However, miR-21 may enhance vascular inflammation in atherosclerosis.89 miR-21 may also shift the Th1/Th2 balance toward the Th2 type90 with miR-21–deficient mice demonstrating enhanced Th1 associated responses.91 Conceivably, overexpression of miR-21 could help to modulate Th1–mediated tissue damage during HSK, an issue that merits further investigation (Table).
An additional approach to modulate the severity of inflammatory diseases is to expand or activate regulatory T cells whose activity is also influenced by expression levels of several miRNAs.92,93 These include miR-10a, miR-146a and miR-155,84,94–96 but their contribution in modulating ocular damage is yet to be analyzed. Conceivably, the above-mentioned miRNAs will help shift the balance toward resolution events in tissue-damaging settings.
In the resolving phase of inflammation, changes in expression of several mediators occur. One prominent change is in the lipid mediators derived from polyunsaturated fatty acids. Thus, whereas during the proinflammatory phase leukotrienes and prostaglandins predominate, during resolution, there is a switch to lipid mediators referred to as specialized pro-resolving mediators (SPM).97 This switch may be regulated, at least in part, by changes in miRNA expression. For example, miR-292-2 levels increase for reasons that include neutrophil death with miR-292-2 targeting the mRNA of the lipoxygenase enzyme responsible for generating the proinflammatory leukotrienes.98 Additionally miR-292-2 increases, by an unknown mechanism, the synthesis of SPM such as resolvins and protectins. In HSK, where in some instances resolution can be delayed, it is conceivable that miR-292-2 levels are suppressed perhaps in situations when viral replication levels are elevated.
The observation that SPMs appear to account for resolution under natural circumstances has led to the development of synthetic formulations that are proving valuable to control inflammatory lesions that include HSK.99,100 It has also become evident in recent studies from the Serhan group that levels of multiple miRNAs change in response to synthetic SPM therapy.101 For example, resolvin administration fine-tunes levels of miRNAs such as miR-21, miR-146b, miR-208a, and miR-292-2 in various tissue-damaging settings.101,102 These miRNAs act by changing the expression of several inflammatory mediators. For example, miR-21 and miR-208a induce IL-10 production during the resolution phase, thus exerting a strong anti-inflammatory effect. miR-146b also regulates levels of IL-8 and RANTES, both powerful chemoattractants that recruit leukocytes into inflamed areas, while miR-292-2 induces the production of more SPM. We speculate that the provision of SPM during HSK may act by inducing the expression of pro-resolution miRNAs that include miR-21, miR-146b, and miR-292-2, while levels of inflammatory and angiogenic miRNAs (miR-155 and miR-132) might diminish (Fig. 4; Table). Our unpublished observations support this claim. Whether any group of miRNAs are sufficient to activate a resolution program is currently not known. If it can be accomplished, the inclusion of pro-resolution miRNAs would be valuable when designing therapeutics to counteract HSK lesions.
Figure 4. .
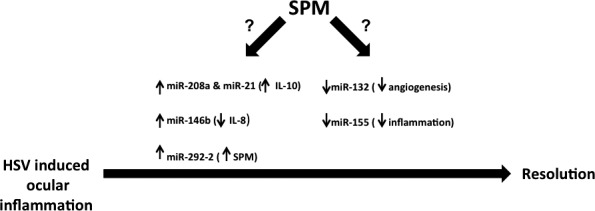
Specialized pro-resolution mediators (SPM) and miRNA-mediated resolution of HSV-induced ocular inflammation: speculations. The provision of SPM during HSK may result in changes in levels of various miRNAs associated with resolution. SPM could increase expression of miR-21 and miR-208a both of which induce anti-inflammatory cytokine, IL-10. SPM can also increase miR-146b expression and miR-146 lowers levels of IL-8, a chemokine crucial for neutrophil recruitment to eyes. miR-292-2 induced by SPM gives positive feedback for more SPM production. The administration of SPM may also lower levels of miR-132 and miR-155 involved in CV and inflammation, respectively.
Potential of miRNA Targeting
Manipulation of disease-associated microRNAs offers unique opportunities, but poses challenges with regard to delivery, target site accessibility, and off-target effects. Progress on this topic has occurred with several reports showing amelioration of diseases via blockade of miRNAs using modified antagomirs, as is the case with locked nucleic acid (LNA) or antagomirs given in nanoparticles.103,104 Our own study evaluated the potential of miR-132 inhibition using antagomir-132 histidine lysine nanoparticle approaches that led to potent reduction of CV. This was also recently confirmed in retinal models by the Martin Friedlander and David Cheresh groups who showed that nanoparticle mediated miR-132 blockade is more efficacious (Westenskow PD, et al. IOVS 2012;53:ARVO E-Abstract 4120) than conventional VEGF inhibition therapies and results in lower off target effects and fewer compensatory increases in angiogenic genes compared with VEGF targeting. Several other recent reports support miRNA inhibition approaches to modulate disease pathologies. For example, miR-122 shows effects against hepatitis C virus infection105; miR-155 is effective in the inflammatory disease models, EAE,106,107 as well as against B cell lymphoma.74,108 Additionally, miR-21 is effective against the inhibition of lupus.28 We are investigating the potential of slow antagomir–released eyedrop-formulated nanoparticles to intercept HSK pathogenesis and believe that the cocktail of miRNAs given in nanoparticles locally may confer therapeutic benefit against HSK.
Concluding Remarks
Although published information about the regulatory role of miRNAs during HSK is minimal, we can surmise that multiple miRNAs are likely to influence all of the many events that occur during HSK pathogenesis. We can also surmise that changing miRNA responses could represent a useful approach to therapy especially because several phases of HSK could be targeted simultaneously using cocktails of multiple miRNAs mimics and antagomirs. The major practical problem would be how to administer the cocktail and how frequently this must be done to achieve therapeutic efficacy. There have been some reports that show the ease of delivery of siRNA to localized areas, such as the eye, lung, and skin, and these siRNAs were shown to be effective in in vivo silencing.109,110 In fact, most of these siRNAs are under clinical trials—although only one such miRNA targeting drug is in clinical trial (miravirsen). It is conceivable many more miRNA intervention therapies could enter clinic in the near future.
It is also likely that the composition of the therapeutic cocktail would need to differ at different phases of HSK lesions. Most likely, treatment close to the onset of lesions would stand the best chance of success. We would speculate that using miRNAs or antagomirs to increase IL-10 expression, to block the recruitment of inflammatory cells to the corneas, and to limit the extent of CV would be successful strategies. At later stages, switching the inflammation to the resolution phase would be desirable as might be achieved by changing the expression of miRNAs involved in SPM production. Although therapy with miRNA has therapeutical advantages, it remains to be seen if the approach would be practical or economical compared to traditional therapies in current use. Answering these questions will require further research. Optimistically, nanoparticles containing multiple miRNAs and antagomirs would be administered once or twice daily and be therapeutically effective.
Acknowledgments
We thank Tamara Veiga-Parga and Fernanda Gimenez for their help with manuscript formatting.
Footnotes
Supported by Grant EY05093 from the National Eye Institute.
Disclosure: S. Mulik, None; S. Bhela, None; B.T. Rouse, None
References
- 1. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75: 843–854 [DOI] [PubMed] [Google Scholar]
- 2. Yang JS, Lai EC. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell. 2011; 43: 892–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pepose JS, Leib DA, Stuart PM, Easty DL. Herpes simplex virus disease: anterior segment of the eye. In: Pepose JS, Holland GN, Wilhelmus KR. eds Ocular Infections and Immunity. St. Louis, MO: Mosby; 1996: 905–932 [Google Scholar]
- 4. Biswas PS, Rouse BT. Early events in HSV keratitis--setting the stage for a blinding disease. Microbes Infect. 2005; 7: 799–810 [DOI] [PubMed] [Google Scholar]
- 5. Karali M, Peluso I, Gennarino VA, et al. miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics. 2010; 11: 715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giménez F, Suryawanshi A, Rouse BT. Pathogenesis of herpes stromal keratitis—a focus on corneal neovascularization [published online ahead of print August 7, 2012] Prog Retin Eye Res. doi:10.1016/j.preteyeres.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stuart PM, Keadle TL. Recurrent herpetic stromal keratitis in mice: a model for studying human HSK. Clin Dev Immunol. 2012; 2012: 728480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deshpande SP, Zheng M, Lee S, Rouse BT. Mechanisms of pathogenesis in herpetic immunoinflammatory ocular lesions. Vet Microbiol. 2002; 86: 17–26 [DOI] [PubMed] [Google Scholar]
- 9. Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol. 1997; 158: 1383–1391 [PubMed] [Google Scholar]
- 10. Tumpey TM, Chen SH, Oakes JE, Lausch RN. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J Virol. 1996; 70: 898–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng M, Schwarz MA, Lee S, Kumaraguru U, Rouse BT. Control of stromal keratitis by inhibition of neovascularization. Am J Pathol. 2001; 159: 1021–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mulik S, Xu J, Reddy PB, et al. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012; 181: 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A. 2002; 99: 978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008; 454: 780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Everett RD. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays. 2000; 22: 761–770 [DOI] [PubMed] [Google Scholar]
- 16. Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997; 71: 5878–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology. 2011; 417: 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cochrane DR, Cittelly DM, Richer JK. Steroid receptors and microRNAs: relationships revealed. Steroids. 2011; 76: 1–10 [DOI] [PubMed] [Google Scholar]
- 19. Rainer J, Ploner C, Jesacher S, et al. Glucocorticoid-regulated microRNAs and mirtrons in acute lymphoblastic leukemia. Leukemia. 2009; 23: 746–752 [DOI] [PubMed] [Google Scholar]
- 20. Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004; 78: 10178–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998; 93: 487–490 [DOI] [PubMed] [Google Scholar]
- 22. Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000; 20: 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noonan EJ, Place RF, Pookot D, et al. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009; 28: 1714–1724 [DOI] [PubMed] [Google Scholar]
- 24. Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006; 38: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimeld C, Hill T, Blyth B, Easty D. An improved model of recurrent herpetic eye disease in mice. Curr Eye Res. 1989; 8: 1193–1205 [DOI] [PubMed] [Google Scholar]
- 26. Pothof J, Verkaik NS, van IJcken W, et al. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J. 2009; 28: 2090–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dziunycz P, Iotzova-Weiss G, Eloranta JJ, et al. Squamous cell carcinoma of the skin shows a distinct microRNA profile modulated by UV radiation. J Invest Dermatol. 2010; 130: 2686–2689 [DOI] [PubMed] [Google Scholar]
- 28. Garchow BG, Bartulos Encinas O, Leung YT, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med. 2011; 3: 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brunner S, Herndler-Brandstetter D, Arnold CR, et al. Upregulation of miR-24 is associated with a decreased DNA damage response upon etoposide treatment in highly differentiated CD8(+) T cells sensitizing them to apoptotic cell death. Aging Cell. 2012; 11: 579–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000; 191: 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reddy PB, Sehrawat S, Suryawanshi A, et al. Influence of galectin-9/Tim-3 interaction on herpes simplex virus-1 latency. J Immunol. 2011; 187: 5745–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schacker T, Zeh J, Hu HL, Hill E, Corey L. Frequency of symptomatic and asymptomatic herpes simplex virus type 2 reactivations among human immunodeficiency virus-infected men. J Infect Dis. 1998; 178: 1616–1622 [DOI] [PubMed] [Google Scholar]
- 33. Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol. 2001; 75: 9828–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anand S, Majeti BK, Acevedo LM, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010; 16: 909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suryawanshi A, Veiga-Parga T, Rajasagi NK, et al. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol. 2011; 187: 1919–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008; 8: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magill ST, Cambronne XA, Luikart BW, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A. 2010; 107: 20382–20387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lagos D, Pollara G, Henderson S, et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat Cell Biol. 2010; 12: 513–519 [DOI] [PubMed] [Google Scholar]
- 39. Shaked I, Meerson A, Wolf Y, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009; 31: 965–973 [DOI] [PubMed] [Google Scholar]
- 40. Lee S, Zheng M, Kim B, Rouse BT. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J Clin Invest. 2002; 110: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan W, Zhang W, Sun L, et al. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res. 2011; 1411: 108–115 [DOI] [PubMed] [Google Scholar]
- 42. Ucar A, Vafaizadeh V, Jarry H, et al. miR-212 and miR-132 are required for epithelial stromal interactions necessary for mouse mammary gland development. Nat Genet. 2010; 42: 1101–1108 [DOI] [PubMed] [Google Scholar]
- 43. Suryawanshi A, Mulik S, Sharma S, Reddy PB, Sehrawat S, Rouse BT. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J Immunol. 2011; 186: 3653–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ambati BK, Nozaki M, Singh N, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006; 443: 993–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A. 2006; 103: 17343–17348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007; 317: 1522–1527 [DOI] [PubMed] [Google Scholar]
- 47. Holm CK, Jensen SB, Jakobsen MR, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012; 13: 737–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muruve DA, Pétrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008; 452: 103–107 [DOI] [PubMed] [Google Scholar]
- 49. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011; 11: 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat Rev Immunol. 2011; 11: 163–175 [DOI] [PubMed] [Google Scholar]
- 51. Biswas PS, Banerjee K, Kinchington PR, Rouse BT. Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp Eye Res. 2006; 82: 46–54 [DOI] [PubMed] [Google Scholar]
- 52. Ward JR, Heath PR, Catto JW, Whyte MK, Milo M, Renshaw SA. Regulation of neutrophil senescence by microRNAs. PLoS One. 2011; 6: e15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Izumi B, Nakasa T, Tanaka N, et al. MicroRNA-223 expression in neutrophils in the early phase of secondary damage after spinal cord injury. Neurosci Lett. 2011; 492: 114–118 [DOI] [PubMed] [Google Scholar]
- 54. Sullivan RP, Leong JW, Schneider SE, et al. MicroRNA-deficient NK cells exhibit decreased survival but enhanced function. J Immunol. 2012; 188: 3019–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008; 451: 1125–1129 [DOI] [PubMed] [Google Scholar]
- 56. O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010; 10: 111–122 [DOI] [PubMed] [Google Scholar]
- 57. O'Connell RM, Rao DS. Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012; 30: 295–312 [DOI] [PubMed] [Google Scholar]
- 58. Trotta R, Chen L, Ciarlariello D, et al. miR-155 regulates IFN-γ production in natural killer cells. Blood. 2012; 119: 3478–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim TD, Lee SU, Yun S, et al. Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood. 2011; 118: 5476–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–659 [DOI] [PubMed] [Google Scholar]
- 61. Daheshia M, Kanangat S, Rouse BT. Production of key molecules by ocular neutrophils early after herpetic infection of the cornea. Exp Eye Res. 1998; 67: 619–624 [DOI] [PubMed] [Google Scholar]
- 62. Bauer D, Mrzyk S, van Rooijen N, Steuhl KP, Heiligenhaus A. Macrophage-depletion influences the course of murine HSV-1 keratitis. Curr Eye Res. 2000; 20: 45–53 [PubMed] [Google Scholar]
- 63. Rogler CE, Levoci L, Ader T, et al. MicroRNA-23b cluster microRNAs regulate transforming growth factor-beta/bone morphogenetic protein signaling and liver stem cell differentiation by targeting Smads. Hepatology. 2009; 50: 575–584 [DOI] [PubMed] [Google Scholar]
- 64. Yuan B, Dong R, Shi D, et al. Down-regulation of miR-23b may contribute to activation of the TGF-β1/Smad3 signalling pathway during the termination stage of liver regeneration. FEBS Lett. 2011; 585: 927–934 [DOI] [PubMed] [Google Scholar]
- 65. Zhu S, Pan W, Song X, et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med. 2012; 18: 1077–1086 [DOI] [PubMed] [Google Scholar]
- 66. Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J Immunol. 2012; 188: 3434–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Blüml S, Bonelli M, Niederreiter B, et al. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum. 2011; 63: 1281–1288 [DOI] [PubMed] [Google Scholar]
- 68. Bhattacharyya S, Balakathiresan NS, Dalgard C, et al. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J Biol Chem. 2011; 286: 11604–11615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kurowska-Stolarska M, Alivernini S, Ballantine LE, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A. 2011; 108: 11193–11198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007; 316: 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007; 316: 604–608 [DOI] [PubMed] [Google Scholar]
- 72. O'Connell RM, Kahn D, Gibson WS, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010; 33: 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jiang S, Zhang HW, Lu MH, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010; 70: 3119–3127 [DOI] [PubMed] [Google Scholar]
- 74. Babar IA, Cheng CJ, Booth CJ, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci U S A. 2012; 109: E1695–E1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Steiner DF, Thomas MF, Hu JK, et al. MicroRNA-29 regulates T-box transcription factors and interferon-γ production in helper T cells. Immunity. 2011; 35: 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009; 10: 1252–1259 [DOI] [PubMed] [Google Scholar]
- 77. Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci U S A. 2012; 109: E1248–E1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cox MB, Cairns MJ, Gandhi KS, et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One. 2010; 5: e12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ding S, Liang Y, Zhao M, et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. 2012; 64: 2953–2963 [DOI] [PubMed] [Google Scholar]
- 80. Stittrich AB, Haftmann C, Sgouroudis E, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. 2010; 11: 1057–1062 [DOI] [PubMed] [Google Scholar]
- 81. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009; 30: 646–655 [DOI] [PubMed] [Google Scholar]
- 82. Takahashi H, Kanno T, Nakayamada S, et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. 2012; 13: 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006; 103: 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu LF, Boldin MP, Chaudhry A, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010; 142: 914–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yang L, Boldin MP, Yu Y, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012; 209: 1655–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sheedy FJ, Palsson-McDermott E, Hennessy EJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010; 11: 141–147 [DOI] [PubMed] [Google Scholar]
- 87. Liu PT, Wheelwright M, Teles R, et al. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med. 2012; 18: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sarangi PP, Sehrawat S, Suvas S, Rouse BT. IL-10 and natural regulatory T cells: two independent anti-inflammatory mechanisms in herpes simplex virus-induced ocular immunopathology. J Immunol. 2008; 180: 6297–6306 [DOI] [PubMed] [Google Scholar]
- 89. Zhou J, Wang KC, Wu W, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci U S A. 2011; 108: 10355–10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lu TX, Munitz A, Rothenberg ME. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009; 182: 4994–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lu TX, Hartner J, Lim EJ, et al. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol. 2011; 187: 3362–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liston A, Lu LF, O'Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008; 205: 1993–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhou X, Jeker LT, Fife BT, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008; 205: 1983–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jeker LT, Zhou X, Gershberg K, et al. MicroRNA 10a marks regulatory T cells. PLoS One. 2012; 7: e36684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009; 30: 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009; 182: 2578–2582 [DOI] [PubMed] [Google Scholar]
- 97. Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010; 177: 1576–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fredman G, Li Y, Dalli J, Chiang N, Serhan CN. Self-limited versus delayed resolution of acute inflammation: temporal regulation of pro-resolving mediators and MicroRNA. Sci Rep. 2012; 2: 639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007; 447: 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rajasagi NK, Reddy PB, Suryawanshi A, Mulik S, Gjorstrup P, Rouse BT. Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J Immunol. 2011; 186: 1735–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN. MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits. FASEB J. 2011; 25: 544–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol. 2012; 180: 2018–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Krützfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs'. Nature. 2005; 438: 685–689 [DOI] [PubMed] [Google Scholar]
- 104. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012; 3: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010; 327: 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011; 187: 2213–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Worm J, Stenvang J, Petri A, et al. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 2009; 37: 5784–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhang Y, Roccaro AM, Rombaoa C, et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012; 120: 1678–1686 [DOI] [PubMed] [Google Scholar]
- 109. Nguyen T, Menocal EM, Harborth J, Fruehauf JH. RNAi therapeutics: an update on delivery. Curr Opin Mol Ther. 2008; 10: 158–167 [PubMed] [Google Scholar]
- 110. Kim B, Tang Q, Biswas PS, et al. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am J Pathol. 2004; 165: 2177–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]