

Abstract
Background
To sustain the effect of rivastigmine, a hydrophilic cholinesterase inhibitor, nanobased formulations were prepared. The efficacy of the prepared rivastigmine liposomes (RLs) in comparison to rivastigmine solution (RS) was assessed in an aluminium chloride (AlCl3)-induced Alzheimer’s model.
Methods
Liposomes were prepared by lipid hydration (F1) and heating (F2) methods. Rats were treated with either RS or RLs (1 mg/kg/day) concomitantly with AlCl3 (50 mg/kg/day).
Results
The study showed that the F1 method produced smaller liposomes (67.51 ± 14.2 nm) than F2 (528.7 ± 15.5 nm), but both entrapped the same amount of the drug (92.1% ± 1.4%). After 6 hours, 74.2% ± 1.5% and 60.8% ± 2.3% of rivastigmine were released from F1 and F2, respectively. Both RLs and RS improved the deterioration of spatial memory induced by AlCl3, with RLs having a superior effect. Further biochemical measurements proved that RS and RLs were able to lower plasma C-reactive protein, homocysteine and asymmetric dimethy-larginine levels. RS significantly attenuated acetylcholinesterase (AChE) activity, whereas Na+/K+-adenosine triphosphatase (ATPase) activity was enhanced compared to the AlCl3-treated animals; however, RLs succeeded in normalization of AChE and Na+/K+ ATPase activities. Gene-expression profile showed that cotreatment with RS to AlCl3-treated rats succeeded in exerting significant decreases in BACE1, AChE, and IL1B gene expression. Normalization of the expression of the aforementioned genes was achieved by coadministration of RLs to AlCl3-treated rats. The profound therapeutic effect of RLs over RS was evidenced by nearly preventing amyloid plaque formation, as shown in the histopathological examination of rat brain.
Conclusion
RLs could be a potential drug-delivery system for ameliorating Alzheimer’s disease.
Keywords: rivastigmine, Alzheimer’s disease, liposomes, rats, gene expression
Introduction
Alzheimer’s disease (AD), the most common form of dementia amongst the elderly, is described by impaired neuronal signaling, leading to a slow and progressive decline in cognitive functions and behavior.1 AD is characterized by the deposition of amyloid-β (Aβ) protein in the form of senile plaque deposits in brain regions, such as cortex and hippocampus. Aβ is derived from the proteolytic cleavage of amyloid precursor protein (APP) by two proteases, referred to as β- and γ-secretases. Neurotoxic Aβ formation is initiated by the rate limiting enzyme β-secretase (beta-site APP cleaving enzyme, BACE), which cleaves APP at the N-terminus of Aβ, and the remnant is then cleaved by γ-secretase to release Aβ. α-Secretase cleaves the APP within the Aβ sequence and precludes Aβ formation.2 Inflammation, a secondary event to the deposition of Aβ in the brain, starts as a host defense response to the damaging effects of Aβ contributing to neuronal degeneration.3 Activation of microglia may contribute to the neurodegenerative process through the release of proinflammatory cytokines, and other toxic products including reactive oxygen species (ROS).4 Furthermore, there is evidence that increased plasma concentration of homocysteine (Hcy) may contribute to the pathology of AD by increasing the plasma concentration of asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of nitric oxide synthase. Hcy has been shown to inhibit the activity of endothelial di methylarginine dimethylaminohydrolase (DDAH), the enzyme involved in ADMA hydrolysis, causing the accumulation of ADMA and the inhibition of nitric oxide synthesis.5
Aluminum (Al) exposure was associated with impairment in the cholinergic system, hence representing the way by which Al contributes to the pathological process in AD.6 Aluminum chloride (AlCl3) accumulated in specific brain regions was correlated with the degenerative changes. Therefore, Alinduced AD in rat is the most commonly utilized animal model to mimic human AD-like symptoms.7
AD is caused by reduced synthesis of the neurotransmitter acetylcholine (ACh).8 Acetylcholinesterase inhibitors (AChEIs) increase the availability of ACh through inhibition of its destruction, hence an enhancement of cholinergic transmission in the brain and improvement in the symptoms of AD.9
Rivastigmine (RIVA), a novel cholinesterase inhibitor, is used for the treatment of AD.10 RIVA was shown to be effective in reversing scopolamine-induced cognitive dysfunction11 and Al-induced biochemical dysfunction in rats.12 RIVA is distinct from other available cholinesterase inhibitors (donepezil and galantamine) in that it is a pseudoirreversible inhibitor of both AChE and butyrylcholinesterase (BuChE) rather than a rapidly reversible inhibitor of AChE alone.13 Extensive, saturable first-pass metabolism leads to reduced oral bioavailability, although it is completely absorbed. Patient compliance with an RIVA regimen might be problematic due to its short half-life (1.5 hours).14 Hence, a sustained-release formulation would improve treatment adherence and compliance, as a result of decreased side effects and simplified dosing regimens, respectively. Several efforts were made to sustain the RIVA effect by encapsulation in liposomes15,16 and nanoparticles.17,18
Liposomes are defined as bilayered lipid vesicles enclosing an aqueous volume.19 Liposomes compose a significant vesicular carrier system for therapeutic effectiveness in terms of duration of action, decrease in dose frequency, and delivering drugs at a higher efficacy and lower toxicity.20
Owing to the lipophilic nature of liposomes, they were considered an ideal brain-targeting drug-delivery system. The mechanisms by which liposomes cross the blood–brain barrier (BBB) could be passive diffusion through the lipophilic endothelial cells, endocytosis, or fusion with brain-capillary endothelial cells.21 Previous research compared the AChE inhibition effect of RIVA solution to that of RIVA liposomes (RLs) using the Ellman method, and reported that the highest AChE% inhibition values in both blood and brain of mice were after administration of RLs.22
The current study aimed to develop and characterize a safe liposomal formulation of RIVA and investigate the possible therapeutic potential of this nanobased formulation in comparison to conventional drug solution in AlCl3-treated rats.
Materials and methods
Materials
RIVA as hydrogen tartarate was a gift from Novartis Pharma (Cairo, Egypt). 1,2-Diacyl-sn-glycero-3-phosphocholine (from egg yolk, ≥99%) (PC), dihexadecyl phosphate (DCP), and cholesterol (Chol) were purchased from Sigma-Aldrich Chemicals (St Louis, MO, USA). AlCl3 was obtained from BDH Laboratories, Poole, UK. All other chemicals were of pure analytical grade.
Methods
Preparation of RIVA liposomes
RLs were prepared by two methods: the lipid layer hydration (LH) method23 and the heating method (HM).24 In both techniques, the lipids (PC, DCP, and Chol) were used in different molar ratios and the liposomes were loaded with different molar drug concentrations (Table 1). In the LH method, the lipids were dissolved in solvent mixture (chloroform:methanol in the ratio 2:1 v/v), attached to a rotary evaporator (Laborata 4000 efficient; Heidolph Instruments, Schwabach, Germany) at 40°C and rotated at 60 rpm under vacuum (HS-3000; Hahn Shin Scientific, Bucheon, South Korea) overnight until all the liquid evaporated to a thin, dry lipid film. Then 7 mL of drug solution (40 mg/mL) in phosphate-buffered saline (PBS, pH 7.4) were added to the dried film, vortexed (VSM-3; PRO Scientific, Oxford, CT, USA) for 5 minutes, and placed in a bath sonicator (S30H Elmasonic; Elma Hans Schmidbauer, Singen, Germany) for another 5 minutes. The final volume was adjusted to 10 mL, and the liposomal suspension was stored at 4°C overnight.
Table 1.
Composition of liposome formulations
| Formula | Ratio between lipids | Concentration of total lipids (mM) | Concentration of rivastigmine (mM) | Method of preparation |
|---|---|---|---|---|
|
| ||||
| PC:DCP:Chol | ||||
| F1 | 4:2:1 | 1.3 | 10 mM | Lipid hydration |
| F2 | 4:2:1 | 1.3 | 10 mM | Heating |
| F3 | 8:2:0 | 1.3 | 10 mM | Heating |
| F4 | 8:0:2 | 1.3 | 10 mM | Heating |
| F5 | 7:2:1 | 1.3 | 2.5 mM | Heating |
| F6 | 7:2:1 | 1.3 | 10 mM | Heating |
| F7 | 7:2:1 | 1.3 | 25 mM | Heating |
Abbreviations: PC, phosphatidylcholine; DCP, dihexadecyl phosphate; Chol, cholesterol.
The other method used to prepare the liposomes was a mod-ification of the HM, as follows. Each of the lipid ingredients was hydrated separately in 2 mL PBS pH 7.4 for 1 hour under nitrogen atmosphere at room temperature. Then, Chol dispersion was heated at 120°C and stirred at 1000 rpm (WiseStir MSH-20D; Daigger, Vernon Hills, IL, USA) for 30 minutes. The temperature was adjusted to 70°C, and glycerol (3% v/v) and the other lipids were added while stirring at 1000 rpm for another 15 minutes. The temperature was then adjusted to 60°C, and drug solution in PBS pH 7.4 was added to the above mixture and stirred for another 30 minutes. Liposome suspension obtained was left at room temperature under nitrogen without stirring to anneal for 1 hour and then the final volume was adjusted to 10 mL and kept overnight at 4°C.
Characterization of liposomes
Percent encapsulation efficiency
The entrapped RIVA inside the liposomes was separated from free drug by ultracentrifugation at 18,000 rpm for 1 hour at 4°C (Megafuge 1.0 R; Heraeus, Haneau, Germany), 0.1 mL of supernatant was diluted with PBS pH 7.4, and drug concentration was estimated by spectrophotometric determination of the second derivative of the drug at 262 nm (UV-1601 PC; Shimadzu, Kyoto, Japan).25 The percent encapsulation efficiency (EE%) of the drug was calculated as follows:
| (1) |
where Ct and Cf are the total and the free RIVA concentrations, respectively.
The intraday precision of the analysis method was evaluated by analyzing samples of different concentrations of RIVA (4, 16, and 40 μg/mL) in triplicates on the same day. The interday precision was evaluated from the same concentration on 3 consecutive days.
Vesicle-size and size-distribution determination
The mean diameter and particle-size distribution of vesicles of each sample was determined using dynamic light-scattering with a Zetasizer ZS (Malvern Instruments, Malvern, UK). The system was equipped with 4 mW helium/neon laser at 633 nm wavelength and measured the sample with noninvasive backscatter technology at a detection angle of 173° using DTS Nano version 6.12 software (Malvern instruments). All measurements were carried out at 25°C assuming 0.8872 cps and 1.330 as medium viscosity and refractive index respectively. Samples were diluted ten times with PBS pH 7.4 before measurement. Results were presented as an average diameter of the liposome suspension (z-average mean) against percent sample volume, and the polydispersity index (PDI), which is a measure of the width of the size distribution, was also deduced.
Optical microscopy
A thin layer of the diluted formulations was examined using an ordinary light microscope (Leica Imaging Systems, Cambridge, UK). Photomicrographs were taken using a digital camera (Victor, Yokohama, Japan).
Transmission electron microscopy
A drop of the diluted sample was placed on a copper grid (200-mesh, Science Services, Munich, Germany), left to dry in the air, and examined under a transmission electron microscope (TEM 1230; JEOL, Tokyo, Japan) at 80 k V.
Zeta potential
The charges on the vesicular surface were determined using the Zetasizer ZS (Malvern Instruments) at 25°C for 120 seconds using a combination of laser Doppler velocimetry and phase-analysis light scattering to measure particle electrophoretic mobility. The average zeta potential and conductivity of three samples of each formulation were determined.
In vitro release study
An in vitro release study was performed using vertical Franz diffusion cells with an effective area of 3.14 cm2. The donor compartment contained 1 mL of reconstituted liposome suspension of different formulations and was covered with parafilm. The receptor compartment contained 25 mL PBS pH 7.4 to maintain sink conditions, and was separated from the donor compartment by cellulose membrane with a molecular weight cutoff of 12,000–14,000 (Spectrum Medica, Los Angeles, CA, USA). The receptor compartment was maintained at 37°C and stirred at 60 rpm. At predetermined time intervals (0.5, 1, 2, 3, 6, 12, and 24 hours), samples (1 mL) were withdrawn from the sampling port and replaced with fresh buffer kept at the same temperature. RIVA concentration in samples was analyzed spectrophotometrically at 262 nm. For each formulation, drug release was studied in triplicate, and the mean cumulative percent of drug released was determined and compared to that from 1 mL control solution of RIVA in PBS pH 7.4 at a concentration of 4 mg/mL.
Acute-toxicity study
An in vivo acute-toxicity study of the optimum RL formulation compared to RIVA solution (RS) was carried out to assess their safety upon administration to rats. Thirty male albino rats of Wistar strain weighing 260 ± 20 g were used. Rats were divided into six groups of fve rats each. Three groups were administered RS subcutaneously, and the other three groups were administered RL subcutaneously. Three dose levels of RS and RL were tested in rats: 1, 10, and 20 mg/kg, and the lethal dose that killed half the number of animals (LD50) for both formulations over 24 hours was deduced. The rats were allowed to acclimatize for 1 week before the study and had free access to rat chow and water. The study was performed in accordance with ethical procedures and policies approved by the Animal Care and Use Committee of the Faculty of Pharmacy, Cairo University, Cairo, Egypt.
In vivo study
Experimental design
Male albino rats of Wistar strain weighing 260 ± 20 g were obtained from the animal house colony, National Research Center, Cairo, Egypt. Before starting the experiments, animals were allowed to acclimatize for 1 week and had access to rat chow and water ad libitum throughout the study. The experiments were performed in accordance with ethical procedures and policies approved by the Animal Care and Use Committee of the Faculty of Pharmacy, Cairo University, Cairo, Egypt, following the 18th World Medical Association General Assembly, Helsinki, June 1964, and updated by the 59th World Medical Association General Assembly, Seoul, October 2008. Rats were assigned into four groups, each containing twelve animals. Group I rats were administered ultrapure water orally and served as normal control. In group II, rats received oral AlCl3 (50 mg/kg/day). Group III and IV rats were treated with AlCl3 (50 mg/kg/day) concomitant with subcutaneous injection of RS and RL, respectively (1 mg/kg/day). The experiment lasted for 3 months.12
Morris water maze
After 3 months, spatial memory was measured by the Morris water-maze (MWM) test.26 The water maze consisted of a circular water tank (160 cm in diameter and 35 cm in height), which was divided by four fixed points on its perimeter to four quadrants. It contained an escape platform of 10 cm in diameter of the same color as the rest of the basin (to eliminate any false-positive results due to vision), placed in a constant quadrant of the basin throughout the trials and kept 1.5 cm below the water surface. Rats were placed at a start point in the middle of the rim of a quadrant not containing the escape area with their face to the wall. Animals had four trials per day separated by 10 minutes for 5 successive days, during which the times required to find the hidden platform were averaged.
Blood and tissue sampling
After an overnight fast and under anesthesia, blood samples were collected from retro-orbital venous plexus of all animals into ethylenediaminetetraacetic acid (EDTA) tubes. Plasma was separated and stored at –80°C until analysis of C-reactive protein (CRP), Hcy, and ADMA. Brains were rapidly dissected and washed thoroughly with ice-cold saline. Whole brains of four rats from each group were formalin-fixed and paraffin-embedded for subsequent histopathological examination. Whole brains of the remaining eight rats were midsagittally divided into two portions. The cerebral cortex of the right portion was microdissected and kept at –80°C for gene expression analysis. The left portion was homogenized in phosphate buffer (0.1 M, pH 7.4) containing 1 mM EDTA, 0.25 M sucrose, 10 mM KCl and 1 mM phenylmethanesulfonyl fluoride and centrifuged at 800× g for 5 minutes at 4°C. The resulting supernatant was recentrifuged at 12,000× g for 15 minutes at 4°C to obtain postmitochondrial supernatant for the assessment of AChE and Na+/K+-adenosine triphosphatase (ATPase).
Biochemical measurements
Plasma level of CRP was measured by latex turbidimetric assay using a Nephstar CRP kit supplied by Goldsite Diagnostics (Shenzhen, China). Plasma Hcy level was assessed using a homocysteine enzyme immunoassay kit (Axis-Shield, Heidelberg, Germany). ADMA was measured using an ADMA enzyme-linked immunosorbent assay kit (Immundiagnostik, Bensheim, Germany). Brain AChE activity was measured colorimetrically using a Quimica Clinica Aplicada SA kit (QCA, Amposta, Spain),27 whereas brain Na+/K+-ATPase activity was assessed following the method of Sovoboda and Mossinger.28 Total protein was measured according to the method of Lowry et al,29 using bovine serum albumin as standard.
Gene-expression analysis
Total RNA was isolated from cerebral cortex using Trizol reagent (Invitrogen, USA) and RNeasy® Mini Kit (Qiagen, Hilden, Germany). A reverse-transcription reaction was performed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time fluorescence-monitored polymerase chain reactions (PCRs) were performed using a StepOne Real-Time PCR System (Applied Biosystems). The detection of AChE (assay ID: Rn00596883_m1), interleukin (IL)-1β (assay ID: Rn00580432_m1), and BACE1 (assay ID: Rn00569988_m1) mRNAs was performed using primers and probes mix (TaqMan probes labeled with 6-carboxyfluorescein dihydrocyclopyrroloindole tripeptide minor groove binder [MGB]) (gene-expression assay from Applied Biosystems). The expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (VIC-MGB-labeled probe) was used as an endogenous control. TaqMan reactions were set up in optical 48-well reaction plates by using 25 μL of a mixture containing 12.5 μL of TaqMan Universal PCR Master Mix 2×, 1.25 μL of primers and probes mix of gene assay, 1.25 μL of primers and probes mix of GAPDH (20×, according to the manufacturer’s instructions), and 5 μL of nuclease-free water. Five microliters of the cDNA samples were subsequently added to each well. The temperature profile was as follows: 95°C for 10 minutes, then 95°C for 15 seconds and 60°C for 1 minute for 45 cycles.
As a relative quantitation, fold changes were calculated following the 2–ΔΔCt method using the manufacturer’s software. Cycle threshold (Ct) is the cycle number at which fluorescence signal, generated by the cleavage of the probe, crosses the threshold. For each sample, the Ct value of target-gene mRNA was normalized to the GAPDH endogenous control as ΔCt (ΔCt = Cttarget gene – CtGAPDH). The fold change of the target-gene mRNA in the experimental sample relative to control sample was determined by 2–ΔΔCt, where ΔΔCt = ACtexperimental control –ACtcontrol.
Statistical analysis
Data were presented as means ± standard error (SE). Statistical differences were assessed by analysis of variance and repeated-measures analysis of variance, followed by the Tukey–Kramer test using the InStat 2.04 statistical package (GraphPad Software, La Jolla, CA, USA). Probability was considered statistically significant at P < 0.05.
Results
Spectrophotometric determination of RIVA second derivative
RIVA concentration in samples was estimated by spectrophotometric determination of its second derivative at 262 nm on a scaling factor of 100 with respect to a constructed calibration curve over concentration range 4–48 μL/mL (n = 3), and the corresponding regression equation was computed with correlation coefficient (R2) of 0.999. The intra- and interday precision showed percent coefficient of variation (CV%) of less than 0.3, thus the adopted analysis method was reproducible and reliable. The components of the formulations did not interfere with determination of RIVA by this method.
Percent encapsulation efficiency
EE% of RIVA in liposomes was found to range from 75.8% ± 1.1% to 97.4% ± 2.5% (Table 2). The method of preparation of liposomes did not affect the percent of RIVA encapsulated in liposomes much, as F1, prepared by the LH method attained EE% of 91.2% ± 2.3%, while F2, prepared using HM, was 92.1% ± 1.4% (P > 0.05). The presence of Chol had an important effect on EE% of RIVA in liposomes, as F3, prepared without Chol, had a significantly lower EE% of RIVA (86.6% ± 0.6%) compared to F6, prepared with Chol (97.4% ± 2.5%) (P < 0.05). Results also showed that increasing the ratio of PC to DCP and Chol in formulation from 4:2:1 in F2 (92.1% ± 1.4%) to 7:2:1 in F6 (97.4% ± 2.4%) led to a significant difference in EE% of the drug in liposomes (P < 0.05). The initial amount of drug used to load the liposomes had a remarkable effect on EE% of liposomes. By increasing the loading RIVA amount from 2.5 mM (F5) to 10 mM (F6) there was a 1.3-fold increase in EE% of the drug from 75.8% ± 1.1% to 97.4% ± 2.5% (P < 0.05). On the other hand by the further increase of RIVA amount used to load the liposomes to 25mM (F7), no more increase in EE% of the drug was recorded (P > 0.05).
Table 2.
Characterization parameters of the prepared liposomes
| Formula | Mean EE ± SE (%) | Mean z-averagea ± SE (d · nm) | PDI | Mean zeta potential ± SE (mV) | Mean conductivity ± SE (mS/cm) |
|---|---|---|---|---|---|
| F1 | 91.2 ± 2.3 | 67.51 ± 14.2 | 0.627 | –24.5 ± 1.3 | 15.7 ± 0.3 |
| F2 | 92.1 ± 1.4 | 528.7 ± 15.5 | 0.706 | –21.7 ± 0.9 | 15.6 ± 0.1 |
| F3 | 86.6 ± 0.6 | 158.7 ± 9.4 | 0.733 | –14.0 ± 1.4 | 16.0 ± 0.1 |
| F4 | 84.4 ± 2.2 | 403.1 ± 11.7 | 0.614 | –6.6 ± 0.3 | 16.5 ± 0.2 |
| F5 | 75.8 ± 1.1 | 167.1 ± 11.4 | 0.755 | –23.0 ± 1.5 | 15.9 ± 0.1 |
| F6 | 97.4 ± 2.5 | 226.1 ± 12.2 | 0.612 | –25.1 ± 0.8 | 16.0 ± 0.1 |
| F7 | 94.6 ± 1.1 | 189.3 ± 14.5 | 0.645 | –24.6 ± 1.1 | 15.6 ± 0.2 |
Note:aAverage diameter of the vesicles.
Abbreviations: EE, encapsulation efficiency; SE, standard error; PDI, polydispersity index.
Vesicle-size and size-distribution determination
Liposomes prepared using the LH method (F1) attained remarkably smaller size (67.51 ± 14.2 nm) and narrower size distribution (PDI 0.627) than those prepared using HM (F2) (528.7 ± 15.5 nm; PDI 0.706) (P < 0.05) (Table 2). The presence of Chol affected the liposome vesicle size considerably, as the size of liposomes lacking Chol (F3) was 158.7 ± 9.4 nm compared to 226.1 ± 12.2 nm of F6, containing Chol (P < 0.05). Increasing the ratio of PC to DCP and Chol in the formulation from 4:2:1 in F2 to 7:2:1 in F6 led to a significant decrease in vesicle size of the liposomes from 528.7 ± 15.5 to 226.1 ± 12.2 nm (P < 0.05). Liposomes with lower drug concentration (F5) acquired vesicle size of 167.1 ± 11.4 nm, while that of higher drug concentration (F7) was 189.3 ± 14.5 nm.
Optical microscopy
The liposomes prepared using the LH method had a smooth surface, were spherical in shape, and existed mainly as single unilamellar vesicles (SUVs) (Figure 1A). Liposomes prepared using the HM also had a smooth surface, were nonaggregated, and spherical in shape, and some of the liposomes existed as multilamellar vesicles (MLVs) with relatively larger size (Figure 1B).
Figure 1.
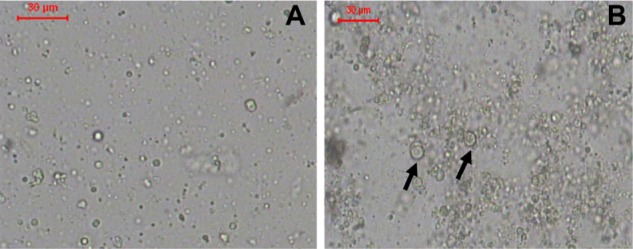
(A and B) Optical photomicrographs of liposomes. (A) F1, prepared by lipid hydration method and (B) F2, prepared by heating method.
Notes: Bar = 30 μm. Arrows point to multilamellar vesicles.
Transmission electron microscopy
TEM of the liposomes showed that both LH and HM generated spherical and discrete liposomes (Figure 2). TEM also showed that in some liposomes, the outer membrane could be obviously detected and enclosed the internal aqueous space.
Figure 2.
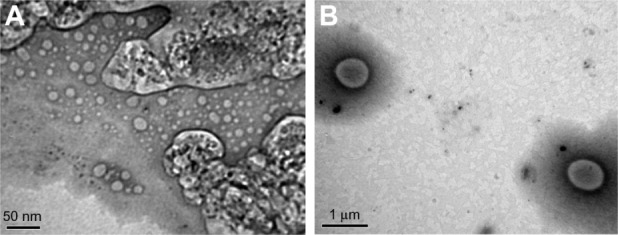
(A and B) Transmission electron micrographs of liposomes. (A) F1, prepared by lipid hydration method (bar = 50 nm) and (B) F2, prepared by heating method (bar = 1 μm).
Zeta potential
Zeta-potential values of the liposomes were found to be in the range of –6.6 ± 0.3 to –25.1 ± 0.8 m V, and the mean conductivity of the samples ranged from 15.6 ± 0.1 to 16.5 ± 0.2 mS/cm (Table 2). Results showed that the composition of liposome was the most prominent factor affecting the zeta-potential values of the vesicles rather than the method of liposome formulation. Formulation F4 (lacking the imparting negative charge agent DCP) attained the lowest zeta potential (–6.6 ± 0.3 mV). Furthermore, the zeta potential of formulations without Chol (F3, –14.0 ± 1.4 mV) was considerably lower than that of other formulations.
In vitro release study
Complete RIVA release from the prepared liposomes was attained after 24 hours compared to only 3 hours in the case of the control RIVA solution (Figure 3). The time taken to release 50% of the drug (t50%) from formulations (F7, 2.520 ± 0.2 hours and F1, 3.404 ± 0.5 hours) was significantly longer than that from control solution (0.585 ± 0.06 hours) (P < 0.05) (Table 3). Drug release from liposomes was fast in the first 3 hours and followed by a slow pattern afterwards, with F1 showing faster drug release than F2 (P < 0.05). After 6 hours, the percent of RIVA released from F1 was 74.2% ± 1.5%, whereas it reached 60.8% ± 2.3% from F2. After 3 hours, the percent of RIVA released from F2 and F6 formulations varied from 51.6% ± 3.2% to 54.1% ± 2.1% depending upon the lipid ratio used (P < 0.05). Besides, faster drug release was observed in the case of F3 and F4, prepared lacking either Chol or DCP, respectively, as after 6 hours, 79.7% ± 3.1% and 89.1% ± 3.6% of RIVA were released, respectively, in comparison to 60.8% ± 0.3% from F2, prepared with Chol and DCP (P < 0.05). By increasing the loading drug amount from 2.5 mM (F5) to 10 mM (F6), there was not much change in release profile of RIVA from liposomes, as the respective drug percentages released after 3 hours were 53.2% ± 1.8% and 54.1% ± 2.2% (P < 0.05). The further increase in loading amount of drug to 25 mM led to a significant increase in percent RIVA released to 86.5% ± 2.9% after 3 hours (P < 0.05).
Figure 3.
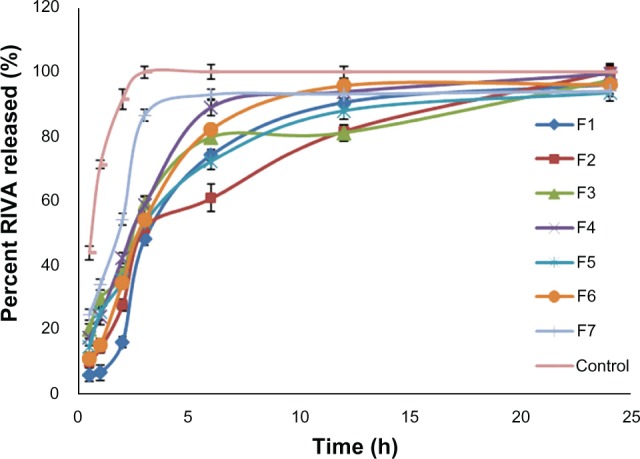
In-vitro release of rivastigmine (RIVA) from different liposome formulations in comparison to control solution (4 mg/mL) through cellulose membrane in phosphate-buffered saline (pH 7.4).
Table 3.
Kinetic analysis of release of rivastigmine from different liposome formulations
| Formulation | t50%a ± SE | R2-valueb of different models | Order of release | ||
|---|---|---|---|---|---|
|
|
|||||
| Zero | First | Diffusion | |||
| F1 | 3.330 ± 0.2 | 0.972 | 0.865 | 0.783 | Zero |
| F2 | 2.818 ± 0.3 | 0.999 | 0.964 | 0.912 | Zero |
| F3 | 2.534 ± 0.5 | 0.961 | 0.956 | 0.922 | Zero |
| F4 | 2.972 ± 0.3 | 0.999 | 0.981 | 0.980 | Zero |
| F5 | 3.404 ± 0.5 | 0.985 | 0.945 | 0.972 | Zero |
| F6 | 2.815 ± 0.4 | 0.989 | 0.985 | 0.954 | Zero |
| F7 | 2.520 ± 0.2 | 0.999 | 0.993 | 0.987 | Zero |
| Control solution | 0.585 ± 0.06 | 0.922 | 0.813 | 0.964 | Diffusion |
Notes:aTime taken for release of 50% rivastigmine from liposome formulations;
bcorrelation coefficient.
Data obtained from the release study were fitted to various kinetic equations, and results showed that drug release followed a zero order, as indicated by the higher (R2) values (Table 3). On the other hand, the release of the control solution of RIVA followed Higuchi diffusion kinetics, where the n-value (an exponent that characterizes the mechanism of release) was less than 0.45.
It was concluded from the in vitro characterization tests above that F6 was the optimum liposome formulation. It was assigned RL and was further utilized for in vivo studies.
Acute-toxicity study
The estimated LD50 value of RS administered subcutaneously in rats was 4.34 mg/kg, while that of RL was 5.26 mg/kg.
In vivo study
It should be noted that during the in vivo studies, no death occurred within animals administered 1 mg/kg RS or RL. No morphological or behavioral changes and no body-weight loss were detected in rats during the study period.
Effect of RS and RL on spatial memory deficits in AlCl3-treated rats
Impairment of spatial memory in the MWM was induced by AlCl3 administration, as indicated by the significant difference in escape latencies compared to control rats on days 2, 3, 4, and 5. Cotreatment with RS to AlCl3-treated rats significantly improved spatial memory, as evidenced by decreased escape latency on day 2 (45.9 ± 3 versus 58.8 ± 2.8), day 3 (38.5 ± 2 versus 51.3 ± 3), day 4 (33 ± 2.7 versus 45.1 ± 2.2), and day 5 (27.2 ± 1.6 versus 41.2 ± 2) when compared to the AlCl3-treated animals (P < 0.05). Restoration of spatial memory was achieved upon treating rats with RL concomitantly with AlCl3 (Figure 4).
Figure 4.
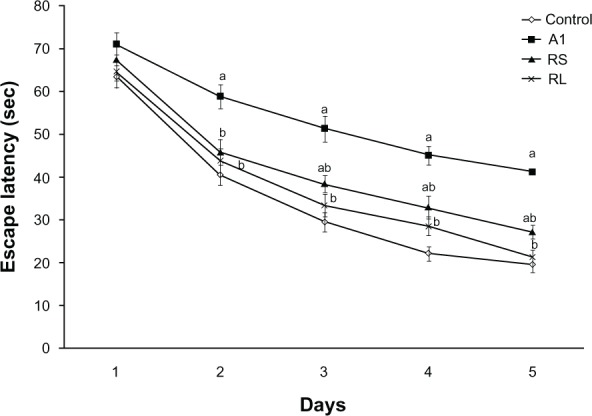
Effect of aluminium chloride (AlCl3) and combined treatment by rivastigmine solution (RS) and rivastigmine liposome formulation (RL) on the time (seconds) required to reach the platform in Morris water-maze test.
Notes: Values represent means ± standard error. aP < 0.05 relative to control group; bP < 0.05 relative to AlCl3-treated group.
Effect of RS and RL on some biochemical parameters in AlCl3-treated rats
AlCl3-treated rats showed significant increase in plasma CRP, Hcy, and ADMA of 112.5%, 142.1% and, 187.2%, respectively, compared to the control values. Also, a significant increase (60.7%) in the enzymatic activity of AChE in brain cortex was observed, whereas Na+/K+-ATPase activity was significantly decreased (58.2%) compared to the control group (P < 0.05). Cotreatment with RS and RL to AlCl3-treated rats provoked significant reduction in plasma level of CRP (30.5% and 32.9%, respectively), Hcy (31.5% and 40.2%, respectively), and ADMA (35.2% and 29.9%, respectively) when compared to the AlCl3-treated animals (P < 0.05), where a comparable effect of the two formulations was noticed. Regarding cortical enzyme activities, RS significantly attenuated AChE activity by 19.1%, whereas Na+/K+-ATPase activity was enhanced by 70.4% compared to the AlCl3-treated animals (P < 0.05). RL succeeded in normalization of AChE and Na+/K+-ATPase activities (Table 4).
Table 4.
Effect of rivastigmine solution (RS) and rivastigmine liposome formulation (RL) on plasma CRP, Hcy, ADMA, and brain AChE and Na+/K+-ATPase activities in AlCl3-treated rats
| Groups | Control | Al | Al + RS | Al + RL |
|---|---|---|---|---|
| Parameters | ||||
| CRP (mg/L) | 4.0 ± 0.42 | 8.5 ± 0.89a | 5.9 ± 0.38a,b | 5.7 ± 0.46a,b |
| Hcy (μmol/L) | 3.8 ± 0.47 | 9.2 ± 0.64a | 6.3 ± 0.64a,b | 5.5 ± 0.6a,b |
| ADMA (μmol/L) | 0.86 ± 0.11 | 2.47 ± 0.21a | 1.6 ± 0.19a,b | 1.73 ± 0.18a,b |
| AChE (U/mg protein) | 419.6 ± 33.2 | 674.3 ± 38.2a | 545 ± 26.6a,b | 494.5 ± 28.7b |
| Na+/K+ATPase (μmol/Pi/hour/mg protein) | 6.41 ± 0.6 | 2.67 ± 0.29a | 4.55 ± 0.37a,b | 5.8 ± 0.5b |
Notes:aSignificantly different from the control group;
bsignificantly different from AlCl3-treated group at P < 0.05. Values are expressed as means ± standard error of eight rats.
Abbreviations: CRP, C-reactive protein; Hcy, homocysteine; ADMA, asymmetric dimethylarginine; AChE, acetylcholinesterase; ATPase, adenosine triphosphatase; AlCl3, aluminium chloride.
Gene-expression profile
AlCl3-treated rats exhibited significant increase in the expression of BACE1 (316%), AChE, (370%), and IL1B (358%) genes compared to control rats (100%). Cotreatment with RS to AlCl3-treated rats succeeded in exerting significant decrease in BACE1 (63.2%), AChE (70.2%), and IL1B (70.3%) gene expression when compared to AlCl3-treated animals. Modulation of the expression of the aforementioned genes to normal levels was achieved by coadministration of RL to AlCl3-treated rats (Figure 5).
Figure 5.
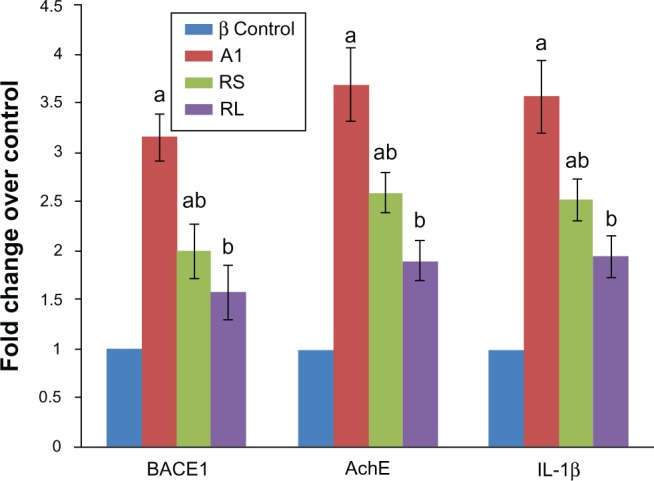
Real-time polymerase chain reaction analysis of beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), acetylcholinesterase (AChE), and interleukin 1β (IL-1β) mRNA in the studied groups (control, Al, AlCl3-treated rats; rivastigmine [RIVA] solution [RS], AlCl3-treated rats + RIVA solution; RIVA liposome formulation [RL], aluminium chloride [AlCl3]-treated rats + RL).
Notes: Changes in mRNA of each one of the target genes relative to GAPDH were determined by 2–ΔΔCt as fold change over control. Results are means ± standard error; n = 5. aSignificantly different from control group; bsignificantly different from AlCl3-treated group at P < 0.05.
Histopathological evaluation
Photomicrographs of brain sections of control rats showed normal nerve cells (Figure 6A). Brain sections of AlCl3-treated rats showed focal area of deep eosinophilic amyloid plaques of different sizes with brain necrosis (Figure 6B). Administration of RS to AlCl3-treated rats decreased the formation of amyloid plaques and necrotic neurons (Figure 6C); however, administration of RL protected rat brain by preventing amyloid plaque formation (Figure 6D).
Figure 6.
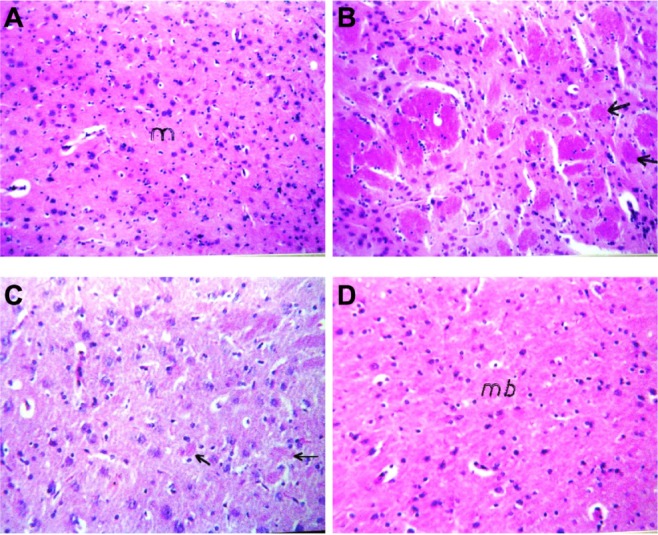
Photomicrographs of brain section of (A) control rats and (B) aluminium chloride (AlCl3)-treated rats. Arrows refer to focal area of deep eosinophilic round of amyloid plaque. (C) AlCl3-intoxicated rats treated with rivastigmine solution. Arrows refer to few small plaques in the mid brain with diffuse gliosis. (D) AlCl3-intoxicated rats treated with rivastigmine liposome formulation (hematoxylin and eosin 40x).
Discussion
A comparison between LH and HM used to prepare RL was set in this study. Liposomes prepared by the LH method suffered from major shortcomings by using volatile organic solvents to solubilize the lipids during their preparation, which might remain, in trace amounts, in the final preparation, contribute to toxicity, and influence the stability of the liposome formulations.30,31 It has been suggested that organic solvents can exert toxicity towards cells at both the molecular and the phase levels. Molecular toxicity caused by organic solvents was evidenced in enzyme inhibition, protein denaturation, and membrane modifications, such as membrane expansion, structure disorders, and permeability changes. Phase toxicity included extraction of nutrients, disruption of the cell components, and formation of emulsions and coating of cells.32 A correlation between the ability of some organic solvents to destabilize membrane proteins and their cytotoxicity was previously shown by Ivanov.33 Techniques such as gel filtration, dialysis, and vacuum could be utilized to decrease the concentration of the residual solvents in liposomes, though they would be difficult to apply on a large scale and time-consuming. Moreover, the level of these solvents in the final formulations must be assessed to ensure the clinical safety of the products.34 The HM, used to prepare liposomes without using toxic solvents, could be considered a safe and scalable method applied on an industrial scale. Mozafari and colleagues35 showed that nanoliposomes prepared by the HM were completely nontoxic toward cultured cells, while nanoliposomes prepared by a conventional method, using volatile solvents, showed significant levels of cytotoxicity using Chinese hamster ovary K1 cells. Moreover, using heat in the HM to formulate liposomes produced sterile liposomes and abolished the need for sterilization, hence reducing the manufacturing time and cost of liposome formulation.36 The phospholipid molecules and cholesterol used in the structure of lipid vesicles were safe ingredients obtained from natural sources, such as egg, soy, or milk. Glycerol used in the HM was a bioacceptable, nontoxic agent serving as an isotonizing agent in the liposomal preparations. Moreover, it acted as a dispersant, preventing coagulation or sedimentation of the vesicles, thereby enhancing the stability of the liposome preparations.37 Besides, the reason for choosing an anionic charge conferring agent (DCP) was due to several reports on the toxicity and complications of cationic charging agents.38,39
Results of drug encapsulation showed that the presence of Chol and the loading RIVA amount were two major factors affecting the EE% of drug inside the liposomes. RIVA was encapsulated in higher amounts inside formulation F6 than in F3, lacking Chol. It is well known that Chol modulates the fluidity of the lipid bilayer by preventing crystallization of the acyl chains of phospholipids and providing steric hindrance to their movement, thus increasing the stability of the vesicles in the presence of blood proteins and reducing the permeability of the liposomal lipid layer to enclosed drug.40,41 The positive effect of loading RIVA amount on EE% could be explained as follows: being a hydrophilic drug, the dissolved RIVA in the solution surrounding the liposomes during formation diffuses into the entrapped volume of the liposomes, so increasing RIVA concentration from 2.5 (F5) to 10 mM (F6), which led to a significant increase in EE% of the drug (P < 0.05). But as the RIVA amount was further increased to 25 mM (F7), no corresponding increase in EE% was observed. This was due to a limitation set by the volume entrapped inside the liposome. It is worth mentioning that the incorporation of the negatively charged DCP increased the interlamellar distance between the consecutive liposomal bilayers, leading to an increased drug EE% of the prepared liposomes.23
The preparation method affected the liposome size, with the largest liposomes produced by the HM. This is surprising, as different techniques produced liposomes with almost the same EE% of RIVA, although there was a large difference between the mean sizes of liposomes produced by the two techniques. This contradiction is attributed to the entrapped volume inside the liposomes, which determines the aqueous EE% of hydrophilic drugs and depends on liposome size, composition, and lamellarity. The hydrophilic drugs are entrapped in the aqueous volume of the liposomes. The HM produced some liposomes that existed as MLVs, which were characterized by the fact that part of their internal volume was occupied by the lipid bilayer existing as multilayers, leaving less aqueous spaces for the encapsulation of hydrophilic drugs. So even though liposomes produced by the HM were larger in size, they could not encapsulate more RIVA inside them than those produced by the LH method. Similar results were reported by Thompson et al,42 who prepared liposomes by different techniques and found that the liposomes prepared by both microfluidization and HMs had high entrapment efficiencies, although liposomes produced by microfluidization were smaller in size than those produced by the HM and a higher proportion of liposomes existing as SUVs. Formulation F2 had a wider size distribution than F1 due to the presence of MLVs in F2, which were known to have a wider size distribution than SUVs in F1.43
The zeta potential is correlated with in vitro and in vivo stability of liposome formulations. A minimum zeta potential of ±30 mV is required to create physically stable liposome suspension, which is stabilized by electrostatic repulsion only.44 High negative charges of zeta potential acquired by the prepared liposomes signified the electrostatic repulsion between vesicles carrying the same electrical charge and preventing their aggregation (Table 2).
By comparing the release of RIVA from liposomes to that from the control solution, it could be deduced that liposomes were able to sustain the release of the drug over 24 hours compared to only 3 hours in the case of the control solution. This was verified by the longer t50% of liposomes compared to that of the control solution (P < 0.05) (Table 3). The RIVA release profile from formulations was biphasic, as an initial fast drug release was noticed within the first 3 hours due to the release of the drug from the small vesicles, and was followed by slow release of the rest of the drug within 24 hours. The effect of method of formulation on the RIVA release profile could be explained as follows: the LH method produced smaller liposomes (F1) than those produced by the HM (F2). It was suggested that drug release from liposomes was controlled by their size, as small liposomes had larger curvature and looser lipid packing than larger ones, making them more readily attacked by blood components and releasing the encapsulated drug in a faster pattern.43 Results also showed that RIVA release increased by increasing the loading drug amount. Bayomi et al45 also reported that diltiazem release was faster from chitosan microspheres of higher drug content. The RIVA release from liposomes followed zero-order kinetics, which meant that drug release was independent on the initial drug concentration inside liposomes.
According to the acute-toxicity study carried out in rats, giving RIVA in liposome formulation decreased the toxicity of the drug compared to the free drug solution. This might be due to the fact that a drug substance in a liposome formulation exhibited a different pharmacokinetic and/or tissue distribution profile from the same drug substance in a nonliposomal formulation given by the same route of administration.
It is well established that Al is a neurotoxic agent that gradually accumulates in the hippocampus and cortex regions of rat brain following chronic exposure, affecting learning and memory functions. It has been reported that Al was found in senile plaques in brains of AD patients.46 Al crosses the BBB via specific high-affinity receptors for transferrin. Upon entering the brain, it affects the slow and fast axonal transports, induces inflammatory responses, and causes synaptic structural abnormalities, resulting in profound memory loss.47 The current results indicated that chronic AlCl3 administration resulted in progressive deterioration of spatial memory, as tested by MWM. It was demonstrated that intracerebral AlCl3 administration caused learning deficits in the MWM task in rabbits,48 which was in agreement with our findings. This phenomenon could be attributed to the ability of Al to interfere with downstream effector molecules, such as cyclic GMP, involved in long-term potentiation,49 thereby explaining the observed memory impairment and neurobehavioral deficits.
Our data revealed that plasma CRP, Hcy, and ADMA were significantly elevated. These findings are in harmony with those of Zaciragic et al50 and Ho et al.51 In brain tissue, the pathophysiology of CRP accumulation is complex because of the requirement of systemically produced CRP to cross the BBB. However, it has been established that during inflammatory conditions, the BBB becomes dysfunctional, enabling proteins normally only found in serum to enter the cerebrospinal fluid, so the transit of circulating CRP across the BBB is the most likely potential source of cerebral CRP.52 Strang et al53 demonstrated that amyloid plaques induced the dissociation of pentameric CRP to individual monomers, which possess strong proinflammatory properties not shared with pentameric CRP, and localizing inflammation to Alzheimer’s plaques. Regarding Hcy, it can penetrate the BBB through carrier receptor-mediated transport, and elevated plasma Hcy compromises the integrity of the BBB. Therefore, the brain concentration of Hcy should be similar to that of blood.54 Hyperhomocysteinemia can induce tau hyperphosphorylation and interrupt DNA repair in hippocampal neurons and make neurons more vulnerable to amyloid toxicity.55 Demethylation of the PS-1 (an important component of γ-secretase) promoter gene stimulates PS-1 expression, which in turn increases the production of Aβ. Zhang et al56 found that the levels of PS-1 mRNA and protein were increased in rats with hyperhomocysteinemia, suggesting that high Hcy may selectively disrupt the methylation of PS-1 promoter, leading to increased Aβ production. ADMA is an endogenous competitive inhibitor of nitric oxide synthase. It was found that Hcy inhibits DDAH, thereby increasing ADMA accumulation and reducing NO production in cultured neurons. This finding may explain the relationship between increased circulating Hcy and the development of cognitive dysfunction in AD.57
It has been shown that AChE activity and phospholipid composition of rat brain synaptic plasma membranes, as well as the Na+/K+-ATPase kinetics, were altered significantly with Al treatment.58 Consistent with the previous finding of Silva et al,59 we observed that AlCl3 administration caused a significant increase in AChE activity, whereas activity of Na+/K+-ATPase was inhibited in rat cerebral cortex. It was suggested that allosteric interaction between Al and the peripheral anionic site of the enzyme might lead to increased enzymatic activity6 Na+/K+-ATPase is a ubiquitous ion-transporter enzyme that is ATP-dependent. The marked inhibition of ATPase activity by Al administration may be attributed to Al-induced oxidative stress in cranial tissue.7
The current results revealed significant upregulation of BACE1 gene expression in brain cortex of AlCl3-treated rats. Castorina et al60 found that AlCl3 affected APP metabolism by enhancing Aβ-induced toxicity through the induction of BACE1 transcript level in differentiated neuroblastoma cells. Al is a known pro-oxidant, which can directly act by production of free radicals or indirectly by downregulating the expression of some genes linked to the antioxidant defense.7 It was concluded that APP transcription level is highly related to the oxidative level. In addition, metal ions may have an effect on APP transcription by directly binding to the 5′-untranslated region of APP transcript, and modulate some transcription factor, such as specificity protein 1, to regulate the expression of APP and/or BACE1.61 Equally important, Chen et al62 demonstrated that increased lipid peroxidation can lead to amyloidogenesis through upregulation of BACE1 expression in vivo.
Chronic inflammatory processes might contribute to the neurodegeneration associated with AD, by overexpression of cytokines such as IL-1β in activated microglia.63 Our results showed significant increase in IL1B and AChE gene expression in rat brains. Al neurotoxicity involved sequestering Al and iron followed by plaque formation in brain. Microglia is then activated in an attempt to destroy the plaques.64 Metal-mediated oxidative stress, such as that induced by Al, provoked significant upregulation of a family of genes known to drive proinflammatory neuropathology65 The promoters of these upregulated genes, such as IL-1β, are enriched in DNA binding sites for two stress-related transcription factors, hypoxia-inducible factor and nuclear factor kappa B (NF-κB), DNA binding proteins known to contribute to pathogenic processes associated with AD.66 Increased IL-1β levels in AD subjects may be responsible for enhancing AChE expression and activity, possibly explaining the cholinergic dysfunction observed in AD.67 Acute stress is known to induce expression of the AChE gene and to increase brain AChE activity. ACh has anti-inflammatory actions. Hence, elevated AChE concentrations will lead to a decrease in the levels of ACh, which could trigger the onset of low-grade systemic inflammation seen in AD. This “cholinergic anti-inflammatory pathway” mediated by ACh acts by inhibiting the production of tumor necrosis factor-a and IL-1β and suppresses the activation of NF-kB expression.68 Alteration of the aforementioned findings were confirmed by histopathological examination that revealed neuronal degeneration and brain necrosis after Al administration.
Increasing ACh concentration in the brain by modulating AChE activity is among the most promising therapeutic strategies. AChEIs may possibly contribute in delaying disease progression by interfering in the delicate balance of the cytokine cascade.69 Our data demonstrated that both RS and RL exerted significant improvement of cognitive effect, decrease in plasma CRP, Hcy, and ADMA, downregulation of BACE1, AChE, and IL1B gene expression and decreased AChE activity as compared to AlCl3-treated animals. The overcoming effect of RL compared to RS was manifested by normalization of cognitive effect as assessed by MWM (Figure 4), gene expression (Figure 5), and AChE activity (Table 4). The anti-inflammatory effect of RL and RS, evidenced by significant decrease in systemic CRP, Hcy and ADMA observed in our study, resulted from its ability to downregulate the inflammatory activation of immune cells through an increased ACh level acting on nicotinic α7 receptors, as shown by Nizri et al.70 It has been reported that administration of AChE inhibitors almost completely blocked the LPS-induced increase in IL-1β production suggesting that increased ACh levels within the brain directly inhibit IL-1β production. It is also possible that the peripheral increase in ACh suppressed the production of pro-inflammatory cytokines by macrophages, which, in turn, decreased the inflammatory signal to the brain, resulting in attenuated production of IL-1β.71 Overexpression of human AChE in neurons of transgenic mice produces progressive cognitive deterioration, as assessed by the MWM, suggesting that downregulation of cholinergic function is detrimental to spatial memory. With positron emission tomography techniques, it was observed that cerebral glucose metabolism declined in untreated AD patients, whereas glucose metabolism in RIVA-treated subjects was stabilized. These differences were also correlated with cognitive performance.72
AChE was shown to regulate APP processing and to accelerate assembly of amyloid peptide into β-amyloid fibrils in vitro,73 suggesting a link between AChE overexpression and β-amyloid formation. RIVA treatment for 21 days shifted APP processing towards the a-secretase pathway in rodents.74 The BACE1 promoter contains a number of transcription factor-binding sites, such as NF-κB. A lack of astrocytic BACE1 expression in the absence of inflammatory signaling was demonstrated whereas under conditions of chronic stress astrocytic BACE1 expression was induced.75 The anti-inflammatory effect of both RS and RL, observed in our study, might explain the downregulation of BACE1 expression.
Our data demonstrated that both RS and RL prevented the decline in Na+/K+-ATPase activity associated with Al treatment, with RL exerting a more profound effect than RS. Peroxynitrite, a reactive oxidant produced by the reaction between NO and superoxide, was found to induce inhibition of membrane Na+/K+-ATPase activity. Kumar and Kumar76 demonstrated that RIVA increases Na+/K+-ATPase activity through decreasing nitrite level, suggesting an antioxidant-like effect of RIVA. Bihaqi et al12 showed that cotreatment with RIVA during Al administration prevented the depletion of GSH level significantly, suggesting that the drug was efficient in preventing oxidative damage and associated alterations. The superior effect of RL over RS, reported in our study, might be attributed to the method of liposome preparation as it contained PC, a natural lipid component adding liphophilicity to the preparation, hence increasing drug permeability through brain tissue with more sustained effect.
Administration of RS succeeded in ameliorating biochemical and molecular aspects, which was reflected on histopathological investigation. Microscopic examination of brain sections showed marked reduction in neurodegeneration with improvement in neuronal features. Surprisingly, nanobased formulation of RL abolished the amyloidogenic effect of AlCl3 administration.
Conclusion
In summary, the HM was successful in preparing safe liposomes, showing high encapsulation efficiency of RIVA and sustaining its release for a prolonged period of time, thus offering patient compliance. The present results indicated that the RL formulation resulted in faster memory regain and amelioration of metabolic disturbances in AlCl3-treated rats. The RIVA nanobased formulation exerted superior influence over conventional drug solutions and could be a potential drug-delivery system for treating Alzheimer’s disease. However, further clinical studies are needed for extrapolating the experimental animal results to humans.
Acknowledgments
The authors would like to thank Dr Adel Bakir (Professor of Pathology, Faculty of Veterinary Medicine, Cairo University, Egypt) for his kind cooperation in the histopathological examinations involved in this research.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mount C, Downton C. Alzheimer disease: progress or proft? Nat Med. 2006;12:780–784. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- 2.Sinha S, Lieberburg I. Cellular mechanisms of beta-amyloid protein production and secretion. Proc Natl Acad Sci U S A. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Scarano F, Baltuch Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 5.Böger RH, Bode-Böger SM, Sydow K, Heistad DD, Lentz SR. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in monkeys with hyperhomocyst(e) inemia or hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:1557–1564. doi: 10.1161/01.atv.20.6.1557. [DOI] [PubMed] [Google Scholar]
- 6.Gulya K, Rakonczay Z, Kása P. Cholinotoxic effects of aluminum in rat brain. J Neurochem. 1990;54:1020–1026. doi: 10.1111/j.1471-4159.1990.tb02352.x. [DOI] [PubMed] [Google Scholar]
- 7.Garcia T, Esparza JL, Nogués MB, Romeu M, Domingo JL, Gomez M. Oxidative stress status and RNA expression in hippocampus of an animal model of Alzheimer’s disease after chronic exposure to aluminum. Hippocampus. 2010;20:218–225. doi: 10.1002/hipo.20612. [DOI] [PubMed] [Google Scholar]
- 8.Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287:2335–2338. doi: 10.1001/jama.287.18.2335. [DOI] [PubMed] [Google Scholar]
- 9.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 10.Raschetti R, Albanese E, Vanacore N, Maggini M. Cholinesterase inhibitors in mild cognitive impairment: a systematic review of randomised trials. PLoS Med. 2007;4:e338. doi: 10.1371/journal.pmed.0040338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang RH, Bejar C, Weinstock M. Gender differences in the effect of rivastigmine on brain cholinesterase activity and cognitive function in rats. Neuropharmacology. 2000;39:497–506. doi: 10.1016/s0028-3908(99)00157-4. [DOI] [PubMed] [Google Scholar]
- 12.Bihaqi SW, Sharma M, Singh AP, Tiwari M. Neuroprotective role of Convolvulus pluricaulis on aluminium induced neurotoxicity in rat brain. J Ethnopharmacol. 2009;124:409–415. doi: 10.1016/j.jep.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 13.Camps P, Muñoz-Torrero D. Cholinergic drugs in pharmacotherapy of Alzheimer’s disease. Mini Rev Med Chem. 2002;2:11–25. doi: 10.2174/1389557023406638. [DOI] [PubMed] [Google Scholar]
- 14.Novartis Pharmaceuticals. Exelon monograph. East Hanover (NJ): Novartis Pharmaceuticals; 2004. [Google Scholar]
- 15.Arumugam K, Subramanian GS, Mallayasamy SR, Averineni RK, Reddy MS, Udupa N. A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm. 2008;58:287–297. doi: 10.2478/v10007-008-0014-3. [DOI] [PubMed] [Google Scholar]
- 16.Degim Z, Mutlu MB, Yilmaz S, Eşsiz D. Investigation of liposome formulation effects on rivastigmine transport through human colonic adenocarcinoma cell line (CACO-2) Pharmazie. 2010;65:32–40. [PubMed] [Google Scholar]
- 17.Joshi SA, Chavhan SS, Sawant KK. Rivastigmine-loaded PLGA and PBCA nanoparticles: preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur J Pharm Biopharm. 2010;76:189–199. doi: 10.1016/j.ejpb.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Sahni JK, Doggui S, Ali J, Baboota S, Dao L, Ramassamy C. Neurotherapeutic applications of nanoparticles in Alzheimer’s disease. J Control Release. 2011;152:208–231. doi: 10.1016/j.jconrel.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Allen TM, McAllister L, Mausolf S, Gyorffy E. Liposome–cell interactions. A study of the interactions of liposomes containing entrapped anti-cancer drugs with the EMT6, S49 and AE1 (transport-deficient) cell lines. Biochim Biophys Acta. 1981;643:346–362. doi: 10.1016/0005-2736(81)90080-8. [DOI] [PubMed] [Google Scholar]
- 20.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 21.Brasnjevic I, Steinbusch H, Schmitz C, Martinez-Martinez P. Delivery of peptide and protein drugs over the blood–brain barrier. Prog Neurobiol. 2009;87:212–251. doi: 10.1016/j.pneurobio.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Mutlu NB, Değim Z, Yilmaz Ş, Eşsiz D, Nacar A. New perspective for the treatment of Alzheimer diseases: liposomal rivastigmine formulations. Drug Dev Ind Pharm. 2011;37:775–789. doi: 10.3109/03639045.2010.541262. [DOI] [PubMed] [Google Scholar]
- 23.Vemuri S, Rhodes CT. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm Acta Helv. 1995;70:95–111. doi: 10.1016/0031-6865(95)00010-7. [DOI] [PubMed] [Google Scholar]
- 24.Mozafari MR, Reed CJ, Rostron C, Kocum C, Piskin E. Formation and characterisation of non-toxic anionic liposomes for delivery of therapeutic agents to the pulmonary airways. Cell Mol Biol Lett. 2002;7:243–244. [PubMed] [Google Scholar]
- 25.Salem MA, El-Kosasy AM, El-Bardicy MG, Abd El-Rahman MK. Spectrophotometric and spectrodensitometric methods for the determination of rivastigmine hydrogen tartrate in presence of its degradation product. Drug Test Anal. 2010;2:225–233. doi: 10.1002/dta.121. [DOI] [PubMed] [Google Scholar]
- 26.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 27.den Blaauwen DH, Poppe WA, Tritschler W. Cholinesterase (EC 3.1.1.8) with butyrylthiocholine-iodide as substrate: references depending on age and sex with special reference to hormonal effects and pregnancy. J Clin Chem Clin Biochem. 1983;21:381–386. [PubMed] [Google Scholar]
- 28.Sovoboda P, Mossinger B. Catecholamines and brain microsomal Na, K-adenosinetriphosphatase - I. Protection against lipoperoxidative damage. Biochem Pharmacol. 1981;30:427–432. doi: 10.1016/0006-2952(81)90626-2. [DOI] [PubMed] [Google Scholar]
- 29.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 30.Kikuchi H, Yamauchi H, Hirota S. A polyol dilution method for mass production of liposomes. J Liposome Res. 1994;4:71–91. [Google Scholar]
- 31.Cortesi R, Esposito E, Gambarin S, Telloli P, Menegatti E, Nastruzzi C. Preparation of liposomes by reverse-phase evaporation using alternative organic solvents. J Microencapsul. 1999;16:251–256. doi: 10.1080/026520499289220. [DOI] [PubMed] [Google Scholar]
- 32.Bar R, Laane C, Tramper J. Phase toxicity in a water-solvent two-liquid phase microbial system. In: Lilly MD, editor. Biocatalysis in Organic Media. Amsterdam, Netherlands: Elsevier Science; 1987. pp. 147–153. [Google Scholar]
- 33.Ivanov IT. Rapid method for comparing the cytotoxicity of organic solvents and their ability to destabilize proteins of the erythrocyte membrane. Pharmazie. 2001;56:808–809. [PubMed] [Google Scholar]
- 34.Dwivedi AM. Residual solvent analysis in pharmaceuticals. Pharm Tech Eur. 2002;14:42–46. [Google Scholar]
- 35.Mozafari MR, Reed CJ, Rostron C. Cytotoxicity evaluation of anionic nanoliposomes and nanolipoplexes prepared by the heating method without employing volatile solvents and detergents. Pharmazie. 2007;62:205–209. [PubMed] [Google Scholar]
- 36.Mozafari MR, Reed CJ, Rostron C, Kocum C, Piskin E. Construction of stable anionic liposome-plasmid particles using the heating method: a preliminary investigation. Cell Mol Biol Lett. 2002;7:923–927. [PubMed] [Google Scholar]
- 37.Mozafari MR. Nanoliposomes: preparation and analysis. In: Weissig V, editor. Liposomes: Methods and Protocols. Volume 1: Pharmaceutical Nanocarriers. Vol. 2010. New York: Springer; p. 48. [Google Scholar]
- 38.Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000;17:521–525. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]
- 39.Omidi Y, Barar J, Akhtar S. Toxicogenomics of cationic lipid-based vectors for gene therapy: impact of microarray technology. Curr Drug Deliv. 2005;2:429–441. doi: 10.2174/156720105774370249. [DOI] [PubMed] [Google Scholar]
- 40.New RRC. Preparation of liposomes. In: New RRC, editor. Liposomes: A Practical Approach. New York: Oxford University Press; 1990. pp. 36–90. [Google Scholar]
- 41.Reineccius G. Liposomes for controlled release in the food industry. In: Risch S, Reineccius G, editors. Encapsulation and Controlled Release of Food Ingredients. Washington: American Chemical Society; 1995. pp. 113–131. [Google Scholar]
- 42.Thompson AK, Mozafari MR, Singh H. The properties of liposomes produced from milk fat globule membrane material using different techniques. Lait. 2007;87:349–360. [Google Scholar]
- 43.Nagayasu A, Uchiyama K, Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev. 1999;40:75–87. doi: 10.1016/s0169-409x(99)00041-1. [DOI] [PubMed] [Google Scholar]
- 44.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 45.Bayomi MA, Al-Suwaych SA, El-Helw AM, Mesnad AF. Preparation of casein chitosan microspheres containing diltiazem hydrochloride by an aqueous coacervation technique. Pharm Acta Helv. 1998;7:187–192. doi: 10.1016/s0031-6865(98)00020-x. [DOI] [PubMed] [Google Scholar]
- 46.McLachlan DR, Bergeron C, Smith JE, Boomer D, Rifat SL. Risk for neuropathologically confirmed Alzheimer’s disease and residual aluminum in municipal drinking water employing weighted residential histories. Neurology. 1996;46:401–405. doi: 10.1212/wnl.46.2.401. [DOI] [PubMed] [Google Scholar]
- 47.Campbell A, Becaria A, Lahiri DK, Sharman K, Bondy SC. Chronic exposure to aluminium in drinking water increases inflammatory parameters selectively in the brain. J Neurosci Res. 2004;75:565–572. doi: 10.1002/jnr.10877. [DOI] [PubMed] [Google Scholar]
- 48.Rabe A, Lee MH, Shek J, Wisniewski HM. Learning deficit in immature rabbits with aluminium-induced neurofibrillary changes. Exp Neurol. 1982;76:441–446. doi: 10.1016/0014-4886(82)90220-5. [DOI] [PubMed] [Google Scholar]
- 49.Canales JJ, Corbalán R, Montoliu C, et al. Aluminium impairs the glutamate-nitric oxide-cGMP pathway in cultured neurons and in rat brain in vivo: molecular mechanisms and implications for neuropathology. J Inorg Biochem. 2001;87:63–69. doi: 10.1016/s0162-0134(01)00316-6. [DOI] [PubMed] [Google Scholar]
- 50.Zaciragic A, Lepara O, Valjevac A, et al. Elevated serum C-reactive protein concentration in Bosnian patients with probable Alzheimer’s disease. J Alzheimers Dis. 2007;12:151–156. doi: 10.3233/jad-2007-12204. [DOI] [PubMed] [Google Scholar]
- 51.Ho YS, Yu MS, Yang XF, So KF, Yuen WH, Chang RC. Neuroprotective effects of polysaccharides from wolfberry, the fruits of Lycium barbarum, against homocysteine induced toxicity in rat cortical neurons. J Alzheimers Dis. 2010;19:813–827. doi: 10.3233/JAD-2010-1280. [DOI] [PubMed] [Google Scholar]
- 52.Stolp HB, Dziegielewska KM. Role of developmental inflammation and blood–brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol. 2009;35:132–146. doi: 10.1111/j.1365-2990.2008.01005.x. [DOI] [PubMed] [Google Scholar]
- 53.Strang F, Scheichl A, Chen YC, et al. Amyloid plaques dissociate pentameric to monomeric C-reactive protein:A novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol. 2012;22:337–346. doi: 10.1111/j.1750-3639.2011.00539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamath AF, Chauhan AK, Kisucka J, et al. Elevated levels of Hcy compromise blood brain barrier integrity in mice. Blood. 2006;107:591–593. doi: 10.1182/blood-2005-06-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruman II, Kumaravel TS, Lohani A, et al. Folic acid deficiency and Hcy impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of AD. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang CE, Wei W, Liu YH, Peng JH, Tian Q, Handy DE. Hyperhomocysteinemia increases β amyloid by enhancing expression of γ-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol. 2009;174:1481–1491. doi: 10.2353/ajpath.2009.081036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selley ML. Homocysteine increases the production of asymmetric dimethylarginine in cultured neurons. J Neurosci Res. 2004;77:90–93. doi: 10.1002/jnr.20070. [DOI] [PubMed] [Google Scholar]
- 58.Swegert CV, Dave KR, Katyare SS. Effect of aluminium induced Alzheimer like condition on oxidative energy metabolism in rat liver, brain and heart mitochondria. Mech Ageing Dev. 1999;112:27–42. doi: 10.1016/s0047-6374(99)00051-2. [DOI] [PubMed] [Google Scholar]
- 59.Silva VS, Nunes MA, Cordeiro JM, et al. Comparative effects of aluminium and ouabain on synaptosomal choline uptake, acetylcholine release and (Na+/K+) ATPase. Toxicology. 2007;236:158–177. doi: 10.1016/j.tox.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 60.Castorina A, Tiralongo A, Giunta S, Carnazza ML, Scapagnini G, D’Agata V Early effects of aluminum chloride on beta-secretase mRNA expression in a neuronal model of beta-amyloid toxicity. Cell Biol Toxicol. 2010;26:367–377. doi: 10.1007/s10565-009-9149-3. [DOI] [PubMed] [Google Scholar]
- 61.Rogers JT, Randall JD, Cahill CM, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 62.Chen L, Na R, Gu M, Richardson A, Ran Q. Lipid peroxidation upregulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer’s disease. J Neurochem. 2008;107:197–207. doi: 10.1111/j.1471-4159.2008.05603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61:255–260. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 64.van Rensburg SJ, Daniels WM, Potocnik FC, van Zyl JM, Taljaard JJ, Emsley RA. A new model for the pathophysiology of Alzheimer’s disease. Aluminum toxicity is exacerbated by hydrogen peroxide and attenuated by an amyloid protein fragment and melatonin. S Afr Med J. 1997;87:1111–1115. [PubMed] [Google Scholar]
- 65.Lukiw WJ, Percy ME. Nanomolar aluminum induces pro-inflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem. 2005;99:1895–1898. doi: 10.1016/j.jinorgbio.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 66.Taylor JM, Crack PJ. Impact of oxidative stress on neuronal survival. Clin Exp Pharmacol Physiol. 2004;31:397–406. doi: 10.1111/j.1440-1681.2004.04017.x. [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Liu L, Kang J, et al. Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000;20:149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pavlov VA, Tracey KJ. Controlling inflammation: the cholinergic antiinflammatory pathway. Biochem Soc Trans. 2006;34:1037–1040. doi: 10.1042/BST0341037. [DOI] [PubMed] [Google Scholar]
- 69.Gambi F, Reale M, Iarlori C, et al. Alzheimer patients treated with an AChE inhibitor show higher IL-4 and lower IL-1 beta levels and expression in peripheral blood mononuclear cells. J Clin Psychopharmacol. 2004;24:314–321. doi: 10.1097/01.jcp.0000125683.74595.2f. [DOI] [PubMed] [Google Scholar]
- 70.Nizri E, Irony-Tur-Sinai M, Faranesh N, et al. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol. 2008;203:12–22. doi: 10.1016/j.jneuroim.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 71.Pollak Y, Gilboa A, Menachem O, Ben-Hur T, Soreq H, Yirmiya R. Acetylcholinesterase inhibitors reduce brain and blood interleukin-1 β production. Ann Neurol. 2005;57:741–745. doi: 10.1002/ana.20454. [DOI] [PubMed] [Google Scholar]
- 72.Stefanova E, Wall A, Almkvist O, et al. Longitudinal PET evaluation of cerebral glucose metabolism in rivastigmine treated patients with mild Alzheimer’s disease. J Neural Transm. 2006;113:205–218. doi: 10.1007/s00702-005-0312-6. [DOI] [PubMed] [Google Scholar]
- 73.Inestrosa NC, Alvarez A, Perez CA, et al. Acetylcholinesterase accelerates assembly of amyloid-b-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 74.Bailey JA, Ray B, Greig NH, Lahiri DK. Rivastigmine lowers Ab and increases sAPPa levels, which parallel elevated synaptic markers and metabolic activity in degenerating primary rat neurons. PLoS One. 2011;6:e21954. doi: 10.1371/journal.pone.0021954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hartlage-Rübsamen M, Zeitschel U, Apelt J, et al. Astrocytic expression of the Alzheimer’s disease beta secretase (BACE1) is stimulus-dependent. Glia. 2003;41:169–179. doi: 10.1002/glia.10178. [DOI] [PubMed] [Google Scholar]
- 76.Kumar P, Kumar A. Protective effect of rivastigmine against 3-nitropropionic acid-induced Huntington’s disease like symptoms: possible behavioural, biochemical and cellular alterations. Eur J Pharmacol. 2009;615:91–101. doi: 10.1016/j.ejphar.2009.04.058. [DOI] [PubMed] [Google Scholar]