

Abstract
Biological network data, such as metabolic-, signaling- or physical interaction graphs of proteins are increasingly available in public repositories for important species. Tools for the quantitative analysis of these networks are being developed today. Protein network-based drug target identification methods usually return protein hubs with large degrees in the networks as potentially important targets. Some known, important protein targets, however, are not hubs at all, and perturbing protein hubs in these networks may have several unwanted physiological effects, due to their interaction with numerous partners. Here, we show a novel method applicable in networks with directed edges (such as metabolic networks) that compensates for the low degree (non-hub) vertices in the network, and identifies important nodes, regardless of their hub properties. Our method computes the PageRank for the nodes of the network, and divides the PageRank by the in-degree (i.e., the number of incoming edges) of the node. This quotient is the same in all nodes in an undirected graph (even for large- and low-degree nodes, that is, for hubs and non-hubs as well), but may differ significantly from node to node in directed graphs. We suggest to assign importance to non-hub nodes with large PageRank/in-degree quotient. Consequently, our method gives high scores to nodes with large PageRank, relative to their degrees: therefore non-hub important nodes can easily be identified in large networks. We demonstrate that these relatively high PageRank scores have biological relevance: the method correctly finds numerous already validated drug targets in distinct organisms (Mycobacterium tuberculosis, Plasmodium falciparum and MRSA Staphylococcus aureus), and consequently, it may suggest new possible protein targets as well. Additionally, our scoring method was not chosen arbitrarily: its value for all nodes of all undirected graphs is constant; therefore its high value captures importance in the directed edge structure of the graph.
Introduction
Methods analyzing biological networks are gaining significant interest because of their availability in large public repositories [1]–[9]. Finding important nodes in these protein-protein interaction or metabolic networks may lead to the identification of novel drug targets. The FDA approved drugs target presently only 324 human and pathogen proteins [10] from at least tens of thousands of possible proteins, therefore any well-founded method that may help to identify new ones has a substantial value.
Selecting important nodes that would serve as drug targets is a difficult task. In the literature, important nodes frequently means nodes with high degree (i.e., with many connecting edges, leading to a great number of neighboring nodes); these nodes are called “hubs” and “superhubs” [11], [12]. The proteins, corresponding to these hubs are mainly catalyzing vital biochemical reactions in metabolic networks [13] or their neighbor-set are robust: they are hardly changed in biological processes [12].
Targeting hub proteins with numerous vital functions with inhibitors may lead to unwanted off-target effects [14], [15] in the living cell, since any interventions involving these hub proteins may effect a large number of other processes and proteins as well.
In the present study we restrict our attention to metabolic networks: here the nodes are biochemical reactions, and reactions A and B are connected with a directed edge (A,B) if a product of reaction A enters reaction B as a substrate or a co-factor. In a given organism reactions can be corresponded to enzymes, catalyzing them. This correspondence can be made easily by inspecting the underlying database: we applied the KEGG database [16] for this mapping.
In the analysis of metabolic networks, large or very large degree nodes (hubs or superhubs, corresponding to “currency metabolites” [17]) usually need special attention if we want to compensate for their overwhelming weight: these nodes are sometimes simply removed from the network in a pre-processing step [18], changing significantly the connectivity properties of the network. We do not remove the high-degree nodes in the networks, since then the whole graph would be changed significantly. We rather introduce a new scoring function, that compensates the important small degree nodes against hubs or superhubs.
Results and Discussion
In the present work we introduce a method for finding relevant nodes (e.g., possible new protein targets) in networks with directed edges, especially in metabolic networks, that is robust and can compensate small degree nodes against large degree nodes, therefore our method does not need pre-processing steps to remove vertices, corresponding to “currency metabolites”. We also show that our method successfully identifies numerous already verified relevant protein targets, and therefore, may be used to identify novel ones in other directed networks as well.
Let us note that proving that several highly scored proteins in our method are new, still unknown protein targets, would require multi-year wet-lab work (i) for developing new inhibitors against the new, suggested protein targets; (ii)proving that the inhibitors have significant biological activity, (iii) proving that the inhibitors inhibit the new target protein, and not some other enzymes. That work is out of scope of the present theoretical paper. Therefore our proof contains references to target proteins, discovered earlier independently from us, that gained high scores in our method, solely by graph theoretic analysis of the underlying metabolic graphs.
We demonstrated in [19] that the PageRank of vertices [20], applied first in the Google web-search engine [20] for identifying important web pages, can also be used in the robust analysis of protein networks to identify important nodes. Here “robustness” means that changes in the less interesting parts of the network will not cause significant changes in the PageRank of the more important nodes (see [19] for a more exact statement).
It is known, however, that large degree nodes usually have large PageRank on the average [21], therefore PageRank alone cannot always compensate the overweight of hubs and superhubs in the identification of important nodes in a network.
Here we suggest to use for the scoring the importance of nodes in metabolic networks the “relativized personalized PageRank”. Let G be a directed graph. The PageRank [20] of graph G is the limit probability distribution of the random walk, defined by the column-stochastic transition matrix.
| (1) |
where A is row-stochastic transition matrix, prepared by normalizing the rows of the adjacency matrix of graph G
[22], 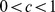 is the damping constant, 1 is the all-1 column-vector, and vector w with non-negative coordinates, satisfying
is the damping constant, 1 is the all-1 column-vector, and vector w with non-negative coordinates, satisfying 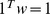 , is the personalization vector. In the original, non-personalized version of the PageRank of an n-vertex graph,
, is the personalization vector. In the original, non-personalized version of the PageRank of an n-vertex graph, 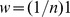 . We use everywhere in this work the value
. We use everywhere in this work the value 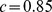 .
.
We must note that the role of the personalization vector, w, was originally to capture the personal interests of the web-surfers to compute a personalized ranking of the web-pages for web-search engines [20]. If no personalization is given (when 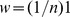 ) that means that in the teleporting step of the walk, each vertex can be visited with the same probability. Personal interests of the web-surfers can be introduced into the random walk by increasing the probability of web-sites interesting to the surfer by increasing their probability in the distribution given in vector w.
) that means that in the teleporting step of the walk, each vertex can be visited with the same probability. Personal interests of the web-surfers can be introduced into the random walk by increasing the probability of web-sites interesting to the surfer by increasing their probability in the distribution given in vector w.
In [19] we have shown that if vector w is personalized to proteins, appearing in higher concentrations in proteomics analysis of certain diseases, then this personalized PageRank may emphasize other closely related proteins to the disease, that eventually did not appear in the proteomics analysis, either because of their low concentration or by their cellular compartmentalization.
It is demonstrated [23] through computational simulations, that in undirected graphs, the PageRank of a node is approximately proportional to the degree of that node; consequently, for undirected graphs, the PageRank will not yield additional information on node relevance, relative to degree.
In [22] we proved that in the case of undirected graphs, the PageRank of the vertices are exactly proportional to their degrees if and only if the coordinates of the personalization vector w are proportional to the degrees of the vertices, that is:
| (2) |
where 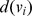 denotes the degree of vertex
denotes the degree of vertex 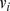 , and
, and 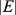 denotes the number of the edges in graph G.
denotes the number of the edges in graph G.
In other words, the PageRank, defined by the limit probability distribution of (1) with w given in (2) is exactly w for undirected graphs. Therefore if we divide coordinate i by 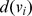 , for
, for 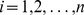 , then we get the same constant for each coordinates.
, then we get the same constant for each coordinates.
This means that dividing the PageRank personalized by vector w, by the degrees, we factor out high- or low degreeness from the score: for every vertex the ratio is the same.
We would like to introduce a similar measure for directed graphs, that factors out the degrees in above sense, and the resulted scores would allow to reach high values for low-degree nodes, too.
More exactly, we define for the directed graph G the vector.
| (3) |
where 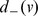 is the in-degree of vertex y (i.e., the number of directed edges pointing to vertex v).
is the in-degree of vertex y (i.e., the number of directed edges pointing to vertex v).
Now we can define the “relativized personalized PageRank” of graphs as follows: Let PPageRank denote the PageRank given by the stationary distribution of the walk of equation (1) computed with w of equation (3), then.
| (4) |
Clearly, in undirected graphs, our relativized PageRank
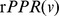 is exactly constant, i.e., it is completely independent from the node (and its degree). Therefore, in undirected graphs, it is the same for large- and small degree nodes, so in directed graphs its high value may describe a sort of “intrinsic” importance of the node, independent from its high- or low degree, and depending only on the directed graph-connectivity structure of the network. We find this to be an important property, since it shows that our score function was not chosen “arbitrarily”, it “factors out” the undirected degree from the scoring.
is exactly constant, i.e., it is completely independent from the node (and its degree). Therefore, in undirected graphs, it is the same for large- and small degree nodes, so in directed graphs its high value may describe a sort of “intrinsic” importance of the node, independent from its high- or low degree, and depending only on the directed graph-connectivity structure of the network. We find this to be an important property, since it shows that our score function was not chosen “arbitrarily”, it “factors out” the undirected degree from the scoring.The PageRank of large degree nodes are on the average, large in any graph (see [21] for a much more exact statement). Dividing the PageRank of node v by its in-degree will compensate the small in-degree nodes, since their PageRank is divided only with a small number. Therefore the small in-degree, relevant nodes may stand out in this scoring function. This scoring function will usually not give high scores for network hubs, but these hubs can easily be identified by simple degree counting, and does not need more sophisticated tools.
As we demonstrate here in the application examples, the new scoring method will choose low degree nodes with proven biological interest. Therefore, the presented approach can effectively be used to find promising drug targets because the reactions (nodes) with high PageRank and low in-degree correspond to essential reactions.
Application Examples in Microbial Networks
For demonstrating the applicability of this new scoring function, we present several examples from much researched pathogen microorganisms. We show that several well known protein targets correspond to highly scored nodes, and this fact may imply that other highly scored nodes may be promising, non-hub, new drug targets. We would like to stress that in identifying new possible drug targets we applied only the rPPR score of us (4), and have not used structural or functional annotations of the proteins, just their positions in the metabolic networks.
Mycobacterium Tuberculosis
Our first example is the mycolic acid metabolic pathway [24] in the Mycobacterium tuberculosis bacterium. Since mycolic acid synthesis is missing in eukaryotes, targeting specific enzymes in this pathway seems to be a natural choice for target search.
rPPR was computed for the mycolic acid pathway, the result is depicted on Figure 1. The size of the nodes are corresponding to the degree of the vertex, and the color of the node to the rPPR of the vertex: the warmer the color the higher the rPPR score.
Figure 1. The network of the mycolic acid synthesis [24].

Node sizes correspond to the degree of the node, node color correspond to the personalized PageRank of the node: warmer colors mean larger PageRank. Note the small but yellow node labeled by InhA in the upper central part of the picture.
The yellow inhA node in the upper right quadrant of Figure 1 has in-degree 1, therefore it is not a hub at all. On the other hand, by Table 1, its rPPR is far the highest in the network. This scoring correlates well with the fact that inhA (long-chain enoyl-acyl carrier protein reductase) is one of the oldest known and most important target of TB drugs isoniazid and ethionamide, and also the prime target of several novel drugs under development today [25]–[27].
Table 1. The list of six nodes with the rPPR scores in the mycolic acid pathway of the Mycobacterium tuberculosis.
| Node | PPR in | Degree | in-degree | PPR in / in-degree |
| inhA | 0.049 | 4 | 1 | 0.049 |
| fabH | 0.058 | 8 | 2 | 0.029 |
| fas | 0.029 | 3 | 1 | 0.029 |
| kasB kasA | 0.045 | 7 | 2 | 0.023 |
| UNK1 | 0.055 | 4 | 3 | 0.018 |
| fabD | 0.133 | 12 | 8 | 0.017 |
The node with the second highest rPPR, labeled by FabH (see Table 1) is also a well-researched possible TB drug target [28]–[31].
Our second example concerns the whole metabolic network (not only the mycolic acid pathway) of the Mycobacterium tuberculosis. The network contains 947 nodes, the rPPR scores and the vertices with non-zero in-degrees are given in Table S1 in the on-line supporting material.
Table 2 shows the list of the nodes with the highest rPPR score. The highest and second highest scoring reactions correspond to the protein pdxH, a putative pyridoxine 5′-phosphate oxidase (Rv2607) is reported [32] having strongly different putative binding pocket than any other member of its enzyme family.
Table 2. The list of the 11 nodes in the metabolic network of the tuberculosis bacterium with the highest rPPR score.
| reaction ID | PPR in | Degree | in-degree | PPR in/in-degree | protein correspondence |
| R00278 | 0.0061 | 3 | 2 | 0.0030 | Rv2607 pdxH |
| R00277 | 0.0061 | 3 | 2 | 0.0030 | Rv2607 pdxH |
| R01209 | 0.0025 | 7 | 1 | 0.0025 | Rv0189c ilvD |
| R03051 | 0.0028 | 3 | 2 | 0.0014 | Rv3001c ilvC |
| R06905 | 0.0013 | 1 | 1 | 0.0013 | bnsG |
| R03968 | 0.0020 | 4 | 2 | 0.0010 | Rv2987c(leuD) Rv2988c(leuC) |
| R04942 | 0.0020 | 3 | 2 | 0.0010 | Rv1077 cysM |
| R04440 | 0.0020 | 4 | 2 | 0.0010 | Rv3001c(ilvC) |
| R05071 | 0.0027 | 5 | 3 | 0.0009 | Rv3001c(ilvC) |
| R01214 | 0.0046 | 12 | 6 | 0.0008 | Rv2210c(ilvE) |
| R01215 | 0.0046 | 12 | 6 | 0.0008 | Rv0337c(aspC) |
The full table is available as Table S1 in the on-line supporting material.
Very recently it is reported [33] that the downregulation of the third largest scoring protein with gene name ilvD (Rv0189c, a dihydroxyacid dehydratase) affects the growth of Mycobacterium tuberculosis in vitro and in mice.
The sixth highest scoring hit, the leuD gene (Rv2987c) is shown to be essential in Mycobacterium tuberculosis even in macrophages [34].
The seventh highest-scoring protein is cysM (Cysteine synthase, Rv1077), is reported [35] to have intermediate protection properties and in sulfur donor selectivity, and also is known to play a main role in a mycobacteria-specific, alternative cysteine biosynthesis pathway [36].
The third, fourth, eighth, ninth and tenth highest scored hits are related to branched chain amino acid (BCCA, comprises leucine, isoleucine and valine) synthesis of the bacterium. Examples were shown in [37] that these proteins may serve as drug targets.
Plasmodium Falciparum
The metabolic network for Plasmodium falciparum contains 450 nodes. Table 3 shows eleven of the highest rPPR scoring vertices, while the full table is available as Table S2 in the supporting on-line material.
Table 3. The list of the eleven nodes with the highest rPPR in Plasmodium falciparum.
| reaction ID | PPR in | Degree | in-degree | PPR in/in-degree | protein correspondence |
| R00173 | 0.0123 | 3 | 2 | 0.0061 | pyridoxal kinases |
| R00174 | 0.0123 | 3 | 2 | 0.0061 | pyridoxal kinases |
| R03316 | 0.0043 | 8 | 2 | 0.0021 | 2-oxoglutarate dehydrogenase |
| R01890 | 0.0024 | 3 | 2 | 0.0012 | cholinephosphate cytidylyltransferase |
| R01021 | 0.0024 | 3 | 2 | 0.0012 | choline kinase |
| R07604 | 0.0020 | 8 | 2 | 0.0010 | branch.-chain alpha keto-acid dehydr. |
| R07602 | 0.0020 | 8 | 2 | 0.0010 | branch.-chain alpha keto-acid dehydr. |
| R07600 | 0.0020 | 8 | 2 | 0.0010 | branch.-chain alpha keto-acid dehydr. |
| R01961 | 0.0018 | 4 | 2 | 0.0009 | hexokinase |
| R01940 | 0.0008 | 3 | 1 | 0.0008 | 2-oxoglutarate dehydrogenase |
| R01626 | 0.0081 | 19 | 10 | 0.0008 | PfMCAT |
The full table with 450 nodes is available as Table S2 in the supporting on-line material.
Reactions of the highest and second highest score (R00174 and R00173, resp.) are corresponded to pyridoxal kinases (EC:2.7.1.35) that are shown to be targets or Roscovitine in [38] and a possible target in the malaria parasite in [39]. It is reported in [40] that inhibiting pyridoxal 5-phosphate-dependent enzymes kills the parasite efficiently.
The fourth highest scoring R01890, corresponding to PfCCT, cholinephosphate cytidylyltransferase, is shown to be the target of a potent experimental malaria drug, PG12 in [41].
The fifth highest scoring hit, R01021, corresponds to choline kinase, that is reported to be the target of hexadecyltrimethylammonium bromide in the malaria parasite in [42].
The sixth, seventh and eights highest scored reactions (R07604, R07602, R07602) are corresponded to branched-chain alpha keto-acid dehydrogenases, and they are shown to be specific in function in Plasmodium falciparum, therefore they may serve as a selective target [43].
In [44] it is shown that the ninth hit R01961, corresponding to hexokinase, can be viable target in Plasmodium falciparum.
The tenth highest scoring reaction is R01940 (EC:1.2.4.2, 2-oxoglutarate dehydrogenase (OGDH) E1 component). The related pathways include vitamin B metabolism, a recently suggested target [45], and relates to alpha-ketoacid dehydrogenase multienzyme complexes, with specificity for Plasmodium falciparum. Glutamate dehydrogenase, catalyzing a closely related reaction, was suggested also as new drug target recently [46].
With unusually high in- and outdegrees, the eleventh highest scoring reaction, R01626 is corresponded to enzyme PfMCAT, that is shown to be essential in fatty acid synthesis of the parasite in [47].
MRSA Staphylococcus Aureus
We applied the metabolic data of the MRSA Staphylococcus aureus SAA strain (USA300_FPR3757 (CA-MRSA)) for the network generation. The network contains 803 network nodes. The data of all the nodes are available as Table S3 in the on-line supporting material, while nine nodes with the largest score is given on Table 4.
Table 4. The list of nine nodes with the highest rPPR score in MRSA Staphylococcus aureus.
| reaction ID | PPR in | Degree | in-degree | PPR in/in-degree | protein correspondence |
| R00174 | 0.0083 | 3 | 2 | 0.0041 | phosphomethylpyrimidine kinase (EC:2.7.4.7) |
| R00173 | 0.0083 | 3 | 2 | 0.0041 | pyridoxal phosphate phosphatase |
| R07600 | 0.0047 | 13 | 2 | 0.0024 | 2-oxoisovalerate dehydrogenase |
| R02272 | 0.0045 | 4 | 2 | 0.0023 | hemL |
| R04109 | 0.0039 | 3 | 2 | 0.0019 | hemA |
| R03316 | 0.0032 | 13 | 2 | 0.0016 | sucA |
| R00036 | 0.0027 | 4 | 2 | 0.0013 | hemB |
| R07604 | 0.0026 | 13 | 2 | 0.0013 | 2-oxoisovalerate dehydrogenase |
| R01209 | 0.0013 | 8 | 1 | 0.0013 | ilvD |
The full table with 450 nodes is available as Table S3 in the supporting on-line material.
The two highest scoring reactions relate to vitamin B metabolism and reported to be important in SAA in [48].
R02272, R04109 and R00036 correspond to the hemL, hemA and hemB genes, respectively, of the heme synthesis. Inactivation of the hemB gene leads to an aberrant form of the bacterium, the small colony variant (SCV) [49]. Most recently, [50] also reports the significance of these genes.
R07600, R07604 and R01209 (ilvD) play a main role in branched-chain amino acids biosynthesis pathway of the bacterium [51].
Materials and Methods
The mycolic acid network was prepared using the pathways published in [24]. The nodes are labeled by the gene names of the enzymes, and two nodes X and Y, corresponding to enzymes denoted by their gene names, are connected by a directed edge from X to Y if and only if there exists a substrate u, leaving the reaction, catalyzed by enzyme X, that enters the reaction, catalyzed by enzyme Y. Substrate u labels the directed edge from X to Y.
For creating Figure 1, we applied Cytoscape [52] for the data published in [24]. A high resolution version of Figure 1 is available as Figure S1 in the on-line supporting material.
The metabolic networks for Mycobacterium tuberculosis, Plasmodium falciparum and MRSA Staphylococcus aureus were generated from the KEGG database [16], data downloaded on December 13, 2010. The network nodes were labeled by the KEGG reaction ID's. The full datasets, containing the degrees and the PageRanks of the nodes are available as on-line supporting material.
PageRank was computed using the NetrworkX Python library [53] (downloadable from the Los Alamos national Laboratory http://networkx.lanl.gov/) with our Python script ppr_pub.py, downloadable from http://uratim.com/rPPR.
Conclusions
Traditionally, the discovery of novel protein targets relies on multi-decade long work on several biochemical reactions in living organisms. New tools and insights make possible that the systems biology would also suggest new possible targets, by examining the protein-protein or protein-metabolite interactions of the cell. We believe that using well-developed methods from graph theory and computer science will yield significant results in biology. In particular, ordinary PageRank can help to evaluate important nodes and pathways in directed networks, especially when relativized with other network properties, like the in-degree of nodes. We think that the present method is capable for identifying low-degree nodes with high intrinsic metabolic functionality in networks, clearly and automatically.
The rPPR measure introduced in Equation (4) has the following remarkable property: its value is the same for each vertex (either with large or small degrees) of an undirected graph, while for directed graphs, rPPR may change significantly from vertex to vertex, and it captures importance due to the directions of the edges in the graph.
Our method gives high scores to nodes that have high PageRank relative to their degrees, therefore clearly and easily identifies important nodes of low-degrees in biological networks. Consequently, the method and the scoring function can be effectively used to find promising drug targets in metabolic networks, because the reactions (nodes) with high PageRank and low in-degree correspond to essential reactions.
Supporting Information
The degree, in-degree, PPR and rPPR data for the metabolic network of Mycobacterium tuberculosis .
(XLS)
The degree, in-degree, PPR and rPPR data for the metabolic network of of Plasmodium falciparum .
(XLS)
The degree, in-degree, PPR and rPPR data for the metabolic network of MRSA bacterium.
(XLS)
Acknowledgments
“Google” and “PageRank” are registered trademarks of Google, Inc.
Funding Statement
This work was supported by NKTH TB-INTER (www.nih.gov.hu), OTKA CNK 77780 (www.otka.hu), and the TÁMOP 4.2.1./B-09/KMR-2010-0003 (http://kp.elte.hu/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, et al. (2010) The intact molecular interaction database in 2010. Nucleic Acids Res 38: D525–D531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prasad TSK, Kandasamy K, Pandey A (2009) Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol Biol 577: 67–79. [DOI] [PubMed] [Google Scholar]
- 3. Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, et al. (2002) Mint: a molecular interaction database. FEBS Lett 513: 135–140. [DOI] [PubMed] [Google Scholar]
- 4. Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, et al. (2002) Dip, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res 30: 303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bader GD, Betel D, Hogue CWV (2003) Bind: the biomolecular interaction network database. Nucleic Acids Res 31: 248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bader S, Kuhner S, Gavin AC (2008) Interaction networks for systems biology. FEBS Lett 582: 1220–1224. [DOI] [PubMed] [Google Scholar]
- 7. Rivas JDL, de Luis A (2004) Interactome data and databases: different types of protein interaction. Comp Funct Genomics 5: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cusick ME, Klitgord N, Vidal M, Hill DE (2005) Interactome: gateway into systems biology. Hum Mol Genet 14 Spec No. 2: R171–R181. [DOI] [PubMed] [Google Scholar]
- 9. Farkas IJ, Korcsmaros T, Kovacs IA, Mihalik A, Palotai R, et al. (2011) Network-based tools for the identification of novel drug targets. Sci Signal 4: pt3. [DOI] [PubMed] [Google Scholar]
- 10. Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5: 993–996. [DOI] [PubMed] [Google Scholar]
- 11. Ideker T, Bafna V, Lemberger T (2007) Integrating scientific cultures. Mol Syst Biol 3: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu X, Jain VV, Finn PW, Perkins DL (2007) Hubs in biological interaction networks exhibit low changes in expression in experimental asthma. Mol Syst Biol 3: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeiffer T, Soyer OS, Bonhoeffer S (2005) The evolution of connectivity in metabolic networks. PLoS Biol 3: e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Russell RB, Aloy P (2008) Targeting and tinkering with interaction networks. Nat Chem Biol 4: 666–673. [DOI] [PubMed] [Google Scholar]
- 15. Hwang WC, Zhang A, Ramanathan M (2008) Identification of information flow-modulating drug targets: a novel bridging paradigm for drug discovery. Clin Pharmacol Ther 84: 563–572. [DOI] [PubMed] [Google Scholar]
- 16. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, et al. (1999) Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holme P (2009) Signatures of currency vertices. Journal of the Physical Society of Japan 78: 034801. [Google Scholar]
- 18. Huss M, Holme P (2007) Currency and commodity metabolites: their identification and relation to the modularity of metabolic networks. IET Syst Biol 1: 280–285. [DOI] [PubMed] [Google Scholar]
- 19. Ivan G, Grolmusz V (2011) When the web meets the cell: using personalized pagerank for analyzing protein interaction networks. Bioinformatics 27: 405–407. [DOI] [PubMed] [Google Scholar]
- 20. Brin S, Page L (1998) The anatomy of a large-scale hypertextual web search engine. COMPUTER NETWORKS AND ISDN SYSTEMS 30: 107–117. [Google Scholar]
- 21.Fortunato S, Boguna M, Flammini A, Menczer F (2008) Approximating pagerank from in-degree. In: Aiello W, Broder A, Janssen J, Milios E, editors, Algorithms and Models for the Web-Graph. Springer Berlin / Heidelberg, volume 4936 of Lecture Notes in Computer Science, 59–71. Available at http://dx.doi.org/10.1007/978-3-540-78808-9-6 . 10.1007/978-3-540-78808-9-6.
- 22.Grolmusz V (2012) A note on the pagerank of undirected graphs. arXiv Computing Research Repository (CoRR) abs/1205.1960.
- 23. Perra N, Fortunato S (2008) Spectral centrality measures in complex networks. Phys Rev E 78: 036107. [DOI] [PubMed] [Google Scholar]
- 24. Takayama K, Wang C, Besra GS (2005) Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev 18: 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Sacchettini JC (1998) Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279: 98–102. [DOI] [PubMed] [Google Scholar]
- 26. Vilcheze C, Av-Gay Y, Attarian R, Liu Z, Hazbon MH, et al. (2008) Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol 69: 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tonge PJ, Kisker C, Slayden RA (2007) Development of modern InhA inhibitors to combat drug resistant strains of Mycobacterium tuberculosis. Curr Top Med Chem 7: 489–498. [DOI] [PubMed] [Google Scholar]
- 28. Brown AK, Taylor RC, Bhatt A, Futterer K, Besra GS (2009) Platensimycin activity against mycobacterial beta-ketoacyl-acp synthases. PLoS One 4: e6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veyron-Churlet R, Molle V, Taylor RC, Brown AK, Besra GS, et al. (2009) The Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase iii activity is inhibited by phosphorylation on a single threonine residue. J Biol Chem 284: 6414–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alhamadsheh MM, Musayev F, Komissarov AA, Sachdeva S, Wright HT, et al. (2007) Alkyl-CoA disulfides as inhibitors and mechanistic probes for FabH enzymes. Chem Biol 14: 513–524. [DOI] [PubMed] [Google Scholar]
- 31. Choi KH, Kremer L, Besra GS, Rock CO (2000) Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase iii (mtFabH) from Mycobacterium tuberculosis. J Biol Chem 275: 28201–28207. [DOI] [PubMed] [Google Scholar]
- 32. Pedelacq JD, Rho BS, Kim CY, Waldo GS, Lekin TP, et al. (2006) Crystal structure of a putative pyridoxine 5′-phosphate oxidase (Rv2607) from Mycobacterium tuberculosis. Proteins 62: 563–569. [DOI] [PubMed] [Google Scholar]
- 33. Singh V, Chandra D, Srivastava BS, Srivastava R (2011) Downregulation of Rv0189c, encoding a dihydroxyacid dehydratase, affects growth of Mycobacterium tuberculosis in vitro and in mice. Microbiology 157: 38–46. [DOI] [PubMed] [Google Scholar]
- 34. Bange FC, Brown AM, Jacobs WR (1996) Leucine auxotrophy restricts growth of Mycobacterium bovis BCG in macrophages. Infect Immun 64: 1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Agren D, Schnell R, Schneider G (2009) The C-terminal of CysM from Mycobacterium tuberculosis protects the aminoacrylate intermediate and is involved in sulfur donor selectivity. FEBS Lett 583: 330–336. [DOI] [PubMed] [Google Scholar]
- 36. Agren D, Schnell R, Oehlmann W, Singh M, Schneider G (2008) Cysteine synthase (CysM) of Mycobacterium tuberculosis is an O-phosphoserine sulfhydrylase: evidence for an alternative cysteine biosynthesis pathway in mycobacteria. J Biol Chem 283: 31567–31574. [DOI] [PubMed] [Google Scholar]
- 37. Grandoni JA, Marta PT, Schloss JV (1998) Inhibitors of branched-chain amino acid biosynthesis as potential antituberculosis agents. J Antimicrob Chemother 42: 475–482. [DOI] [PubMed] [Google Scholar]
- 38. Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, et al. (2005) Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem 280: 31208–31219. [DOI] [PubMed] [Google Scholar]
- 39. Manhani KK, Arcuri HA, da Silveira NJF, Uchoa HB, de Azevedo WF, et al. (2005) Molecular models of protein kinase 6 from Plasmodium falciparum. J Mol Model 12: 42–48. [DOI] [PubMed] [Google Scholar]
- 40. Muller IB, Wu F, Bergmann B, Knockel J, Walter RD, et al. (2009) Poisoning pyridoxal 5-phosphate-dependent enzymes: a new strategy to target the malaria parasite Plasmodium falciparum. PLoS One 4: e4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonzalez-Bulnes P, Bobenchik AM, Augagneur Y, Cerdan R, Vial HJ, et al. (2011) PG12, a phospholipid analog with potent antimalarial activity, inhibits Plasmodium falciparum CTP:phosphocholine cytidylyltransferase activity. J Biol Chem 286: 28940–28947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choubey V, Maity P, Guha M, Kumar S, Srivastava K, et al. (2007) Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism. Antimicrob Agents Chemother 51: 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McMillan PJ, Stimmler LM, Foth BJ, McFadden GI, Muller S (2005) The human malaria parasite Plasmodium falciparum possesses two distinct dihydrolipoamide dehydrogenases. Mol Microbiol 55: 27–38. [DOI] [PubMed] [Google Scholar]
- 44. Olafsson P, Matile H, Certa U (1992) Molecular analysis of Plasmodium falciparum hexokinase. Mol Biochem Parasitol 56: 89–101. [DOI] [PubMed] [Google Scholar]
- 45. Muller IB, Hyde JE, Wrenger C (2010) Vitamin B metabolism in Plasmodium falciparum as a source of drug targets. Trends Parasitol 26: 35–43. [DOI] [PubMed] [Google Scholar]
- 46. Aparicio IM, Marin-Menendez A, Bell A, Engel PC (2010) Susceptibility of Plasmodium falciparum to glutamate dehydrogenase inhibitors–a possible new antimalarial target. Mol Biochem Parasitol 172: 152–155. [DOI] [PubMed] [Google Scholar]
- 47. Prigge ST, He X, Gerena L, Waters NC, Reynolds KA (2003) The initiating steps of a type ii fatty acid synthase in Plasmodium falciparum are catalyzed by pfACP, pfMCAT, and pfKASIII. Biochemistry 42: 1160–1169. [DOI] [PubMed] [Google Scholar]
- 48. Muller IB, Bergmann B, Groves MR, Couto I, Amaral L, et al. (2009) The vitamin B1 metabolism of Staphylococcus aureus is controlled at enzymatic and transcriptional levels. PLoS One 4: e7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McNamara PJ, Proctor RA (2000) Staphylococcus aureus small colony variants, electron transport and persistent infections. Int J Antimicrob Agents 14: 117–122. [DOI] [PubMed] [Google Scholar]
- 50. Haley KP, Skaar EP (2012) A battle for iron: host sequestration and Staphylococcus aureus acquisition. Microbes Infect 14: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lei T, Yang J, Zheng L, Markowski T, Witthuhn BA, et al. (2012) The essentiality of staphylococcal gcp is independent of its repression of branched-chain amino acids biosynthesis. PLoS One 7: e46836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagberg AA, Schult DA, Swart PJ (2008) Exploring network structure, dynamics, and function using NetworkX. In: Proceedings of the 7th Python in Science Conference (SciPy2008). Pasadena, CA USA, 11–15.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The degree, in-degree, PPR and rPPR data for the metabolic network of Mycobacterium tuberculosis .
(XLS)
The degree, in-degree, PPR and rPPR data for the metabolic network of of Plasmodium falciparum .
(XLS)
The degree, in-degree, PPR and rPPR data for the metabolic network of MRSA bacterium.
(XLS)