

Abstract
The expression of nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) is regulated by the p53 and Egr-1 tumor suppressor pathways. Many anti-cancer drugs and chemicals induce NAG-1 expression, but the mechanisms are not fully understood. Transgenic mice expressing human NAG-1 are resistant to intestinal and prostate cancer, suggesting that NAG-1 is a tumor suppressor. Proteasome inhibitors exhibit anti-glioblastoma activities in preclinical studies. Here, we show that the proteasome inhibitors MG132 and bortezomib induced NAG-1 expression and secretion in glioblastoma cells. MG132 increased NAG-1 expression through transcriptional and post-transcriptional mechanisms. At the transcriptional level, the induction of NAG-1 required the −133 to +41 bp region of the promoter. At post-transcriptional levels, MG132 stabilized NAG-1 mRNA by increasing the half-life from 1.5 h to > 8 h. Because of the dramatic increase in mRNA stability, this is likely the major contributor to MG132-mediated NAG-1 induction. Further probing into the mechanism revealed that MG132 increased phosphorylation of the p38 MAPK pathway. Consequently, inhibiting p38 phosphorylation blocked activation of the NAG-1 promoter and decreased mRNA stability, indicating that p38 MAPK activation mediates both MG132-dependent promoter activation and mRNA stabilization of NAG-1. We propose that the induction of NAG-1 by p38 MAPK is a potential contributor to the anti-glioblastoma activity of proteasome inhibitors.
Keywords: NAG-1/GDF15, MG132, glioblastoma, p38 MAPK
1. Introduction
Glioblastoma multiforme (GBM) is the most common primary brain tumor characterized by aggressive invasive behavior and a poor prognosis [1]. There is an urgent need to identify novel drugs targeting GBM and to develop more effective therapeutic strategies.
Proteasomes degrade misfolded proteins, control the turnover of many regulatory proteins, and are essential for many cellular processes including regulation of gene expression, cell cycle progression, cellular differentiation, cell death, and tumor growth [2]. Preclinical studies have demonstrated that the proteasome inhibitors MG132 and bortezomib inhibit proliferation and promote cell death in glioblastoma cells [3,4]. In a single Phase 1 clinical trial, bortezomib did not improve prognosis in patients with GBM [5]. Thus, a better understanding of how the molecular pathways of proteasome inhibitors act in glioblastoma cells is needed to harness their therapeutic potential.
Nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) represents a divergent member of the TGF-β superfamily. NAG-1 is also known as macrophage inhibitory cytokine-1, placental transforming growth factor-β, prostate-derived factor, placental bone morphogenetic protein, or growth differentiation factor-15 (GDF-15) [6,7]. NAG-1 is synthesized as a 308-amino acid pro-peptide and secreted as a mature protein after proteolytic cleavage [6,7,8,9,10]. The secreted form is present in the blood of humans and media of cultured cells expressing NAG-1 [9]. Both the pro-form and secreted form are likely to play central roles in the biological activity of NAG-1 [6,7,9,10]. There are conflicting reports concerning the tumor suppressor and oncogenic activities of NAG-1 [10]. NAG-1 expression is upregulated by the tumor suppressors p53 and Egr-1 [6,7,9]. Several drugs with anti-inflammatory and anti-tumorigenic activities increase NAG-1 expression [6,7,9]. Many in vitro studies suggest that expression of NAG-1 inhibits tumor cell growth [6,7,8,9,11,12,13]. The most compelling evidence for anti-tumorigenic activity comes from studies with transgenic mice expressing human NAG-1 [14]. It has not been investigated if proteasome inhibitors induce the expression of this potential tumor suppressor protein.
Here we report that the proteasome inhibitor MG132 stimulates NAG-1 expression in glioblastoma cells. MG132-induced NAG-1 expression was dependent on the p38 MAPK pathway and involved both transcriptional (promoter activation) and post-transcriptional mechanisms (mRNA stability). Our data suggest that NAG-1 expression provides a novel pathway for anti-glioblastoma activity of proteasome inhibitors.
2. Materials and Methods
Total cell lysates from cultured cells were isolated in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease and phosphatase inhibitors for Western blot analysis. Equal amounts of lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed. The Human GDF-15 Duo Set Enzyme-Linked Immunosorbent Assay (ELISA) Development kit (R&D Systems, Minneapolis, MN, USA) was used to determine NAG-1 protein levels in the cell culture medium. Total RNA from cultured cells was isolated using the RNeasy MINI kit (Qiagen, Valencia, CA). After cDNA synthesis, RT-PCR assays were performed using the MyiQ Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA). PCR amplification levels were normalized to those of β-actin. PCR primers are shown in Supplementary Materials and Methods. The human NAG-1 promoter constructs used in the luciferase assay were described previously [15]. The two-tailed unpaired Student’s t-test was used for statistical assessment between experimental groups. A p < 0.05 was considered significant. Cell culture, luciferase reporter assays, small interfering RNA (siRNA) transfection, and flow cytometric analysis of propidium iodide-stained cells were performed using standard procedures (see details in Supplementary Materials and Methods).
3. Results
3.1. Proteasome inhibitor MG132 induces NAG-1 expression in glioblastoma cells and causes accumulation of U87MG cells in the sub-G1 phase
We measured NAG-1 mRNA and protein levels after treatment with MG132 in the U87MG, T98G, A172, and U138MG glioblastoma cell lines to investigate whether the proteasome inhibitor MG132 could induce NAG-1 expression. NAG-1 mRNA and protein levels remained undetectable after DMSO vehicle treatment (Fig. 1A, B). Treatment with MG132 increased NAG-1 mRNA expression in all four cell lines (Fig. 1A). MG132 increased protein expression of pro-form NAG-1 in U87MG and A172 cells (Fig. 1B). Furthermore, bortezomib, a proteasome inhibitor in clinical use, also induced NAG-1 mRNA and protein expression in U87MG and A172 cells (Supplementary Fig. 1). Thus, MG132 and bortezomib increased NAG-1 expression in glioblastoma cells. MG132 and U87MG cells were used for all subsequent experiments because of the large increase in NAG-1 expression observed after treatment. Next, we sought to examine MG132 activity in U87MG cells. The flow cytometric analysis showed that treatment with MG132 for 24 h increased the proportion of cells in the sub-G1 phase (Fig. 1C). Importantly, U87MG cells transfected with siRNA to knockdown NAG-1 were less sensitive than control cells to the action of MG132 (Fig. 1C, D).
Fig. 1. Proteasome inhibitor MG132 induces NAG-1 expression in glioblastoma cells and causes accumulation of U87MG cells in the sub-G1 phase.
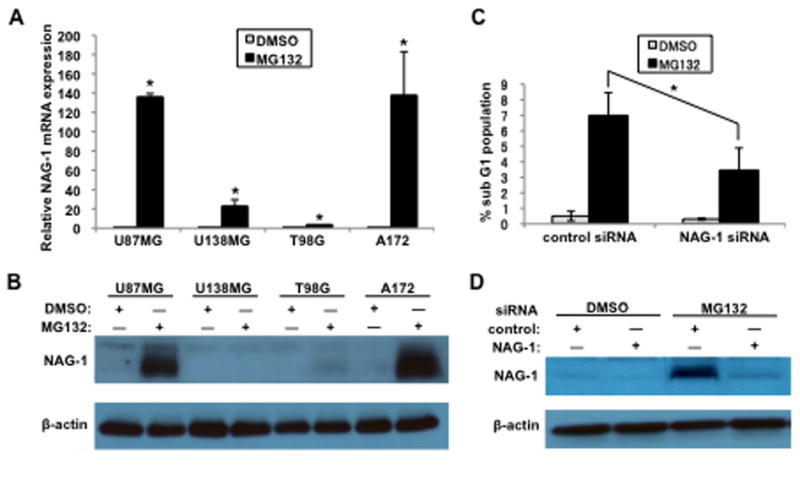
(A and B) Glioblastoma cells (U87MG, U138MG, T98G, and A172 cells) were treated with 1 μM MG132 for 12 h. (A) NAG-1 mRNA levels were determined by quantitative RT-PCR. Values are mean ± standard deviation (SD) of three independent experiments. * p < 0.05. (B and D) Cell lysates were subjected to Western blotting analysis, and the 35-kDa pro-form of the NAG-1 protein and β-actin are shown. (C and D) Control nontarget siRNA or NAG-1 siRNA were transfected into U87MG cells for 48 h. Cells were then treated with 1 μM MG132 for 24 h. (C) The population of sub-G1 DNA content cells was detected by flow cytometry with propidium iodide staining. Values are mean ± SD of three independent experiments. * p < 0.05.
3.2. MG132 induces NAG-1 expression in a concentration and time-dependent manner
U87MG cells were treated with increasing concentrations (0.25–5 μM) of MG132 for 12 h. NAG-1 mRNA levels increased 100 to 175-fold in U87MG cells treated with 1, 2, or 5 μM MG132 (Fig. 2A). NAG-1 protein levels increased steadily with MG132 concentrations of 0.25–1 μM and then reached a plateau (Fig. 2B). Next, U87MG cells were treated with 1 μM MG132 for 0–24 h. NAG-1 mRNA expression peaked at 18 h followed by a decline in expression, although expression remained high even at 24 h compared to that in DMSO-treated cells (Fig. 2D). Similarly, NAG-1 protein expression increased in a time-dependent manner, with a steady increase in expression at 0–12 h after exposure to MG132 (Fig. 2E). Expression of the secreted form of NAG-1 was also induced by MG132 in U87MG cells in a concentration and time-dependent manner (Fig. 2C, F).
Fig. 2. MG132 induces NAG-1 expression in a concentration and time-dependent manner.
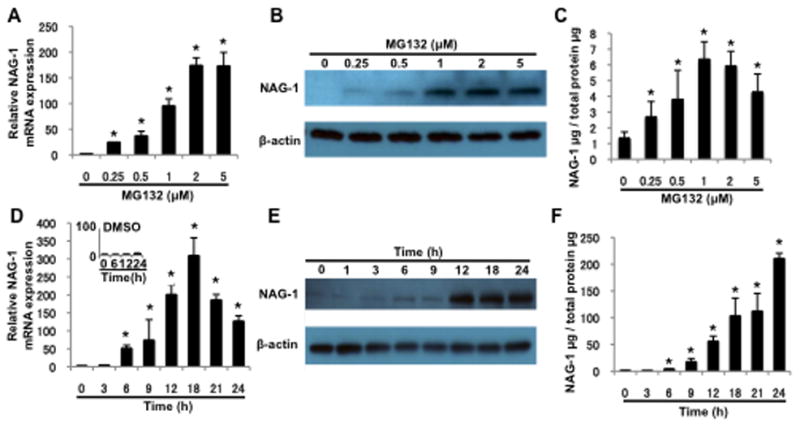
(A–C) U87MG cells were treated with the indicated concentrations of MG132 for 12 h. (D–F) U87MG cells were treated with 1 μM MG132 for the indicated amount of time. (A and D) Quantitative RT-PCR shows the NAG-1 mRNA levels. * p < 0.05. (B and E) Western blotting analysis depicts the 35-kDa pro-form of the NAG-1 protein and β-actin expression. (C and F) ELISA analysis was used to measure NAG-1 secreted into the media. Values are mean ± SD of three independent experiments. * p < 0.05.
3.3. MG132 increases NAG-1 promoter activity
The NAG-1 promoter construct pNAG3500/LUC [15] was transfected into U87MG cells, and the luciferase assay was performed after MG132 treatment to determine if transcriptional activation was involved in MG132-mediated NAG-1 upregulation. MG132 significantly increased pNAG3500/LUC promoter activity (Fig. 3C), indicating that MG132 increases NAG-1 expression at the transcriptional level. To explore if MG132 directly activates the NAG-1 promoter or indirectly acts by increasing expression of specific transcriptional factors, cells were pretreated with the translation inhibitor cycloheximide (CHX) and incubated with MG132. As shown in Fig. 3A and B, MG132 did not increase NAG-1 mRNA or protein levels in the presence of CHX, suggesting that MG132-induced NAG-1 expression at least requires new intermediate regulatory protein synthesis. We performed the luciferase assay with the deletion constructs pNAG1086/LUC, pNAG474/LUC, and pNAG133/LUC to determine the transcriptional response element(s) in the NAG-1 promoter. As shown in Fig. 3C, MG132 treatment activated all of the NAG-1 promoter constructs. These data confirmed that MG132 stimulates NAG-1 promoter activity and suggest that the critical response elements were located in the −133 to +41 bp region of the NAG-1 promoter. Our previous studies on the regulation of NAG-1 expression revealed the importance of the Sp-1 binding sites (Sp-1 A and Sp-1 BC), which overlap with Egr-1 binding sites in this NAG-1 promoter region to mediate the induction of NAG-1 by many drugs and chemicals [6,7,9,11,15]. U87MG cells were transfected with wild-type or deletion constructs lacking Sp-1/Egr-1 binding sites and treated with MG132 to determine if the Sp-1/Egr-1 binding sites are required for the MG132 effect. As shown in Fig. 3D, deletion of the Sp-1/Egr-1 binding sites did not affect luciferase activity. To further rule out the requirement for Egr-1 and Sp-1 proteins in NAG-1 induction by MG132, Egr-1 mRNA and Egr-1 and Sp-1 protein expression were measured after MG132 treatment. As shown in Supplementary Fig. 2, no change in Egr-1 and Sp-1 expression was observed after MG132 treatment. Thus, the site responsible for the MG132 effect was located between −133 and +41 bp, and activation did not require the Sp-1/Egr-1 binding site. The promoter region from −133 to +41 bp was further analyzed for other potential binding sites, and only a putative C/EBPα site was evident. However, knocking down C/EBPα did not inhibit the expression of MG132-induced NAG-1 mRNA or protein (Supplementary Fig. 3). Further study will be required to elucidate the detailed mechanism of NAG-1 promoter activation by MG132.
Fig. 3. MG132 increases NAG-1 promoter activity and mRNA stability.
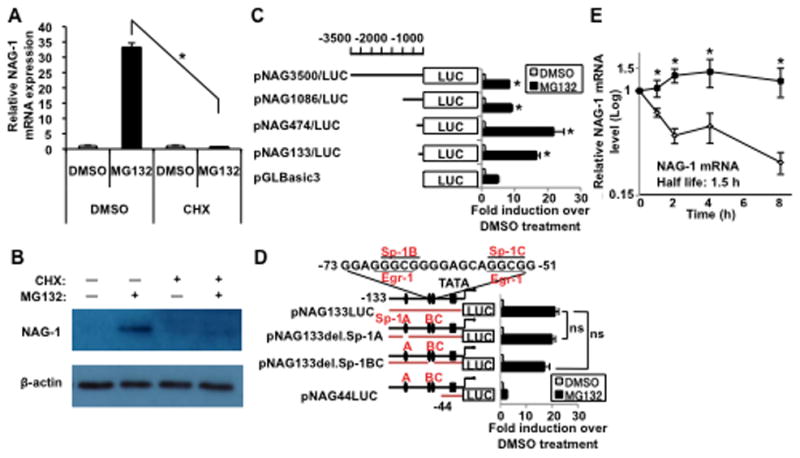
(A and B) U87MG cells were treated with cycloheximide (5 μg/ml) for 30 min, followed by treatment with 1 μM MG132 for 12 h. (A) Quantitative RT-PCR analysis depicts the induction of NAG-1 mRNA levels. Values are mean ± SD of three independent experiments. * p < 0.05. (B) Western blotting analysis depicts the 35-kDa pro-form of the NAG-1 protein and β-actin expression. (C) The indicated human NAG-1 promoter regions were fused to the luciferase (LUC) reporter gene. Each construct was co-transfected with pRL null vector into U87MG cells for 48 h, followed by treatment with 1 μM MG132 for an additional 18 h. Promoter activities were measured by luciferase activity. The y-axis shows fold induction (relative luciferase activity of MG132 normalized DMSO treated cells). * p < 0.05. (D) NAG-1 promoter constructs with internal deletion mutations are described. The internal deletion is shown by a gap in the bar. Wild-type (pNAG133/LUC and pNAG44/LUC) or Sp-1 site deletion (pNAG133del.Sp-1) reporters and pRL-null were co-transfected into U87MG cells and followed as in (C). The results are mean ± SD of four independent transfections. ns indicates p > 0.05. (E) U87MG cells were treated with 1 μM MG132 for 18 h. Nascent RNA synthesis was inhibited with 10 μM actinomycin D, and RNA was harvested at the indicated times after treatment. NAG-1 mRNA levels were adjusted to those of β-actin and measured by quantitative RT-PCR analysis. Results are plotted as the ratio of the mRNA level present when actinomycin D was added at time 0 h. * p < 0.05.
3.4. MG132 increases NAG-1 mRNA stability
MG132 increased NAG-1 promoter luciferase activity 20-fold above that of the DMSO (Fig. 3C), whereas NAG-1 mRNA induction by MG132 increased more than 100-fold (Fig. 2A, D), suggesting that other mechanisms may be involved in MG132-induced NAG-1 mRNA/protein expression. MG132 has been reported to regulate adenylate/uridylate-rich elements (AREs)-containing mRNAs such as p21WAF1/CIP1 by increasing mRNA stability [16]. NAG-1 contains four AUUUA motifs in its 3′ untranslated region that may destabilize NAG-1 mRNA [12]. The effect of MG132 on NAG-1 mRNA stability was examined with the transcription inhibitor actinomycin D. The half-life of NAG-1 mRNA was estimated to be approximately 1.5 h in DMSO-treated cells. After treatment with MG132, the half-life of NAG-1 mRNA increased dramatically to > 8 h (Fig. 3E). MG132 also stabilized NAG-1 mRNA in A172 cells (Supplementary Fig. 4). Thus, MG132 increased NAG-1 mRNA stability, which contributed to the increase in NAG-1 protein expression. Next, we explored specific AREs-binding proteins that contribute to MG132-mediated NAG-1 mRNA stability. HuR, an AREs-binding protein that stabilizes mRNA, is overexpressed in malignant gliomas [17]. HuR expression increased in both the nucleus and the cytoplasm of U87MG cells following MG132 treatment (Supplementary Fig. 5A). However, HuR knockdown did not alter NAG-1 expression level or NAG-1 mRNA stability (Supplementary Fig. 5B, C). Further studies will focus on which AREs-binding proteins are involved in MG132-induced stabilization of NAG-1 mRNA.
3.5. MG132 induces NAG-1 expression through a p38 MAPK-dependent mechanism
We sought to determine which signaling pathways mediate MG132-induced NAG-1 expression in U87MG cells. NAG-1 expression is controlled by various signaling pathways including Sp-1, Egr-1, p53, GSK-3, PKC, PKA, MEK, and p38 MAPK [6,7,8,9,12,15]. MG132 alters several signaling pathways by activating or inhibiting NFκβ, PI3K/Akt, p53, p21WAF1, p27Kip, JNK, and p38 MAPK in glioblastoma cells [4]. Several pharmacological inhibitors were used to determine the potential inhibition of MG132-induced NAG-1 expression. p38 MAPK inhibitors blocked MG132-induced NAG-1 expression, whereas the other inhibitors failed to inhibit MG132-induced NAG-1 expression (Supplementary Fig. 6). This result suggests that the p38 MAPK pathway mediates MG132-induced NAG-1 expression in U87MG cells. U87MG cells were treated with MG132, and p38 MAPK phosphorylation was examined by Western blotting analysis to determine if MG132 induces phosphorylation of p38 MAPK in U87MG cells. As shown in Fig. 4A, phosphorylation of p38 MAPK increased following MG132 treatment, although total p38 MAPK expression did not change. Phosphorylation of p38 MAPK increased as early as 3 h after treatment of cells with MG132, and a large increase was observed at 24 h in U87MG cells. Phosphorylation of p38 MAPK occurred earlier than the increase in NAG-1 expression (Fig. 4A). In the presence of SB203580, a p38 MAPK inhibitor, phosphorylation of p38 MAPK and expression of the pro-form and secreted form of NAG-1 mediated by MG132 was attenuated (Fig. 4A, B). MG132-induced NAG-1 mRNA was inhibited by adding a p38 MAPK inhibitor (Fig. 4E). Thus, MG132 activated the p38 MAPK pathway through p38 phosphorylation leading to the induction of NAG-1. These experiments were repeated using A172 cells to assess whether the regulation of NAG-1 induction by this pathway was a cell line-specific response. Induction of NAG-1 in response to MG132 treatment was observed but was significantly suppressed by SB203580 pretreatment (data not shown). We used p38 MAPK-specific siRNA to knockdown p38 to obtain further evidence for the role of this pathway. U87MG cells transfected with p38 siRNA prior to MG132 treatment prevented induction of NAG-1 in comparison with that in control siRNA-transfected cells, confirming the role of the p38 MAPK pathway in MG132-induced NAG-1 activation (Fig. 4C). We next sought to determine if transcriptional and post-transcriptional mechanisms were dependent on p38 MAPK activation. NAG-1 promoter activity was examined in the presence of a p38 MAPK inhibitor. U87MG cells were transfected with the NAG-1 promoter construct pNAG133/LUC and then treated with SB203580, followed by treatment with MG132. As shown in Fig. 4D, inhibiting p38 MAPK suppressed MG132-induced NAG-1 promoter activity. The contribution of the p38 MAPK inhibitor on the MG132-mediated increase in NAG-1 mRNA half-life was also tested. Inhibiting p38 MAPK phosphorylation blocked the MG132 increase in the half-life of mRNA but did not alter mRNA stability in the absence of MG132 (Fig. 4F). These results suggest that transcriptional and post-transcriptional induction of NAG-1 by MG132 requires activation of the p38 MAPK signaling pathway.
Fig. 4. MG132 induces NAG-1 expression through a p38 MAPK-dependent mechanism.
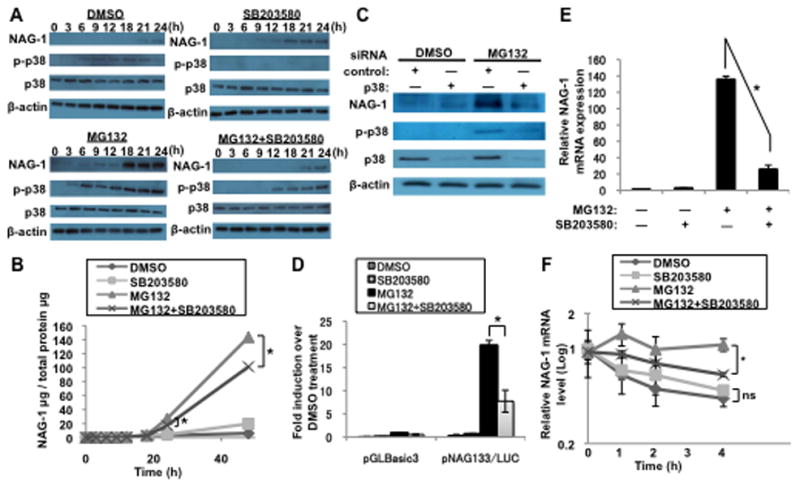
(A and B) U87MG cells were pretreated with 20 μM SB203580 for 1 h and then treated with 1 μM MG132 for the indicated times. (A and C) Western blotting analysis depicts the 35-kDa pro-form of the NAG-1 protein, phospho-p38 MAPK (p-p38), p38 MAPK (p38), and β-actin expression. (B) The NAG-1 protein secreted into the media was measured by ELISA. Values are mean ± SD of three independent experiments. * p < 0.05. (C) Control nontarget siRNA or p38 siRNA were transfected into U87MG cells for 48 h. Cells were then treated with 1 μM MG132 for 24 h. (D) U87MG cells were transfected with the NAG-1 promoter construct pNAG133/LUC. At 48 h after transfection, the cells pretreated with 50 μM SB203580 for 1 h and then treated with 1 μM MG132 for an additional 18 h. Firefly luciferase activity was normalized to that of Renilla luciferase. * p < 0.05. (E and F) U87MG cells were pretreated with 20 μM SB203580 for 1 h. The cells were then incubated with 1 μM MG132 for an additional 18 h. (E) NAG-1 mRNA levels were determined by quantitative RT-PCR. * p < 0.05. (F) Nascent RNA synthesis was inhibited with 10 μM actinomycin D, and RNA harvested at the indicated times after treatment. NAG-1 mRNA levels were measured by quantitative RT-PCR analysis and adjusted to those of β-actin. Results are plotted as the ratio of the mRNA level present at time 0 h when actinomycin D was added. * p < 0.05. ns indicates p > 0.05.
4. Discussion
We have shown that the proteasome inhibitors MG132 and bortezomib, members of a novel class of anti-tumorigenic agents, induced NAG-1 expression in glioblastoma cell lines. Our results indicate that both transcriptional and post-transcriptional mechanisms were responsible for the robust increase in NAG-1 mRNA and protein in the presence of MG132. Post-transcriptional stabilization of NAG-1 mRNA appeared to be a major contributor to the increase in NAG-1 protein expression. Activation of the p38 MAPK pathway mediated both transcriptional and post-transcriptional induction of NAG-1 by MG132.
Previous studies have revealed a number of transcription factors that act on the NAG-1 promoter. The Sp-1/Egr-1 binding site is responsible for maintaining expression in a basal state [6,7,9]. Increases in NAG-1 expression after treatment with anti-cancer drugs are mediated primarily by Sp-1, Egr-1, and p53 [6,7,9]. Activation of the p53 pathway causes a dramatic increase in NAG-1 expression; however, p53 is mutated in about 35% of GBM cases [3,4]. This makes glioblastoma cells with p53 mutations resistant to drugs that depend on p53 activity. These findings underscore the importance of developing therapies that do not rely on the p53 pathway. A recent study reported that treating glioblastoma cells with MG132 stimulates several signaling pathways including p38 MAPK, resulting in cell death and inhibited cell growth [4]. While transient induction of p38 MAPK may provide a survival signal, persistent activation induces cell death [18]. After MG132 treatment of U87MG cells, NAG-1 expression began to increase after 12 h, and the levels remained high for at least 24 h after treatment. The time course for NAG-1 expression after MG132 treatment may be consistent with the notion that persistent activation of p38 MAPK induces cell death. Taken together, the NAG-1/p38 MAPK signaling pathway may play a critical role in the anti-glioblastoma activity of MG132.
The observation that MG132 induced NAG-1 significantly by stabilizing NAG-1 mRNA is important when considering the effects of proteasome inhibitors on glioblastoma cells. The NAG-1 promoter region is highly methylated in many glioblastomas and their derived cell lines [13]. This hypermethylation is frequently detected in CpG islands located near the Sp-1/Egr-1 and p53 binding sites of the NAG-1 promoter [13]. A previous study showed that CpG promoter methylation blocks NAG-1 induction by nonsteroidal anti-inflammatory drugs (NSAIDs) and could cause resistance to NSAID-based therapy [13]. Because MG132 increases NAG-1 expression through a post-transcriptional mechanism, hypermethylated cell lines such as A172 continue to respond to MG132. Glioblastoma characterized by hypermethylation of the NAG-1 promoter could be sensitive to the effects of MG132-mediated NAG-1 induction.
NAG-1 acts as a tumor suppressor and induces cell death in the early stages of cancer. NAG-1 expression may be involved in cell death induced by MG132 or inhibit transformation during the early stage of glioma tumorigenesis similar to other TGF-β superfamily members [19]. The expression of NAG-1 is lower in glioblastoma cell lines than that in benign glioma cell lines and normal human astrocytes [13]. NAG-1 expression leads to glioblastoma cell death and inhibits the growth of glioblastoma cells on soft agar [11,13]. NAG-1 overexpression in human glioblastoma LN-Z308 cells inhibits tumor growth in nude mice xenografts [20]. In contrast, NAG-1 promotes proliferation of glioblastoma cells in murine SMA-560 glioblastoma cells, and depleting NAG-1 delays growth of these cells in syngeneic mice [21]. Elevated levels of NAG-1 in cerebrospinal fluid of patients are associated with GBM and shorter survival [22]. The elevated NAG-1 levels are most likely a reflection of late stage tumor growth rather than a driving force for tumor development [9]. However, no transgenic mouse studies have determined if NAG-1 inhibits glioblastoma development, although some studies clearly indicate that NAG-1 has tumor suppressor activity [14]. Future studies will be needed to address the apparent dichotomous role of NAG-1 in tumorigenesis.
In conclusion, understanding the induction of NAG-1 by the p38 MAPK pathway may provide alternative and improved therapeutic strategies for the use of proteasome inhibitors in glioblastoma cells.
Supplementary Material
Highlights.
Proteasome inhibitor MG132 induces NAG-1 expression in glioblastoma cells.
MG132 increases NAG-1 promoter activity and mRNA stability.
MG132 induces NAG-1 expression through a p38 MAPK-dependent mechanism.
Acknowledgments
We thank Carl Bortner, Maria Sifre, and the Flow Cytometry Center, NIEHS, for assistance with the flow cytometry analysis and Dr. Xingya Wang, Kaliopi C. Chrysovergis, and Justin Kosak for providing technical assistance. This research was supported, in part, by the NIEHS, NIH intramural research program.
Abbreviations
- NAG-1
nonsteroidal anti-inflammatory drug-activated gene-1
- GDF-15
growth differentiation factor-15
- GBM
glioblastoma multiforme
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Polivka J, Jr, Polivka J, Rohan V, Topolcan O, Ferda J. New molecularly targeted therapies for glioblastoma multiforme. Anticancer Res. 2012;32:2935–2946. [PubMed] [Google Scholar]
- 2.Frankland-Searby S, Bhaumik SR. The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta. 2012;1825:64–76. doi: 10.1016/j.bbcan.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM) Oncogene. 2005;24:344–354. doi: 10.1038/sj.onc.1208225. [DOI] [PubMed] [Google Scholar]
- 4.Zanotto-Filho A, Braganhol E, Battastini AM, Moreira JC. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Invest New Drugs. 2012 doi: 10.1007/s10637-012-9804-z. [DOI] [PubMed] [Google Scholar]
- 5.Kubicek GJ, Werner-Wasik M, Machtay M, Mallon G, Myers T, Ramirez M, Andrews D, Curran WJ, Jr, Dicker AP. Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys. 2009;74:433–439. doi: 10.1016/j.ijrobp.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baek SJ, Eling TE. Changes in gene expression contribute to cancer prevention by COX inhibitors. Prog Lipid Res. 2006;45:1–16. doi: 10.1016/j.plipres.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Eling TE, Baek SJ, Shim M, Lee CH. NSAID activated gene (NAG-1), a modulator of tumorigenesis. J Biochem Mol Biol. 2006;39:649–655. doi: 10.5483/bmbrep.2006.39.6.649. [DOI] [PubMed] [Google Scholar]
- 8.Shim M, Eling TE. Vitamin E succinate induces NAG-1 expression in a p38 kinase-dependent mechanism. Mol Cancer Ther. 2008;7:961–971. doi: 10.1158/1535-7163.MCT-07-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Baek SJ, Eling T. COX inhibitors directly alter gene expression: role in cancer prevention? Cancer Metastasis Rev. 2011;30:641–657. doi: 10.1007/s10555-011-9301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimeault M, Batra SK. Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer. J Cell Physiol. 2010;224:626–635. doi: 10.1002/jcp.22196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshioka H, Kamitani H, Watanabe T, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) expression is increased by the histone deacetylase inhibitor trichostatin A. J Biol Chem. 2008;283:33129–33137. doi: 10.1074/jbc.M805248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi K, Lee SH, Eling TE, Baek SJ. A novel peroxisome proliferator-activated receptor gamma ligand, MCC-555, induces apoptosis via posttranscriptional regulation of NAG-1 in colorectal cancer cells. Mol Cancer Ther. 2006;5:1352–1361. doi: 10.1158/1535-7163.MCT-05-0528. [DOI] [PubMed] [Google Scholar]
- 13.Kadowaki M, Yoshioka H, Kamitani H, Watanabe T, Wade PA, Eling TE. DNA methylation-mediated silencing of nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) in glioma cell lines. Int J Cancer. 2012;130:267–277. doi: 10.1002/ijc.26082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baek SJ, Okazaki R, Lee SH, Martinez J, Kim JS, Yamaguchi K, Mishina Y, Martin DW, Shoieb A, McEntee MF, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology. 2006;131:1553–1560. doi: 10.1053/j.gastro.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Baek SJ, Horowitz JM, Eling TE. Molecular cloning and characterization of human nonsteroidal anti-inflammatory drug-activated gene promoter. Basal transcription is mediated by Sp1 and Sp3. J Biol Chem. 2001;276:33384–33392. doi: 10.1074/jbc.M101814200. [DOI] [PubMed] [Google Scholar]
- 16.Mazroui R, Di Marco S, Kaufman RJ, Gallouzi IE. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol Biol Cell. 2007;18:2603–2618. doi: 10.1091/mbc.E06-12-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine- and uridine-rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- 18.Weng MS, Ho YS, Lin JK. Chrysin induces G1 phase cell cycle arrest in C6 glioma cells through inducing p21Waf1/Cip1 expression: involvement of p38 mitogen-activated protein kinase. Biochem Pharmacol. 2005;69:1815–1827. doi: 10.1016/j.bcp.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Jennings MT, Pietenpol JA. The role of transforming growth factor beta in glioma progression. J Neurooncol. 1998;36:123–140. doi: 10.1023/a:1005863419880. [DOI] [PubMed] [Google Scholar]
- 20.Albertoni M, Shaw PH, Nozaki M, Godard S, Tenan M, Hamou MF, Fairlie DW, Breit SN, Paralkar VM, de Tribolet N, Van Meir EG, Hegi ME. Anoxia induces macrophage inhibitory cytokine-1 (MIC-1) in glioblastoma cells independently of p53 and HIF-1. Oncogene. 2002;21:4212–4219. doi: 10.1038/sj.onc.1205610. [DOI] [PubMed] [Google Scholar]
- 21.Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, Tabatabai G, Wick W, Weller M, Wischhusen J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res. 2010;16:3851–3859. doi: 10.1158/1078-0432.CCR-10-0705. [DOI] [PubMed] [Google Scholar]
- 22.Shnaper S, Desbaillets I, Brown DA, Murat A, Migliavacca E, Schluep M, Ostermann S, Hamou MF, Stupp R, Breit SN, de Tribolet N, Hegi ME. Elevated levels of MIC-1/GDF15 in the cerebrospinal fluid of patients are associated with glioblastoma and worse outcome. Int J Cancer. 2009;125:2624–2630. doi: 10.1002/ijc.24639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.