

Abstract
Purpose.
To determine whether soluble CD44 (sCD44), a likely biomarker of primary open-angle glaucoma (POAG), is internalized in cultured human trabecular meshwork (TM) cells and trafficked to mitochondria.
Methods.
In vitro, 32-kD sCD44 was isolated from human sera, biotinylated, and dephosphorylated. TM cells were incubated for 1 hour at 4°C with biotinylated albumin (b-albumin), biotin-labeled sCD44 (b-sCD44), or hypophosphorylated biotin-labeled sCD44 (–p b-sCD44) in the presence or absence of unlabeled sCD44, hyaluronic acid (HA), and a selected 10-mer HA binding peptide. The slides were warmed for 1 or 2 hours at 37°C, and 125 nM MitoTracker Red was added for the last 20 minutes of the incubation. The cells were washed, fixed, incubated with anti-biotin antibody and FITC-labeled goat anti-mouse antibody, and examined under a confocal microscope.
Results.
TM cell membranes were positive for b-sCD44 after 4°C incubation. When the temperature was raised to 37°C, b-sCD44 or –p b-sCD44 appeared in the cytoplasm. The internalization of b-sCD44 was blocked by excess unlabeled sCD44, HA, and a 10-mer HA-binding peptide. Double label experiments with b-sCD44 or –p b-sCD44 and MitoTracker Red indicated partial overlap. The percent co-localization of MitoTracker Red at 2 hours and FITC –p b-sCD44 was 17.4% (P < 0.001) and for FITC b-sCD44 was 11.7% (P < 0.001) compared with b-albumin. The influence of putative CD44 phosphorylation sites on mitochondrial trafficking was determined by TargetP 1.1.
Conclusions.
sCD44 is internalized by TM cells and trafficked in part to mitochondria, which may be a factor in the toxicity of sCD44 in the POAG disease process.
sCD44 was internalized and then localized in part to the mitochondria in human TM cells treated with sCD44.
Introduction
Primary open-angle glaucoma (POAG) is one of the four major causes of blindness and visual disability in the United States.1 Previous studies from our laboratory have identified soluble CD44 (sCD44) as a likely protein marker and causative factor of POAG, especially in African American individuals.2–5 CD44 is an 80- to 250-kD transmembrane protein; its ectodomain is released as 32-kD sCD44 and remains biologically active.6,7 The liberated sCD44 binds ligands acting as a decoy receptor and also binds homotypically. CD44 plays major roles in multiple physiological processes, including innate8,9 and adaptive immunity,10,11 phagocytosis,12,13 and cell survival pathways.14–16 sCD44 is elevated in the aqueous humor of POAG versus non-POAG patients,3,5,17,18 and its concentration correlates with the extent of visual field loss in POAG.5 A hypo-phosphorylated form of sCD44 in POAG aqueous humor further distinguishes POAG and normal pressure glaucoma from normal eyes and other types of glaucoma.19 Moreover, sCD44, particularly the hypo-phosphorylated form of sCD44, is a potent and specific cytotoxin to trabecular meshwork (TM) and retinal ganglion cells.4
CD44 is a member of a heterogeneous group of proteins designated hyaladherins, which are linked by their common ability to bind to hyaluronic acid (HA).20,21 The hyaladherins can be classified into two major groups: membrane-bound forms, such as CD44, receptor ligand C1q, and receptor for HA-mediated motility (RHAMM), and media forms such as versican, aggrecan, brain HA-binding protein, brevican, tumor necrosis factor α stimulated gene-6 (TSG-6), and link protein.20–22 A number of hyaladherins—a recombinant version of sCD44,23 sCD44,24 RHAMM,25 TSG-6,26 C1q,27 a synthetic version of trypsin fragments of aggrecan and link protein named metastatin,28 and a synthetic polypeptide P429—have been documented to exhibit antitumor activity. Notably, the cytotoxic activity of sCD444 and metastatin28 is inhibited by HA, and cells that have higher pericellular concentrations of HA are resistant to metastatin.
A common concept in neurodegenerative diseases is that a toxic protein alters mitochondrial function (e.g., β-amyloid in Alzheimer's disease and parkin in Parkinson's disease).30 It is now recognized that mitochondria are porous, allowing proteins, such as HA-binding proteins, to enter and leave. Exactly why some HA-binding proteins are trafficked into the mitochondria and become toxic is unclear. Multiple apoptotic pathways emanate from the mitochondria,31 resulting in membrane depolarization and release of cytochrome C. Given the emerging role of sCD44 as a biomarker and a possible factor in cell death in POAG, we examined the internalization of sCD44 into TM cells and explored possible ways to prevent its internalization and toxic effects on TM cells.
Materials and Methods
Isolation of sCD44
The 32-kD sCD44 was isolated from human serum (Sigma-Aldrich, St. Louis, MO) by a two-step isolation procedure. The first step utilized an HA affinity column. Human umbilical cord HA (Sigma-Aldrich) was standardized by size exclusion chromatography on a Sepharose CL-4B column (1.6 × 22.0 cm; GE Healthcare, Piscataway, NJ) equilibrated with 0.1 M ammonium acetate and standardized by the elution of blue dextran (molecular weight = 2 × 106; Sigma-Aldrich). HA was bound to EAH-Sepharose (15 mL; product number 17-0569-04; GE Healthcare) according to the manufacturer's instructions, and rinsed with 40 mL of 10 mM sodium phosphate-150 mM sodium chloride, pH 7.2 (PBS) (Sigma-Aldrich) in a sintered glass filter (Fisher Scientific, Pittsburg, PA) to remove azide from the storing buffer. The sepharose was then activated by incubation with 5 g N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Sigma-Aldrich) for 24 hours, and washed with 0.5 M NaCl, 0.1 M sodium acetate, pH 4.0, and 0.5 M NaCl, 0.1 M TRIS, pH 8.3 (Sigma-Aldrich). The activated sepharose was loaded into a XK-20 column (GE Healthcare) and was packed using PBS. Human serum (10 mL; Sigma-Aldrich) was added to the column, incubated overnight at 4°C, and the column was rinsed with 100 mL PBS. The bound proteins were eluted with 0.5 M NaCl and PBS (Sigma-Aldrich). A protein assay (Bio-Rad, Hercules, CA) and sCD44 ELISA (Bender Medsystems, San Diego, CA) were performed according to the manufacturer's instructions to determine the protein and sCD44 concentration.
In the second step of the isolation, a CD44 immunoprecipitation method was used to purify sCD44. The bound proteins were incubated with 100 μg monoclonal anti-CD44 antibody (BU52; Ancell, Bayport, MN) overnight at 4°C and incubated with a goat anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA) conjugated to sepharose for 2 hours. The complex was centrifuged at 2500g for 5 minutes, and the supernatant was aspirated. The resulting pellet was washed three times with PBS, and the bound proteins were eluted with 0.2 M glycine, pH 2.5 (Sigma-Aldrich), and neutralized with 1 M TRIS, pH 8.0 (Sigma-Aldrich). CD44 Western blots of graded amounts of sCD44 were determined by densitometry and the results were compared to Western blot densitometry of human serum. The isolated sCD44 was subjected to one-dimensional 4% to 11% polyacrylamide gradient blue native gel electrophoresis32 to determine aggregation and homotypic binding of sCD44.
Hypophosphorylation and Biotinylation of sCD44
sCD44 was hypophosphorylated by incubation of 4 μg of sCD44 with 30 units alkaline phosphatase (ICN Biochemicals, Irvine, CA) for 1 hour at 37°C and the process was repeated. Hyperphosphorylated sCD44 (+p sCD44) was phosphorylated as previously described.4 To verify the results of phosphatase treatment, the hypophosphorylated sCD44 (–p sCD44) was immunoblotted with phosphospecific anti-serine/threonine antibody (Upstate USA, Charlottesville, VA) as previously described.16 sCD44 and –p sCD44 were biotinylated (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. In brief, sCD44 was dissolved in 100 mM HEPES buffer (Sigma-Aldrich) so that the final concentration of protein was not greater than 10 mg/mL. An aliquot of the biotinylating reagent (10 mg/mL solution of the biotin reagent) was added, incubated for 2 hours at room temperature, and the reaction was stopped by adding 5 μL of ethanolamine. The biotinylated protein was dialyzed (product number 68,100 dialysis tubing; ThermoScientific, Rockford, IL) against three changes of 2 L of buffer to remove unreacted biotin. The biotinylation of sCD44 (b-sCD44) and of hypophosphorylated (–p b-sCD44) was verified by Western blot analysis.
Cell Culture and Immunofluorescence
Human TM cells derived from donors 19, 34, and 49 years of age were plated at 5000 cells per well on 8-well chamber slides in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (FBS) (Sigma-Aldrich). The medium was changed to 0.1% FBS for 2 hours before treatment. Biotinylated albumin (b-albumin; molar ratio of biotin to albumin was 8:1; Sigma-Aldrich), b-sCD44, or –p b-sCD44 (0.1 ng) was added to the wells and the slides were incubated at 4°C for 1 or 2 hours. Excess b-albumin, b-sCD44, or –p b-sCD44 not bound to the cell surface was removed by aspiration and fresh media was added. Cells were also treated with 1 μg of HA or with 1 ng of the 10-mer HA binding peptide, KNGRYSISRT, corresponding to the first HA binding site of sCD44 (amino acid residues 38 through 47). The 10-mer HA binding peptide was synthesized by Genemed Synthesis, Inc. (San Francisco, CA), and acetylated on N-terminus and amidated on C-terminus to prevent protease degradation. Control peptides were a scramble of the 10-mer sequence, or 0.1 ng of unlabeled sCD44. The slides were then warmed to 37°C for 1 or 2 hours. During the last 20 minutes, MitoTracker Red (MitoTrackerCMX Ros; Invitrogen, Carlsbad, CA) was added to the wells at a final concentration of 125 nM. For immunostaining, the cells were washed three times with PBS and fixed in cold methanol for 15 minutes. After membrane permeabilization with 0.1% saponin (Sigma-Aldrich) in PBS for 15 minutes at room temperature, the cells were incubated for 90 minutes with a mouse anti-biotin antibody (diluted 1:500; Sigma-Aldrich). The cells were washed and incubated for 45 minutes with secondary FITC-labeled goat anti-mouse antibody (diluted 1:200 in 10% normal goat serum; Jackson ImmunoResearch). The cells were then washed and mounted in Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector, Burlingame, CA). The staining was examined under fluorescence and confocal microscopes on previously masked slides. Images were obtained by conventional epifluorescence microscopy using a Zeiss Axiovert 100 microscope (Carl Zeiss, Thornwood, NY). Confocal microscopy was conducted using a Leica SP2 confocal microscope (Wetzlar, Germany). Images were obtained in the middle of cells by optical sectioning. The images were captured using a ×40 or ×63 objective and were taken as single image or as a Z-series. The Z-series was created with 20 images at 0.1-μm spacing. Fluorescence intensity per cell, percent of co-localization, and integrated optical density were quantitated by computer-assisted densitometry (Metamorph 7.5; MDS Analytical Technologies, Sunnyvale, CA).
Cell Viability
Primary cultures of third to fourth passage normal human TM cells were plated at a density of 40,000 cells per well in 24 multiwell plates (BectonDickinson, Franklin Lakes, NJ) as previously described4 and were treated with 0.1 μg of sCD44, +p sCD44 and –p sCD44 for 2 hours. Cell viability was determined as previously described.4
Statistical Analysis
All data were expressed as the mean ± SD. Student's t-test and linear regression were performed using StatSoft (Tulsa, OK). P less than 0.05 was considered to be statistically significant.
Results
A two-step isolation procedure using HA affinity columns and immunoprecipitation was performed to isolate sCD44. By Western blot analysis under reducing conditions, the isolated sCD44 preparation was strongly positive for a 32-kD band and positive for a 55-kD band (Fig. 1). The isolated sCD44 preparation was also positive for a 32- and 55-silver stained bands. The relative ratio of sCD44 concentration measured by ELISA to the protein concentration by protein assay was 141,357:1 for human sera. The ratio of the isolated sCD44 concentration measured by ELISA to protein concentration by protein assay was 10.39:1.00. Thus, the relative degree of enrichment was 13,605. To determine if sCD44 binds homotypically, the isolated sCD44 was subjected to blue native gel electrophoresis under nonreducing conditions. sCD44 was detected as a 600- to 1200-kD aggregate (Fig. 1).
Figure 1. .

SDS-PAGE under reducing conditions, blue native gel electrophoresis, and immunoblot analysis of isolated sCD44 from human sera by HA affinity chromatography and immunoprecipitation. Lanes from left to right: lane 1, SDS-PAGE; Silver stain of human sera; lane 2, CD44 Western blot of isolated sCD44 under reducing conditions; lane 3, silver stain of isolated sCD44 under reducing conditions; lane 4, blue native gel electrophoresis, CD44 Western blot of aggregated sCD44. Note the 32-kD sCD44 band and approximately 55-kD CD44 band in lane 2.
An sCD44 standard curve was generated by Western blot densitometry using monoclonal anti-CD44 antibody. The concentration of sCD44 in human serum had been previously determined by ELISA, and volume equivalents of each concentration, 5 to 80 pg, were loaded into each lane. The bands were analyzed by densitometry and the integrated optical density was determined for each load of sCD44. The results showed a linear relationship (R2 = 0.9837), with 2.52 densitometric units per pg of sCD44 (Fig. 2A). Graded amounts of b-sCD44 and –p b-sCD44 were analyzed by Western blot analysis and quantitated using human serum sCD44 as a standard. Each of the preparations was also subjected to Western blot analysis using anti-biotin antibody to verify biotinylation and antiphosphorylation antibodies to verify phosphorylation of b-sCD44 and hypophosphorylation of –p b-sCD44 (Figs. 2B, 2C). The results indicated that the preparations were biotinylated and that alkaline phosphatase treatment decreased the phosphorylation of –p b-sCD44.
Figure 2. .

SDS-PAGE and immunoblot analysis of standard biotinylated sCD44 (b-sCD44) and hypophosphorylated biotinylated sCD44 (–p b-sCD44). (A) Western blot standard curve generated from 5 to 80 pg sCD44 isolated from human serum using BU-52 monoclonal antibody for CD44. (B) Western blot analysis of isolated b-sCD44 and (C) –p b-sCD44. Lanes 1 to 3 are 20, 40, and 80 pg of isolated b-sCD44 and –p b-sCD44 immunoblots using BU-52 monoclonal antibody for CD44; lane 4 is 40 pg of b-sCD44 and of –p b-sCD44 and using anti-biotin antibody; lane 5 is 20 pg for the phosphorylation status of b-sC44 and –p b-sCD44 using phosphospecific anti-serine/threonine antibody. The data are from a representative of three individual experiments that had consistent results.
To determine whether the isolated sCD44, +p sCD44, or –p sCD44 were biologically active, cell viability was determined. The sCD44 and –p sCD44 treatment for 2 hours significantly reduced cell viability, whereas +p sCD44 had a lesser effect on cell viability (Fig. 3).
Figure 3. .

Cell viability of TM cells treated with sCD44. Legends: Control, PBS; sCD44 (0.1 μg); +p sCD44 (0.1 μg); –p sCD44 (0.1 μg) for 2 hours. The data represent the mean of results (n = 6) in a representative experiment. Error bars denote SD. Significance of results: *P < 0.05, **P < 0.01, in comparison with control; †P < 0.05, ††P < 0.01 in comparison with sCD44.
To detect internalization of b-sCD44 into TM cells, the cells were exposed to 0.1 ng b-albumin, 0.001 to 0.1 ng of b-sCD44 or –p b-sCD44 at 4°C for 1 hour. Confocal microscopy results of b-sCD44–treated cells showed that only the TM cell membranes were stained and the cytoplasm was negative (data not shown). In all subsequent experiments, TM cells were rinsed with fresh media to remove unbound biotinylated proteins, and incubated for an additional 1 or 2 hours. MitoTracker Red was added for the last 20 minutes of incubation. MitoTracker Red is internalized into mitochondria and its accumulation is dependent on mitochondrial membrane potential. The cells were fixed with methanol, subjected to 0.1% saponin permeabilization, and stained with anti-biotin antibody and DAPI. The internalization of b-albumin was limited, although there was a substantial internalization and accumulation of MitoTracker Red (Fig. 4A). The lowest dose of b-sCD44, 0.001 ng, resulted in minimal staining in the cytoplasm (data not shown). A 0.01-ng dose (Fig. 4B) or 0.1-ng dose (Fig. 4C) of b-sCD44 resulted in patchy areas of staining on some of the TM cell processes, diffuse cytoplasmic staining, and perinuclear staining, which appeared to overlap in some areas with MitoTracker Red (Fig. 4C). Similar results were also obtained with –p b-sCD44 (data not shown).
Figure 4. .
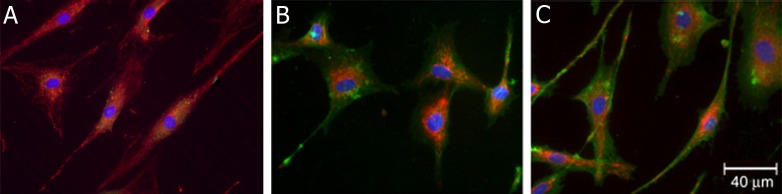
Internalization of b-sCD44 in TM cells. TM cells were treated with (A) 0.1 ng b-albumin, (B) 0.01 ng b-sCD44, or (C) 0.1 ng b-sCD44 for 1 hour at 4°C, rinsed with fresh media to remove unbound biotin-labeled albumin or sCD44, and incubated for 2 hours at 37°C. For the last 20 minutes of incubation, 125 nM MitoTracker Red was added, then fixed with methanol, permeabilized with 0.1% saponin and treated with mouse anti-biotin and FITC-labeled goat anti-mouse IgG antibodies and counterstained with DAPI for nuclear staining. Positive mitochondria staining with MitoTracker Red appears as red, b-albumin or b-sCD44 staining appears in green, and nuclei are in blue. Note higher doses of b-sCD44 resulted in cell detachment. Cells were photographed with an epifluorescence microscope. The data are representative of three individual experiments with consistent results.
Confocal microscopy was used to document the overlap of b-albumin, b-sCD44, or –p b-sCD44 with MitoTracker Red (Fig. 5A). In the control, minimal co-localization was observed with b-albumin and MitoTracker Red (Fig. 5A). Partial co-localization was observed with b-sCD44 and –p b-sCD44 (Fig. 5A). By aligning sequential Z-stacks, a three-dimensional representation of the co-localization of b-sCD44 or –p b-sCD44 with MitoTracker Red was determined (Figs. 5B, 5C). A partial co-localization of b-sCD44 and –p b-sCD44 and MitoTracker Red was noted in the perinuclear areas. In addition, mitochondria in control TM cells appeared to be more evenly dispersed throughout the cytosol than in b-sCD44 or –p b-sCD44–treated TM cells.
Figure 5. .

Confocal microscopy of the internalization of b-sCD44 and –p b-sCD44 in TM cells. TM cells were treated with 0.1 ng b-sCD44 for 2 hours, treated with 125 nM MitoTracker Red, fixed in methanol, permeabilized with 0.1% saponin, and stained with mouse anti-biotin and FITC-labeled goat anti-mouse IgG antibodies. Cells were photographed with a confocal microscope at the mid-level by optical sectioning. In (A), cells were treated with b-albumin (control), 0.1 ng b-sCD44, or 0.1 ng –p b-sCD44 and stained with biotin and goat FITC-labeled anti-mouse IgG antibodies (to label b-sCD44 green) and MitoTracker Red (to label mitochondria red). Arrows indicate areas of co-localization. Z-stacks were generated for an enlarged merged image of b-sCD44 (B) and –p b-sCD44 (C). Co-localization of b-sCD44 and –p b-sCD44 and MitoTracker Red in the YZ and XZ image planes appear as yellow.
To further analyze co-localization of b-albumin, sCD44, and MitoTracker Red, image analysis software was used to quantitate the fluorescence staining of b-albumin, b-sCD44, –p b-sCD44, and MitoTracker Red (Fig. 6A). At 2 hours, there was a highly significant increase in both b-sCD44 and –p b-sCD44 staining compared with b-albumin control (P < 0.001, for both b-sCD44 and –p b-sCD44). There was a statistically significant decrease in FITC green fluorescence intensity for –p b-sCD44 compared with b-sCD44 (P < 0.01). There was also a statistically significant increase in MitoTracker Red intensity in comparison of b-sCD44 with b-albumin control (P < 0.001) and –p sCD44 to b-albumin (P < 0.01). MitoTracker Red staining in –p b-sCD44 cells compared with b-sCD44–treated cells (P < 0.001) was also significant. Using Metamorph imaging software, the percent of co-localization for b-albumin on MitoTracker Red was determined to be 4.0%, FITC –p b-sCD44 on MitoTracker Red was calculated to be 17.4%, and for FITC b-sCD44 on MitoTracker Red was 11.7% (Fig. 6B). The increase in co-localization was highly statistically significant comparing b-albumin and b-sCD44 and –p b-sCD44 (P < 0.001) and –p b-sCD44 (P < 0.001), and significant in comparing b-sCD44 and –p b-sCD44 (P < 0.001).
Figure 6. .

Quantitation of fluorescence and percent of co-localization of b-albumin, b-sCD44, –p b-sCD44, and MitoTracker Red in TM cells at 1 and 2 hours. Confocal microscopy images were obtained in the middle of cells by optical sectioning and fluorescence intensity per cell was quantitated by computer-assisted densitometry. (A) Fluorescence expressed on y-axis as integrated optical densitometry of anti-biotin FITC fluorescence and MitoTracker Red fluorescence in TM cells treated with b-albumin, b-sCD44 or –p b-sCD44. (B) Co-localization expressed in percent of b-albumin, p b-sCD44, or b-sCD44 with MitoTracker Red. The data (n = 30) are respective of three individual experiments with consistent results. Error bars indicate the SEM. Significance of results: ***P < 0.001, comparing b-albumin control and b-sCD44. ††P < 0.01, †††P < 0.001, comparing b-albumin control and –p b-sCD44. §P < 0.05, §§P < 0.01, §§§P < 0.001, comparing b-sCD44 and –p b-sCD44.
To further examine the specificity of internalization of b-sCD44 and partial overlap with MitoTracker Red (Fig. 7A), b-sCD44 was co-administered with HA (Fig. 7B). No b-sCD44 staining was observed on HA treatment, indicating that HA blocked the binding of b-sCD44 to the cell surface and also blocked the internalization of b-sCD44. A more specific test of the internalization of sCD44 was the use of the synthetic 10-mer HA binding peptide, KNGRKYSISRT, corresponding to residues 38 through 47 of CD44, which blocked both the binding of b-sCD44 to the cell surface and the internalization (Fig. 7C). A final test of the internalization of sCD44 was to administer unlabeled sCD44 to competitively block the internalization of b-sCD44. Excess unlabeled sCD44 blocked the internalization of b-sCD44 (Fig. 7D); however, intense staining was observed on the cell surface of TM cells.
Figure 7. .

Internalization of b-sCD44 in TM cells. TM cells were treated with biotinylated-sCD44 for 2 hours, treated with 125 nM MitoTracker Red, fixed in methanol, permeabilized with 0.1% saponin, and incubated with mouse anti-biotin and FITC-labeled goat anti-mouse IgG antibodies. Cells were photographed with a confocal microscope at the mid-level by optical sectioning. (A) 0.1 ng b-sCD44. (B) 0.1 ng b- sCD44 co-administered with 1 μg HA. (C) Initial administration of 1 ng of selected 10-mer HA binding peptide (KNGRKYSISRT) and then 5 minutes later followed with 0.1 ng b-sCD44. The molar ratio of 10-mer to b-sCD44 was 200:1. (D) Initial administration of 0.1 ng of unlabeled sCD44 and then 5 minutes later followed with 0.1 ng b-sCD44.
Discussion
The present study demonstrates that b-sCD44 and –p b-sCD44 are internalized into TM cells. sCD44 was isolated from human sera, biotinylated, and dephosphorylated in vitro. Each of the preparations was bioactive in a cell viability assay. A prior study from our laboratory demonstrated that sCD44 toxicity can be blocked by CD44 neutralizing antibody, excess HA, or the pan-caspase inhibitor Z-VAD-FMK.4
Table. .
CD44 Cleavage and Phosphorylation Sites and Prediction of Mitochondrial Trafficking
| Phosphorylated Cytosolic CD44 Amino Acid* |
Prediction for the Trafficking S1 Cleavage of ICD† by γ-Secretase to Mitochondria |
Prediction for the Trafficking S2 Cleavage of Alternate ΔICD‡ by γ-Secretase to Mitochondria |
| None | 0.410 | 0.588 |
| Ser672 | 0.264 | 0.331 |
| Ser686 | 0.306 | 0.525 |
| Ser672 + Ser686 | 0.200 | 0.295 |
| Ser697 | 0.355 | 0.573 |
| Ser706 | 0.397 | 0.583 |
| Thr720 | 0.412 | 0.588 |
| Ser697 + Thr720 | 0.353 | 0.570 |
| Ser706 + Thr720 | 0.399 | 0.581 |
| Ser697 + Ser706 + Thr720 | 0.470 | 0.568 |
The amino acid sequence of cytosolic CD44 conventional ICD and alternate ICD (ΔICD) were analyzed by TargetP 1.1 Server.71 TargetP is a neural network–based tool that predicts the subcellular location of a protein using its N-terminal sequence. Negatively charged amino acid residues were used as a substitute for phosphorylated residues.
ICD is amino acids 669 to 742, which translocates to the nucleus7 via an identified nuclear targeting signal.43
ΔICD is amino acids 660 to 742, created by an alternate cleavage site of γ-secretase.39 It contains a putative mitochondrial targeting sequence in addition to the known nuclear targeting signal of the conventional ICD.
Confocal microscopy indicated that TM cell membranes were immunopositive for b-sCD44 after a 4°C incubation. When the temperature was raised to 37°C, b-sCD44 and –p b-sCD44 were observed in the cytoplasm, particularly in the perinuclear region. In control b-albumin–treated cells, minimal FITC staining was observed; however, MitoTracker Red staining of mitochondria was evenly dispersed throughout the cytosol and a time-dependent increase in staining was observed from to 1 to 2 hours. In contrast, cells treated with sCD44 displayed large, brightly stained mitochondria, concentrated in the perinuclear area. In both b-sCD44 and –p b-sCD44–treated cells, there was no notable increase in either FITC or MitoTracker Red staining from 1 to 2 hours. Double labeling experiments indicated that b-albumin co-localization (4%) of FITC staining of b-albumin and MitoTracker Red was minimal. In contrast, co-localization between b-sCD44 (11.7%) or –p b-sCD44 (17.4%) with MitoTracker Red was increased, suggesting that both b-sCD44 and –p b-sCD44 were trafficked in part to the mitochondria. Phosphorylation of an amino acid may introduce a conformational change in sCD44 and change the internalization and tracking pattern of sCD44, as shown in Figure 6. The internalization of b-sCD44 and –p b-sCD44 was blocked by co-administering HA, by pretreating the TM cells with a 10-mer HA peptide, or pretreating the TM cells with excess unlabeled sCD44. These results suggest b-sCD44 or –p b-sCD44 bind homotypically to the CD44 receptor. These results also suggest that HA or a 10-mer peptide binding to CD44 receptor blocks sCD44 internalization. Internalization of exogenous biotinylated ligands binding to receptor is temperature dependent.33 The internalized receptor cargo is trafficked by localization signals within the ligand/receptor complex.34 sCD44 internalization may involve mitochondrial dysfunction in a fashion similar to that of β-amyloid fragments,35,36 causing abnormal mitochondrial function and cell death. The extent of exogenous β-amyloid co-localization in mitochondria is variable from partial without quantification36–38 and 1% to 9%.39
sCD44 is released by proteolytic cleavage40 (shedding) from membrane-anchored CD44, which influences CD44-mediated HA binding to cell surfaces.41 In addition, full-length CD44 is endocytosed and is trafficked to multiple intracellular locations. When CD44 is proteolytically cleaved, with a release to the ectodomain as sCD44, the transmembrane portion is cleaved by gamma-secretase to release an intracytoplasmic domain (ICD), which trafficks to the nucleus and creates a positive feedback loop.42 Nuclear CD44 regulates STAT3 and binds to cyclin D1 promoter, leading to cyclin D1 expression and cell proliferation.43 This suggests that CD44 nuclear trafficking results in an autocrine mechanism.
CD44 is highly polymorphic due to alternate splicing and variable N- and O-linked glycosylation sites, which regulate HA binding in some cell types but not in others.44 The activation states of a single cell also influence CD44 binding to HA so that some cells bind HA while others do not.41 Recent evidence indicates that ligand binding using anti-CD44 antibody results in cell death by apoptosis and mitochondrial depolarization.45 CD44 antibody binds to CD44 and results in mitochondrial dysfunction and cell death. These results indicate that ligand binding to CD44 can not only act in a positive feedback loop but also result in cell death.
Exactly how the internalization process and trafficking of sCD44 occurs in TM cells is not well understood. CD44 receptor internalization occurs by clathrin-independent endocytosis and it trafficks to endosomes or recycles via endocytic recycling compartment.46 Albumin is internalized mainly by a caveolin-dependent endocytosis.47 Protein trafficking patterns are complicated; abnormal protein trafficking commonly occurs in neurodegenerative diseases.48 Cell stress–induced modifications of proteins commonly involve phosphorylation and proteolytic cleavage, which may redirect some proteins to the mitochondria.49–51 sCD44 has a putative mitochondrial localization sequence consisting of a highly hydrophobic and basic amino acid–rich sequence.52,53 sCD44 may interfere with normal mitochondrial function, as is seen with the synthetic peptide P429 and RHAMM,54 and trigger the intrinsic pathway of apoptosis. The importance of phosphorylation and mitochondrial import of cofilin was demonstrated by Chua et al.55 Cells stressed by kinase inhibitor import cofilin, an actin-binding protein, into the mitochondria before cell death by apoptosis. Dephosphorylation of cofilin was essential and necessary for trafficking to mitochondria.
The likelihood of mitochondrial trafficking and the effect of phosphorylation and cleavage site of three gamma-secretase substrates (CD44, Notch, and amyloid precursor protein) are shown in Figure 8. Analysis by the TargetP 1.1 program on various phosphorylation modifications of the CD44 at different residues remarkably changed the likelihood of mitochondrial localization (Table). Alternate cleavage site by γ-secretase in the transmembrane also remarkably changed the likelihood of mitochondrial localization. Phosphorylation of Ser672 and Ser686 have the largest effect. Because PKC is a known to phosphorylate Ser672 and modulate the interaction of CD44 and the cytoskeletal linker ezrin in vivo,56 phosphorylation of the Ser672 may prevent mitochondrial trafficking. The γ-secretase cleavage at S1 or S2 influence mitochondrial trafficking. The alternate S2 cleavage of CD44 is likelier to go to the mitochondria than the S1 fragment, which has a nuclear targeting signal.43 Further studies are required for CD44 modification via phosphorylation or alternate cleavage and mitochondrial trafficking.
Figure 8. .

Schematic representation of three γ-secretase substrates, CD44, Notch, amyloid precursor protein (APP), and generation of ICD from portions of the transmembrane (blue), juxtamembrane (red), and cytosolic topological domain (yellow). γ-secretase cleavage sites are within the transmembrane sequences, which liberates an extracellular domain (green) and ICD. The mitochondrial targeting sequence (purple) and nuclear targeting signal (orange) were determined by Euk-mPLoc 2.0 computational program67–69 for CD44. Note sCD44 binds homotypically as a ligand to its CD44 receptor. The internalization of b-sCD44 and CD44 and subsequent trafficking is regulated by phosphorylation and conformation changes in the complex.34 The mitochondrial intermediate peptidase cleavage site (open arrow) is only known for Notch-1.70 AA, amino acid. S1 common γ-secretase cleavage site. S2 alternate γ-secretase cleavage site. S3 alternate γ-secretase cleavage site for APP.42 The nuclear target signal for APP is not known.
Abnormalities in mitochondria have been reported in neurodegenerative diseases57,58 and in glaucoma.59 Patients with POAG were found to have a variety of mutations in mitochondrial DNA as well as decreased mitochondrial respiratory activity.60 Mitochondria are well known mediators of apoptosis, with mitochondrial dysfunction often resulting in apoptotic cell death. Recently, mitochondrial permeability pore was shown to be opened by p53 in response to oxidative stress.61 Intriguingly, p53 forms a complex with cyclophilin D; p53 is also known to form a complex with CD44 via a noncanonical p53 binding sequence in its promoter.62 In a rat model of glaucoma, increased IOP caused the cleavage and constitutive activation of the protein phosphatase calcineurin. This cleavage leads to dephosphorylation of the proapoptotic protein Bad, and subsequent increase of apoptotic cell death of retinal ganglion cells.63 In a model of hypoxia-ischemic brain injury, cytochrome C is released from the brain into cerebrospinal fluid. Notably, exogenous biotinylated cytochrome C is internalized into mitochondria of neurons exposed to anoxia.64
Alterations in the phosphorylation of sCD44 in POAG19 may involve metabolic stress, free radicals, or age-related modification of kinase/phosphatase. The resulting change in phosphorylation may alter CD44 shedding and/or endocytosis and subsequent subcellular localization. If sCD44 is indeed trafficked in part to the mitochondria, this may be a factor in the toxicity of sCD44 in certain cells, such as the TM and retinal ganglion cells in the POAG disease process. Given the emerging role of sCD44 as a marker and possible cause of cell death in POAG, a 10-mer HA peptide was effective in preventing sCD44 internalization and possible toxic effects on TM cells. The development of synthetic techniques may allow for application of peptide, peptide-like drugs, or derivatives to block the internalization of toxic proteins for the treatment of neurodegenerative diseases,65,66 such as POAG.
Acknowledgments
The authors thank Ruth Zelkha for image processing, Xiang Sheen for assistance with cell culture, and Beatrice Yue for reviewing the manuscript.
Footnotes
Supported by American Health Assistance Foundation; National Eye Institute Grant EY12043, core Grant EY01792; Rosemary O'Meara and Kathleen F. Connelly Memorial Funds; Illinois Society for the Prevention of Blindness; Midwest Eye-Banks and Transplantation Center; and an unrestricted grant from Research to Prevent Blindness.
Disclosure: M.J. Nolan, None; T. Koga, None; L. Walker, None; R. McCarty, None; A. Grybauskas, None; M.C. Giovingo, None; K. Skuran, None; P.V. Kuprys, None; P.A. Knepper, None
References
- 1. Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 363: 1711–1720 [DOI] [PubMed] [Google Scholar]
- 2. Knepper PA, Goossens W, Mayanil CSK. CD44H localization in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1998; 39: 673–680 [PubMed] [Google Scholar]
- 3. Knepper PA, Mayanil CS, Goossens W, et al. Aqueous humor in primary open-angle glaucoma contains an increased level of CD44S. Invest Ophthalmol Vis Sci. 2002; 43: 133–139 [PubMed] [Google Scholar]
- 4. Choi J, Miller AM, Nolan MJ, et al. sCD44 is cytotoxic to trabecular meshwork and retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci. 2005; 46: 214–222 [DOI] [PubMed] [Google Scholar]
- 5. Nolan MJ, Giovingo MC, Miller AM, et al. Aqueous humor sCD44 concentration and visual field loss in primary open-angle glaucoma. J Glaucoma. 2007; 16: 419–429 [DOI] [PubMed] [Google Scholar]
- 6. Cichy J, Pure E. The liberation of CD44. J Cell Biol. 2003; 161: 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okamoto I, Kawano Y, Murakami D, et al. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001; 155: 755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor KR, Yamasaki K, Radek KA, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem. 2007; 282: 18265–18275 [DOI] [PubMed] [Google Scholar]
- 9. Knepper PA, Samples JR, Yue BYJT. Biomarkers of primary open-angle glaucoma. Expert Rev Ophthalmol. 2010; 5: 731–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naor D, Nedvetzki S, Walmsley M, et al. CD44 involvement in autoimmune inflammations: the lesson to be learned from CD44-targeting by antibody or from knockout mice. Ann N Y Acad Sci. 2007; 1110: 233–247 [DOI] [PubMed] [Google Scholar]
- 11. Eshkar Sebban L, Ronen D, Levartovsky D, et al. The involvement of CD44 and its novel ligand galectin-8 in apoptotic regulation of autoimmune inflammation. J Immunol. 2007; 179: 1225–1235 [DOI] [PubMed] [Google Scholar]
- 12. Vachon E, Martin R, Plumb J, et al. CD44 is a phagocytic receptor. Blood. 2006; 107: 4149–4158 [DOI] [PubMed] [Google Scholar]
- 13. Vachon E, Martin R, Kwok V, et al. CD44-mediated phagocytosis induces inside-out activation of complement receptor-3 in murine macrophages. Blood. 2007; 110: 4492–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghatak S, Misra S, Toole BP. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J Biol Chem. 2002; 277: 38013–38020 [DOI] [PubMed] [Google Scholar]
- 15. Singleton PA, Salgia RL, Moitra J, et al. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem. 2007; 282: 30643–30657 [DOI] [PubMed] [Google Scholar]
- 16. Zhang M, Wang MH, Singh RK, Wells A, Siegal GP. Epidermal growth factor induces CD44 gene expression through a novel regulatory element in mouse fibroblasts. J Biol Chem. 1997; 272: 14139–14146 [DOI] [PubMed] [Google Scholar]
- 17. Mokbel TH, Ghanem AA, Kishk H, Arafa LF, El-Baiomy AA. Erythropoietin and soluble CD44 levels in patients with primary open-angle glaucoma. Clin Experiment Ophthalmol. 2010; 38: 560–565 [DOI] [PubMed] [Google Scholar]
- 18. Budak YU, Akdogan M, Huysal K. Aqueous humor level of sCD44 in patients with degenerative myopia and primary open-angle glaucoma. BMC Res Notes. 2009; 2: 224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knepper PA, Miller AM, Choi J, et al. Hypophosphorylation of aqueous humor sCD44 and primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2005; 46: 2829–2837 [DOI] [PubMed] [Google Scholar]
- 20. Knudson CB, Knudson W. Hyaluronan-binding proteins in development, tissue homeostasis, and disease. FASEB J. 1993; 7: 1233–1241 [PubMed] [Google Scholar]
- 21. Day AJ, Prestwich GD. Hyaluronan-binding proteins: tying up the giant. J Biol Chem. 2002; 277: 4585–4588 [DOI] [PubMed] [Google Scholar]
- 22. Tammi MI, Day AJ, Turley EA. Hyaluronan and homeostasis: a balancing act. J Biol Chem. 2002; 277: 4581–4584 [DOI] [PubMed] [Google Scholar]
- 23. Sy MS, Guo YJ, Stamenkovic I. Inhibition of tumor growth in vivo with a soluble CD44-immunoglobulin fusion protein. J Exp Med. 1992; 176: 623–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med. 1997; 186: 1985–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mohapatra S, Yang X, Wright JA, Turley EA, Greenberg AH. Soluble hyaluronan receptor RHAMM induces mitotic arrest by suppressing Cdc2 and cyclin B1 expression. J Exp Med. 1996; 183: 1663–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wisniewski HG, Naime D, Hua JC, Vilcek J, Cronstein BN. TSG-6, a glycoprotein associated with arthritis, and its ligand hyaluronan exert opposite effects in a murine model of inflammation. Pflugers Archiv. 1996; 431: R225–226 [DOI] [PubMed] [Google Scholar]
- 27. Rubinstein DB, Stortchevoi A, Boosalis M, et al. Receptor for the globular heads of C1q (gC1q-R, p33, hyaluronan-binding protein) is preferentially expressed by adenocarcinoma cells. Int J Cancer. 2004; 110: 741–750 [DOI] [PubMed] [Google Scholar]
- 28. Liu N, Lapcevich RK, Underhill CB, et al. Metastatin: a hyaluronan-binding complex from cartilage that inhibits tumor growth. Cancer Res. 2001; 61: 1022–1028 [PubMed] [Google Scholar]
- 29. Liu N, Xu XM, Chen J, et al. Hyaluronan-binding peptide can inhibit tumor growth by interacting with Bcl-2. Int J Cancer. 2004; 109: 49–57 [DOI] [PubMed] [Google Scholar]
- 30. Schapira AH. Mitochondrial diseases. Lancet. 2012; 379: 1825–1834 [DOI] [PubMed] [Google Scholar]
- 31. Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001; 106: 27–36 [DOI] [PubMed] [Google Scholar]
- 32. Giorgio V, Bisetto E, Soriano ME, et al. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009; 284: 33982–33988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pelchen-Matthews A, Armes JE, Griffiths G, Marsh M. Differential endocytosis of CD4 in lymphocytic and nonlymphocytic cells. J Exp Med. 1991; 173: 575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gardino AK, Smerdon SJ, Yaffe MB. Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin Cancer Biol. 2006; 16: 173–182 [DOI] [PubMed] [Google Scholar]
- 35. Wang X, Su B, Siedlak SL, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008; 105: 19318–19323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cha MY, Han SH, Son SM, et al. Mitochondria-specific accumulation of amyloid-β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One. 2012; 7: e34929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosales-Corral S, Acuna-Castroviejo D, Tan DX, et al. Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: a protective role for melatonin. Oxid Med Cell Longev. 2012; 2012: 843649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hansson Petersen CA, Alikhani N, Behbahani H, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008; 105: 13145–13150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bateman DA, Chakrabartty A. Cell surface binding and internalization of aβ modulated by degree of aggregation. Int J Alzheimers Dis. 2011; 2011: 962352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stamenkovic I, Yu Q. Shedding light on proteolytic cleavage of CD44: the responsible sheddase and functional significance of shedding. J Invest Dermatol. 2009; 129: 1321–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cichy J, Pure E. Cytokines regulate the affinity of soluble CD44 for hyaluronan. FEBS Lett. 2004; 556: 69–74 [DOI] [PubMed] [Google Scholar]
- 42. Lammich S, Okochi M, Takeda M, et al. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J Biol Chem. 2002; 277: 44754–44759 [DOI] [PubMed] [Google Scholar]
- 43. Lee SF, Srinivasan B, Sephton CF, et al. Gamma-secretase-regulated proteolysis of the Notch receptor by mitochondrial intermediate peptidase. J Biol Chem. 2011; 286: 27447–27453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thorne RF, Legg JW, Isacke CM. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J Cell Sci. 2004; 117: 373–380 [DOI] [PubMed] [Google Scholar]
- 45. Rajasagi M, von Au A, Singh R, Hartmann N, Zöller M, Marhaba R. Anti-CD44 induces apoptosis in T lymphoma via mitochondrial depolarization. J Cell Mol Med. 2010; 14: 1453–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eyster CA, Higginson JD, Huebner R, et al. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic. 2009; 10: 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marin MP, Esteban-Pretel G, Ponsoda X, et al. Endocytosis in cultured neurons is altered by chronic alcohol exposure. Tox Sci. 2010; 115: 202–213 [DOI] [PubMed] [Google Scholar]
- 48. Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropath Exp Neurol. 2007; 66: 873–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bolender N, Sickmann A, Wagner R, Meisinger C, Pfanner N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Reports. 2008; 9: 42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamm LK, Bartoldus I. Secondary structure of a mitochondrial signal peptide in lipid bilayer membranes. FEBS Lett. 1990; 272: 29–33 [DOI] [PubMed] [Google Scholar]
- 51. Tanudji M, Sjoling S, Glaser E, Whelan J. Signals required for the import and processing of the alternative oxidase into mitochondria. J Biol Chem. 1999; 274: 1286–1293 [DOI] [PubMed] [Google Scholar]
- 52. Demory ML, Boerner JL, Davidson R, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009; 284: 36592–36604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W. Pink1 and its {Delta}ψ-dependent cleavage product both localize to the outer mitochondrial membrane by a unique targeting mode. J Biol Chem. 2012; 287: 22969–22987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lynn BD, Turley EA, Nagy JI. Subcellular distribution, calmodulin interaction, and mitochondrial association of the hyaluronan-binding protein RHAMM in rat brain. J Neurosci Res. 2001; 65: 6–16 [DOI] [PubMed] [Google Scholar]
- 55. Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol. 2003; 5: 1083–1089 [DOI] [PubMed] [Google Scholar]
- 56. Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM. A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat Cell Biol. 2002; 4: 399–407 [DOI] [PubMed] [Google Scholar]
- 57. Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci. 2012; 107: 355–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: relevance to mitochondrial dysfunction in Alzheimer's and Parkinson's diseases. Biochim Biophys Acta. 2010; 1802: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Izzotti A, Longobardi M, Cartiglia C, Saccà SC. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma. PLoS One. 2011; 6: e14567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Abu-Amero KK, Morales J, Bosley TM. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006; 47: 2533–2541 [DOI] [PubMed] [Google Scholar]
- 61. Vaseva AV, Marchenko ND, Ji K, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012; 149: 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008; 134: 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang W, Fileta JB, Dobberfuhl A, et al. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci U S A. 2005; 102: 12242–12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu H, Sarnaik SM, Manole MD, et al. Increased cytochrome c in rat cerebrospinal fluid after cardiac arrest and its effects on hypoxic neuronal survival. Resuscitation. 2012; 83: 1491–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Witt KA, Davis TP. CNS drug delivery: opioid peptides and the blood-brain barrier. AAPS J. 2006; 8: 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Egleton RD, Davis TP. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx. 2005; 2: 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chou KC, Shen HB. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mPLoc 2.0. PLoS One. 2010; 5: e9931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chou KC, Shen HB. Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008; 3: 153–162 [DOI] [PubMed] [Google Scholar]
- 69. Chou KC, Shen HB. Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res. 2010; 6: 1728–1734 [DOI] [PubMed] [Google Scholar]
- 70. Lee SF, Srinivasan B, Sephton CF, et al. Gamma-secretase-regulated proteolysis of the Notch receptor by mitochondrial intermediate peptidase. J Biol Chem. 2011; 286: 27447–27453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000; 300: 1005–1016 [DOI] [PubMed] [Google Scholar]