

Abstract
We previously reported that nitric oxide (NO) reduces the rate of bacteremia and maternal mortality in pregnant rats with uterine infection by Escherichia coli expressing the Dr Fimbria (Dr+). The epithelial invasion of Dr+ E. coli is dependent on the expression level of its cellular receptor decay accelerating factor (DAF). NO reduces the rate of bacteremia by down-regulating the expression of DAF. In this study, we elucidated the role of transcription factor Sp1 and RNA binding protein HuR in the down-regulation of human DAF by NO. We generated a series of deletion mutant constructs of DAF gene 5′-untranslated region and mapped NO-response region upstream to the core promoter region of the DAF gene. One of the several Sp1 binding sites in the DAF 5′-untranslated region was located within the NO-response region. The binding of Sp1 to this site was inhibited by NO. Furthermore, NO also promoted the degradation of DAF mRNA. The 3′-untranslated region of DAF harbors an AU-rich element and this element destabilized the mRNA transcript. The NO promoted the rapid degradation of DAF mRNA by inhibiting the binding of mRNA stabilizing protein HuR to this AU-rich region. The inhibition of binding of HuR to AU-rich region was due to the S-nitrosylation of one or more cysteine residues by NO. Thus, these data reveal the molecular mediators of transcriptional and post-transcriptional regulation of DAF by NO with implications in pathophysiology related to DAF.
Keywords: Nitric oxide, Decay Accelerating Factor, mRNA stability, Sp1, S-nitrosylation
Introduction
The urogenital microbial infection in pregnancy is an important cause of maternal and neonatal morbidity and mortality [1–3]. Previous studies in our laboratory have suggested that NO has a protective role in the urogenital tract against Escherichia coli infection during pregnancy. We have shown that inhibition of NO synthesis in pregnant rats with an intrauterine infection increases maternal death [4]. Further, sensitivity of female rat or mouse urinary tract to E. coli infection increases with inhibition of NO [4;5]. We have also shown that the expression of inducible NO synthase (NOS II) was up-regulated in response to intra uterine infection resulting in a spontaneous, localized increase in NO production [6]. Uropathogenic E. coli strains of the O75 serotype which express Dr adhesins account for about 40% of pyelonephritis cases in the third trimester and adherence and invasion of these E. coli in human cervical epithelial cells depends on the presence of Dr adhesins [7–9]. The epithelial cell entry of Dr-fimbriated E coli was shown to be facilitated by a cellular receptor, decay accelerating factor (DAF) [10;11]. The binding of Dr+ E. coli to the short consensus repeat 3 of DAF, expressed in Chinese hamster overy (CHO) cells was found to be critical for internalization to occur [10]. Recently we have shown that in vitro invasion of Dr+ E. coli of an epithelial cell line is directly related to NO-regulated expression of DAF [12]. Elevated NO production significantly decreased DAF protein and mRNA levels in endometrial epithelial cell line, Ishikawa, in a time- and dose-dependent manner [12].
DAF is a glycosylinositol anchored complement regulatory membrane protein which is constitutively expressed at high levels on all serum exposed cells including epithelial cells. DAF protects the cells from autologous complement-mediated injury by accelerating the dissociation of preformed C3/C5 convertase complexes of both the classical and alternative pathways of the complement system [13]. The deficiency or over expression of DAF leads to inefficient complement control and excess complement inhibition respectively. Deficiency of DAF is linked to many disease conditions like nocturnal hemoglobinuria, autoimmune diseases and pregnancy loss due to luteal phase defect [14;15]. Very often these conditions lead to pregnancy related complications such as preeclampsia. In contrast, higher levels of DAF expression are associated with tumorigenesis, decreased tumor cell lysis and metastasis of many types of cancers [16;17]. The transcription factors and the post-transcriptional events that lead to the induced up-regulation of DAF by various factors such as TNF-α, IL4, IL1β, VEGF, HB-EGF and PGE2 have been characterized [18–21]. But there is a gap in the knowledge on the mechanism of DAF down-regulation by NO.
In the present study, we elucidated the role of transcription factor Sp1 and RNA binding protein HuR in the down-regulation of DAF by NO. Human endometrial cell line Ishikawa, which expresses DAF and susceptible to invasion of Dr+ E. coli was used to elucidate the mechanism of NO mediated down-regulation of DAF. We identified a NO-regulatory region in the 5′-UTR upstream to the DAF gene. The binding of transcription factor Sp1 to this NO-regulatory region was inhibited by NO. Furthermore, we identified an AU-rich mRNA destabilizing element in the 3′-untranslated region of DAF gene. The binding of mRNA stabilizing protein HuR to this AU-rich region was inhibited by NO. We found that S-nitrosylation of one or more cysteine residues of HuR was responsible for its reduced binding to mRNA.
Results
DAF gene upstream sequence between nucleotides − 437 and − 277 contains NO regulatory region
To determine whether NO alters the DAF promoter activity, reporter assay was performed. A 0.734 kb fragment of DAF gene upstream sequence was attached to β-galactosidase reporter system and transfected into Ishikawa cells. Transfected Ishikawa cells were then treated with NO donor diethylenetriamine-NO([Z]-1-[2-aminoethyl]-N-[2-ammonioethyl]amino diazen-1-ium-1,2-diolate (DTNO, 1 mM) or spent DTNO (1 mM) or the substrate for NOS enzyme L-arginine (0.3 mM) or L-arginine combined with NOS inhibitor NG-nitro-L-argininemethyl ester (L-NAME, 3 mM). The β-galactosidase activity in cells treated with DTNO and L-arginine were significantly less compared to that of control cells (Fig. 1A). Treatment with spent DTNO did not alter the β-galactosidase activity compared to control. The L-NAME which is a non selective inhibitor of NOS enzyme reversed the effect of L-arginine. Taken together theses results indicated that NO either supplied exogenously in the form of DTNO or produced endogenously by NOS enzyme utilizing the substrate L-arginine decreased the DAF promoter activity. Next, deletion mutagenesis was performed to generate a series of 5’-deletions in the DAF gene upstream sequence in order to identify the regions essential for the regulation of DAF gene (Fig. 1B). Transfection of these deletion mutant plasmids into Ishikawa cells revealed that the upstream sequence between the nucleotides − 602 to −437 and −191 to −108 are essential for DAF gene expression (Fig. 1C). The results also revealed that a negative regulatory element is present in the DAF upstream sequence between the nucleotides − 437 to − 277. The transfected cells harboring theses deletion mutants were exposed to DTNO to identify the region regulated by NO. The results demonstrated that the deletion mutants DAF 0.734, DAF 0.602 and DAF 0.437 of the 5’-upstream region exhibited reduced β-galactosidase activity upon exposure to DTNO (Fig. 1C). This indicated that the region between nucleotides − 437 and − 277 has the sequence regulated by NO.
FIGURE 1.
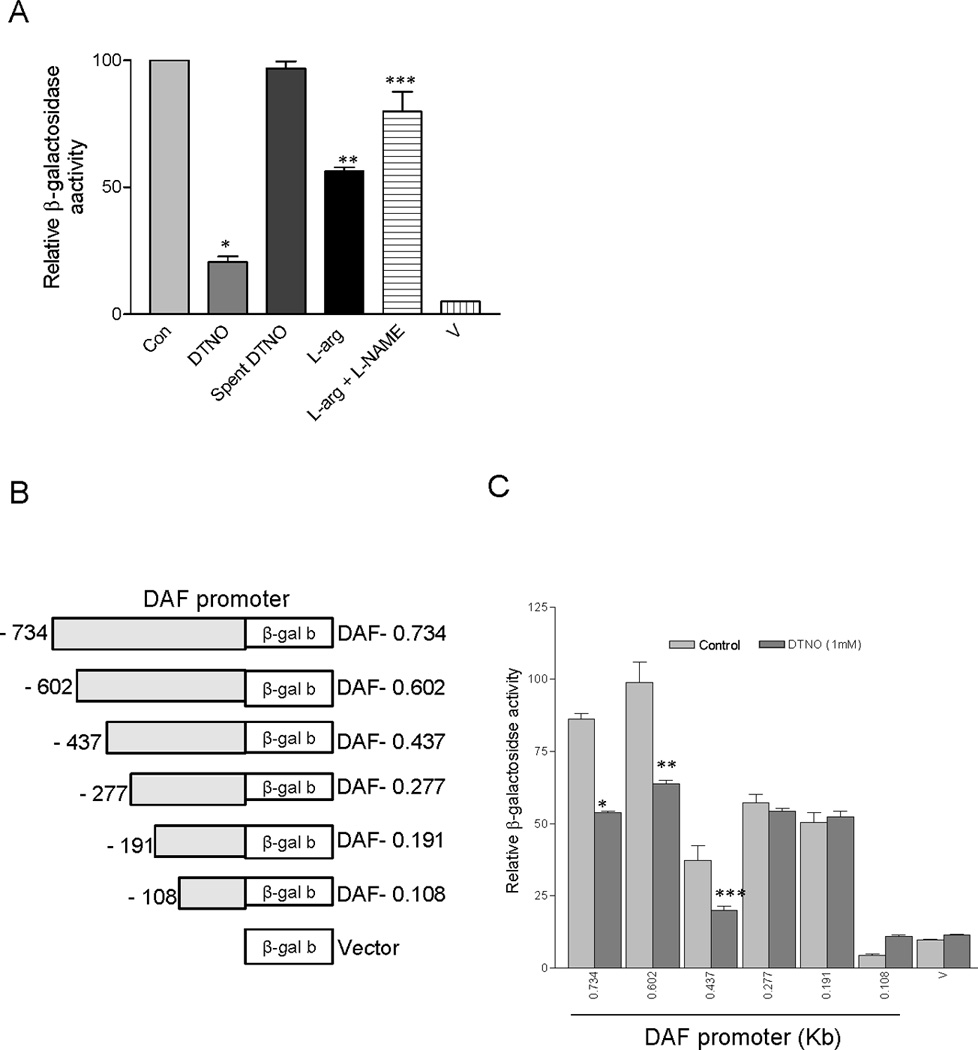
DAF promoter fragment between nucleotides − 437 and − 277 harbors NO regulatory region. (A) Ishikawa cells were transiently transfected with DAF promoter-β- galactosidase reporter plasmid, treated with DTNO (1 mM) or spent DTNO (1 mM) or the substrate for NOS enzyme L-arginine (L-arg, 0.3 mM) or L-arginine combined with NOS inhibitor L-NAME (3 mM) for 24h and the β- galactosidase activity was measured. Vector control (V) devoid of DAF promoter was also used as negative control. All experiments were performed three times, and one set of data is presented. Error bars indicate S.E of triplicate samples (*, **, p < 0.001versus control; *** p < 0.05 versus L-arg). (B) Systematic representation and cloning strategy of DAF gene promoter into pβgalb reporter vector. (C) DAF promoter deletions were cloned into pβgalb reporter vector and transfected to Ishikawa cells. Cells were treated with DTNO (1mM) for 24 h, and β-galactosidase activity was measured. All experiments were performed three times, and one set of data is presented. Error bars indicate S.E of triplicate samples (*, p < 0.0002; **, p < 0.041; ***, p < 0.0027 versus control in each group).
Binding of transcription factor Sp1 to DAF gene upstream sequence between nucleotides − 424 and − 416 is altered by NO
NO inhibits the binding of Sp1 to DNA through a cAMP-PKA dependent pathway [22]. Therefore, we analyzed NO-regulated region for the Sp1 binding site and found that a putative binding site for Sp1 was present between nucleotides − 424 to − 416 (Fig 2A). Transcription factor ELISA was performed to examine whether NO alters the binding of Sp1 to this site. Nuclear fractions were extracted from control and DTNO treated cells and the binding of Sp1 was assessed by ELISA. The binding of Sp1 present in the nuclear fraction of control cells was more compared to that in the DTNO exposed cells (Fig. 2B). The transcription factor NFκB which lacks a binding site in the NO-regulated region did not show binding indicating the specificity of transcription factor ELISA. One can interpret this data in two ways, first, the reduced binding may be due to a decrease in the Sp1 expression by NO and second, the reduced binding may due to a decrease in Sp1 binding affinity to DNA because of post-translational modification induced by NO. Western blot analysis of Sp1 expression in Ishikawa cells exposed to DTNO for 4 h revealed that NO has no effect on the Sp1 expression (Fig. 2C). Therefore, reduced binding of Sp1 to DNA was due to a decrease in binding affinity. The inhibition of binding of Sp1 to − 424 to − 416 site was further confirmed by Chromatin Immunoprecipitation (ChIP) assay. The extent of association between Sp1 and its binding site in DTNO treated and non-treated cells was assessed by immuno-precipitation of Sp1 followed by quantitative real time PCR of Sp1 bound DNA fragment of NO-regulated region (− 424 to − 416 region). The results revealed that the association of this DNA with Sp1 was 7 fold less in DTNO exposed cells compared to control cells (Fig. 2D). The cAMP can also regulate transcription through cAMP response element (CRE) [23]. But there was no putative CREB binding site on the NO-regulated region of DAF gene 5′-UTR. However, there was a CRE site in the core promoter region of DAF gene between the nucleotides −166 to −127 along with an additional Sp1 binding site (Fig. 2E). If NO has an effect on the binding of CRE and Sp1 to these sites, was assessed by transcription factor ELISA. The results indicated that the binding of Sp1 and CRE from the nuclear extracts of control and DTNO treated cells was not altered (Fig 2F). Taken together, these data demonstrated that NO inhibited the binding of Sp1 to the NO-regulated region and thus decreased DAF expression.
FIGURE 2.
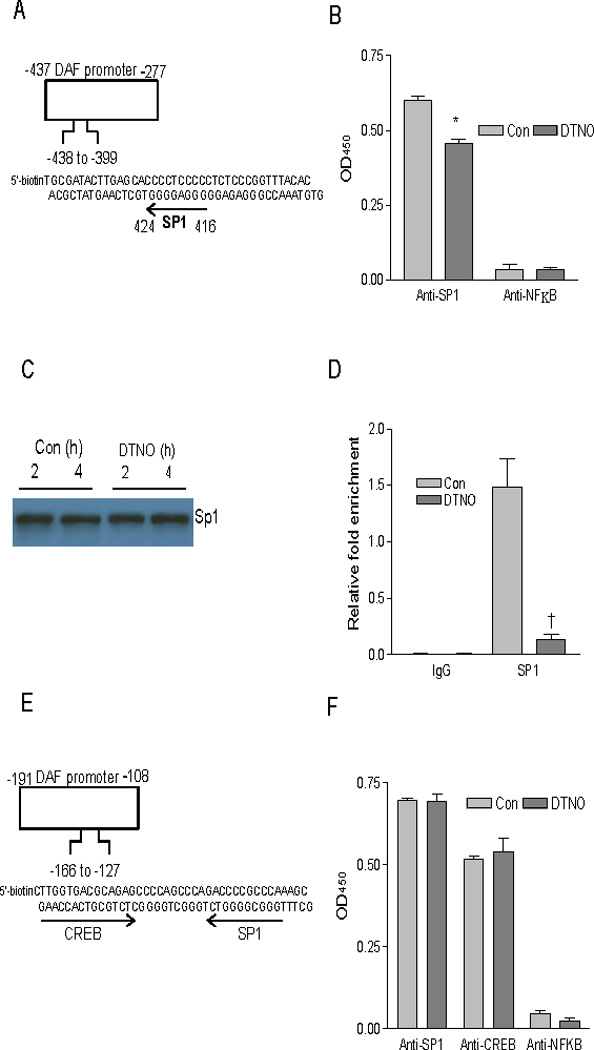
Binding of transcription factor Sp1 to DAF promoter between nucleotides − 424 and − 416 is altered by NO. (A) Nucleotide sequence of 5’-biotin tagged DAF promoter fragment between nucleotides − 438 to − 399 containing one putative Sp1 binding site. (B) Ishikawa cells were treated with DTNO (1 mM) for 4 h. Nuclear fractions from control and DTNO treated cells equivalent to 1.75 µg of protein were added to DAF promoter fragment (−438 to −399) attached to streptavidin coated wells in a micro plate. The amount of Sp1 protein bound to DAF promoter was measured using anti-Sp1 antibody and secondary antibody conjugated to HRP. (*, p < 0.0003 versus control). (C) Expression of Sp1 protein in nuclear extracts of control and DTNO treated cells was assessed by western blotting. (D) ChIP and quantitative qRT-PCR. The control and DTNO treated cells were fixed with formaldehyde, and cross-linked, and the chromatin was sheared. The chromatin was immunoprecipitated with anti-Sp1 antibody or IgG. The binding of Sp1 to the DAF promoter fragments − 438 to − 399 was measured by quantitative real time PCR using specific primers and relative quantitation method. The amounts of immunoprecipitated DNA were normalized to the inputs and plotted (†, p < 0.0063 versus control). (E) Nucleotide sequence of 5’-biotin tagged DAF promoter fragment between nucleotides − 166 to − 127 containing one putative Sp1 binding site and one putative CREB binding site. (F) Nuclear fractions from control and DTNO treated cells equivalent to 1.75 µg of protein were added to DAF promoter fragment (−166 to −127) attached to streptavidin coated wells in a micro plate. The amount of Sp1 or CREB protein bound to DAF promoter was measured using anti-Sp1 or anti-CREB antibody and secondary antibody conjugated to HRP. All experiments were performed three times, and one set of data is presented. Error bars indicate S.E of triplicate samples.
DAF 3′-UTR contains destabilizing elements between nucleotides 1860 and 2100
The adenosine/uracil-rich element (ARE), an mRNA sequence of the types WUAUUUAUW, and WWAUUUAWW has been implicated in mRNA stability [24]. The 3′-UTR analysis of DAF mRNA has revealed a region very rich in AU content between the nucleotides 1860 and 2100 from the transcription start site. This region harbors three overlapping nonameric sequences of the type WUAUUUAUW between nucleotides 1909 and 1928. One more nonameric sequence of the type WWAUUUAWW was found in the region between nucleotides 2052 and 2061 (Fig. 3A). However, the AU content of a stretch of 62 nucleotides sequence including the flanking upstream and downstream sequences of WUAUUUAUW (1909–1928) was high (82%) compared to that of WWAUUUAWW (2052–2061) with the similar length (76%). These features suggest that the sequence between the nucleotides 1909 and 1928 may play a major role in the destabilization of DAF mRNA. To assess the role of this AU-rich region in DAF mRNA expression, a series of DAF 3′-UTR deletions were generated and attached downstream to β-galactosidase reporter gene in pβgalcontrol plasmid under the SV40 promoter (Fig. 3B). Transfection of these plasmids into Ishikawa cells revealed that deletion of ARE in the Δ0.99 plasmid increased β-galactosidase activity significantly (p < 0.001 compared to UTR 1.35) indicating a negative role of ARE in DAF gene expression (Fig. 3C). Further, we also observed a decreased β-galactosidase activity of the plasmid Δ0.72 in which ARE upstream flanking sequence was deleted suggesting the involvement of additional elements in the regulation. These data together demonstrate that the ARE region present down-stream to DAF gene between nucleotides 1860 and 2100 reduces the expression of DAF gene.
FIGURE 3.
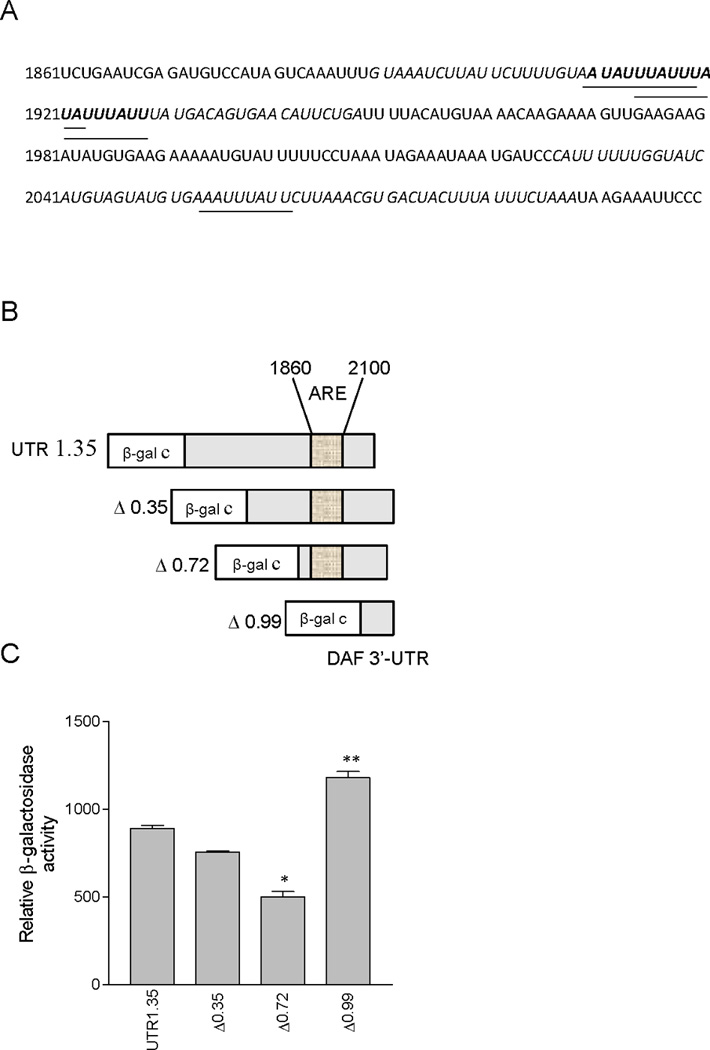
DAF 3’-UTR contains destabilizing elements between nucleotides 1860 and 2100. (A) DAF 3’-UTR was examined for AU rich elements (ARE) using the programme AREsite (http://rna.tbi.univie.ac.at/AREsite). The AU rich regions are indicated by italicized letters. The putative binding sites for elav family protein HuR are indicated by underscores and bold letters. Letters with only underscores indicate WWAUUUAWW type of HuR binding sites. Letters with underscores and bold indicate WUAUUUAUW type of HuR binding sites. (B) Systematic representation and cloning strategy of DAF gene 3’-UTR into pβgalc reporter vector. (C) DAF 3’-UTR deletions were cloned into pβgalc reporter vector and transfected to Ishikawa cells. Cells were incubated for 48 h, and β- galactosidase activity was measured. Experiment was performed three times, and one set of data is presented. Error bars indicate S.E of triplicate samples (*, **, p < 0.001 versus UTR 1.35).
DAF mRNA half-life is altered by NO
To examine whether NO promotes the degradation of DAF mRNA, transcription was arrested in Ishikawa cells by treating them with Actinomycin D and the relative amount of DAF mRNA was measured as a function of time (the addition of DTNO or L-arg was considered as 0 hour). The decay rate of DAF mRNA was less in DTNO and L-arginine treated cells compared to untreated cells (Fig. 4A). The DAF mRNA decay rate in the cells treated with spent DTNO or L-arginine combined with L-NAME was comparable to that of untreated cells. In order to quantify the rate of DAF mRNA decomposition, decay curves were constructed by plotting percentage remaining mRNA versus time (Fig. 4B). The mRNA half-life calculated using the decay constant derived from decay curves indicated 3 and 2.5 fold decrease in the DAF mRNA half-life with DTNO and L-arginine treatment respectively (Fig. 4C). The DAF mRNA half-life in the cells treated with spent DTNO and L-arginine combined with L-NAME did not alter compared to control.
FIGURE 4.
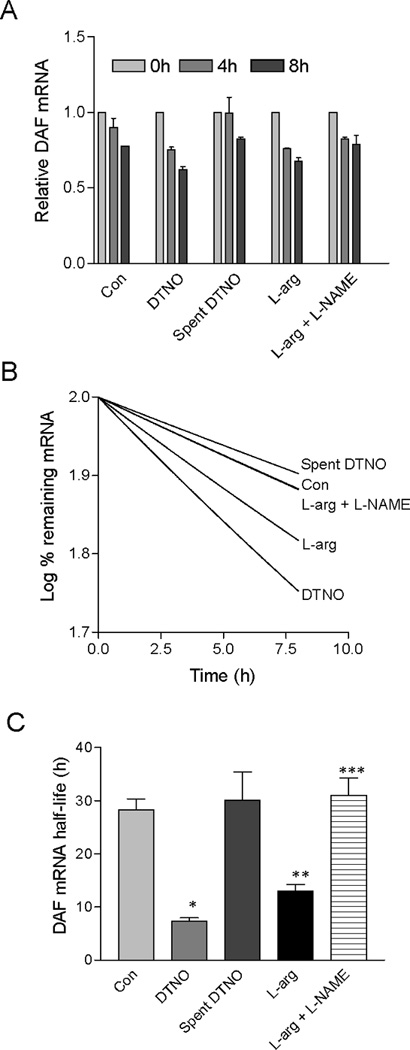
DAF mRNA half-life is altered by NO. (A) Ishikawa cells were treated with Actinomycin D (1 µg/ml) for 4 h and then exposed to DTNO (1 mM) or spent DTNO (1 mM) or the substrate for NOS enzyme L-arginine (0.3 mM) or L-arginine combined with NOS inhibitor L-NAME (3 mM) for different time periods. The fold changes in the DAF mRNA were measured for each treatment by RT-qPCR. Experiment was performed three times and one set of data is presented. Error bars indicate S. E. of triplicate samples. (B) Log % remaining DAF mRNA versus time was plotted for control and treated cells. Decay constants were calculated from non-linear single exponential decay curves. (C) The mRNA half-life was calculated using the formula t1/2 = ln2/kdecay, where t1/2 is DAF mRNA half life and kdecay is decay constant. Error bars indicate S. E. of three experiments. (*, **, p < 0.05 versus control; ***, p < 0.05 versus L-arg).
Binding of HuR protein to ARE was inhibited by NO
The human ELAV family protein HuR is an RNA binding protein which binds to ARE at the 3′-UTR of many mRNA’s and protects them from degradation by competing with destabilizing proteins for the binding site [24]. To examine whether NO inhibits the association of HuR with ARE at 3′-UTR of the DAF gene, RNA immunoprecipitation assay was performed. The HuR protein was immunoprecipitated and then the quantity of DAF mRNA bound to HuR protein was measured by qPCR. The results demonstrated that exposure of Ishikawa cells to DTNO for 4 hours resulted in 10 fold decrease in the quantity of DAF mRNA bound to HuR (Fig. 5A). One can argue that this decrease in the quantity of DAF mRNA bound to HuR may be because of a decrease in the DAF mRNA itself or a decrease in expression of HuR protein mediated by NO. To further assess whether NO significantly reduces the DAF mRNA expression and HuR protein levels, the relative amount of DAF mRNA and HuR mRNA in Ishikawa cells exposed to DTNO was measured by qPCR. The results indicated that DAF mRNA was in fact reduced by two fold and HuR mRNA was reduced significantly only after 8 hours of exposure to DTNO (Fig. 5B, C). However, HuR protein levels remained same even after 24 hours of exposure to DTNO (Fig. 5D). Although exposure of cells to DTNO for 4 hours reduced DAF mRNA levels by 2 fold, the magnitude of decrease in the HuR bound DAF mRNA due to DTNO exposure was much greater (10 fold) suggesting that NO actually inhibits the association between HuR and DAF mRNA.
FIGURE 5.
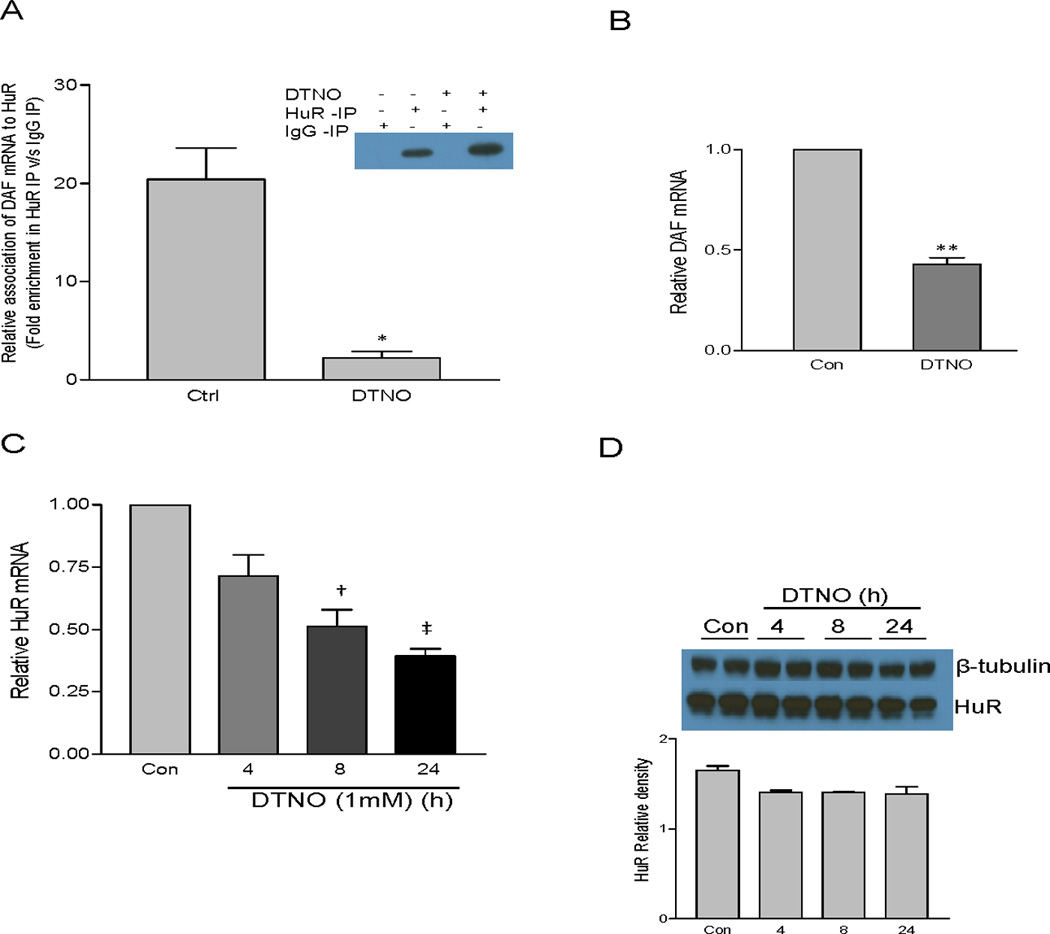
Binding of elav family protein HuR to DAF mRNA is altered by NO. (A) Ishikawa cells were treated with DTNO (1 mM) for 4 h. Amount of DAF mRNA bound to HuR was measured by RNA immunoprecipitation (RIP) assay and presented as fold change in HuR IP versus IgG IP. Experiment was performed three times. Error bars indicate S.E. of triplicate experiments. (*, p < 0.012). Efficiency of IP was checked by western blot (Inset). (B) Ishikawa cells were treated with DTNO (1 mM) for 4 h and the expression of DAF mRNA was measured by RT-qPCR. (**, p < 0.01) (C) Ishikawa cells were treated with DTNO (1 mM) for different periods of time and the relative expression of HuR mRNA was measured by RT-qPCR. Error bars indicate S.E. of triplicate samples (†, p < 0.01; ‡, p < 0.001). (D) Ishikawa cells were treated with DTNO (1 mM) for different periods of time and the expression of HuR protein was measured by western blot (upper panel). Densitometric measurement of HuR protein relative to β-actin is shown (lower panel).
The ability of NO to inhibit the association between HuR and DAF mRNA was further confirmed by an in vitro binding assay using recombinant HuR and 5’-biotin tagged ARE sequence derived from DAF mRNA (Fig. 6A). Different amounts of biotin tagged ARE were allowed to bind to avidin coated wells. A fixed amount of rHuR protein which was either exposed to DTNO in vitro or not exposed was then allowed to bind to this ARE. The amount of HuR bound to ARE was measured using HuR antibody and HRP conjugated secondary antibody. A plot of DAF ARE amount versus optical density at 450nm gave a hyperbolic curve (Fig. 6B). The hyperbolic curve of rHuR exposed to DTNO shifted to the right compared to that of control rHuR. This indicated that the amount of untreated rHuR bound to DAF ARE at any given concentration is more compared to DTNO exposed rHuR and therefore, the binding of DTNO exposed rHuR is weaker than the control rHuR.
FIGURE 6.
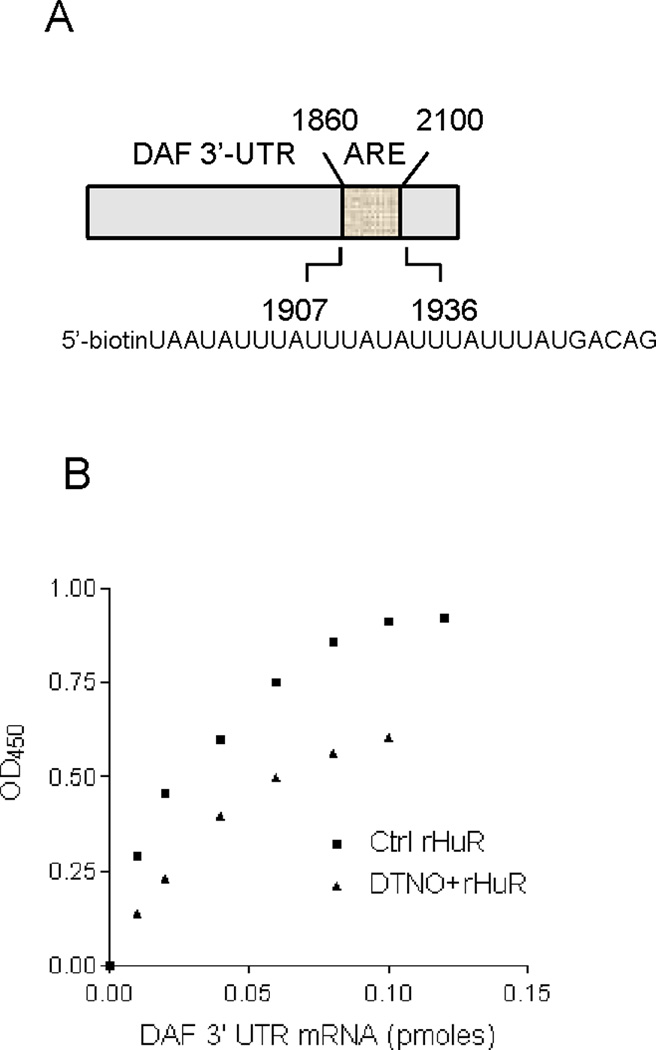
Invitro binding of rHuR to DAF mRNA ARE is altered by NO. (A) Nucleotide sequence of 5’-tagged fragment of DAF mRNA in the 3’-UTR-AU rich region containing putative HuR binding sites. (B) Recombinant HuR (8.7 nM) was exposed to DTNO (1 mM) for 1 h and excess and spent DTNO was removed by passing through desalting column. Equal volume (20 µl) of control and DTNO exposed rHuR was added to DAF mRNA fragment attached to streptavidin coated wells in a microplate. rHuR bound to DAF mRNA was measured using anti-HuR antibody and secondary antibody conjugated to HRP. Experiment was performed three times and one set of data is presented.
S-nitrosylation of HuR by NO reduces its binding to DAF mRNA
Next, the HuR protein was probed for NO induced posttranslational modification that affects its mRNA binding behavior. NO is known to activate many protein kinases through cGMP pathway and phosphorylates a number of target proteins. In order to examine whether phosphorylation of HuR is the mechanism of NO induced binding inhibition, HuR was immunoprecipitated from the Ishikawa cell lysate exposed to DTNO. The western blot with antiphospho amino acid antibody did not show any band corresponding to HuR. Akt, which is constitutively phosphorylated in Ishikawa cells, was used as positive control (Fig. 7A). The p38 MAP kinase can phosphorylate HuR and enhance its binding to ARE [25]. In Ishikawa cells exposed to NO, the phosphorylation of p38 MAP kinase was not enhanced compared to control cells indicating that p38 MAP kinase pathway is not activated in these cells (Fig. 7B). The nitrosylation of cysteine residues is another functionally important posttranslational modification mainly induced by NO. The S-nitrosylation of endogenous HuR in NO exposed Ishikawa cells was investigated by biotin switch assay in which S-nitrosylated cysteins in proteins were converted into cysteine-biotin using a biotinylating reagent (biotin-HPDP). The biotinylated proteins were then isolated using neutravidin coated agarose beads. The presence of HuR in the neutravidin pull-down fraction was investigated by western blot. The unbound fraction that was the supernatant left after neutravidin pull-down, had HuR protein irrespective of DTNO treatment. In contrast, HuR protein was detected in the neutravidin pull-down fraction only after DTNO treatment along with positive control β-actin which is known to be nitrosylated by NO in various cells. This clearly indicated that exposure of Ishikawa cells to DTNO, nitrosylates one or more cysteine residues in HuR (Fig. 7B). Thus there is a close association between S-nitrosylation of HuR and its reduced binding to 3′-UTR of DAF mRNA. Taken together, these results demonstrated that S-nitrosylation of HuR by NO inhibits its binding to DAF-ARE and thus reduces the mRNA half-life.
FIGURE 7.

S-nitrosylation of HuR by NO reduces its binding to DAF mRNA. (A) Ishikawa cells were exposed to DTNO (1 mM) for 4 h and the HuR protein was immunoprecipitated. Phosphorylation status of HuR in the IP sample was examined by western blot using anti-phosphoaminoacids antibody. Akt, which is constitutively phosphorylated in Ishikawa cells, was used as positive control. (B) Ishikawa cells were exposed to DTNO (1 mM) for 4, 8 and 12 h, and the expression of p38 MAP kinase and phospho-p38 MAP kinase was assessed by western blot. Bleomycin exposed cells were used as positive control. (C) Ishikawa cells were exposed to DTNO (1 mM) for 4 h, S-nitrosylation of HuR was examined by biotin switch assay. The eluate fraction contains neutravidin pull-down biotinylated proteins in the cell lysates including S-nitrosylated proteins that were biotinylated in the presence of biotin-HPDP. The unbound fraction contains non-biotinylated proteins existing in the supernatant of neutravidin pull-down. Presence of HuR in both the fractions was analyzed by western blot. The β-actin was used as positive control for S-nitrosylated protein.
Discussion
We have shown that NO down-regulates the DAF gene expression by inhibiting its promoter activity possibly through a decreased binding of Sp1 to one of its consensus binding sites in the distal promoter region. Further, decreased association of HuR with the 3′-UTR-ARE due to the S-nitrosylation of HuR by NO and consequent decrease in the stability of DAF mRNA also contributes to the down-regulation of DAF expression. The finding that DAF expression is regulated by NO is of interest, because previous reports describing the regulation of DAF have focused on cytokines and growth factors. Invariably all these factors up-regulate the DAF expression [18–21]. However, sodium butyrate has been shown to down-regulate DAF expression in colonic cancer cells by inhibiting basal promoter activity of DAF gene [26]. Unlike ubiquitosouly produced NO, butyric acid is produced in the colon by bacterial fermentation of dieteray fibers [27]. Although butyric acid is readily absorbed in to the blood it is rapidly cleared limiting its reach to various tissues and cells [28]. Compared to butyric acid the down-regulation of DAF by NO could be more prevalent in various cells because the activity of NO is not restricted to the site of its production and NO production is ubiquitous. The low molecualr weight thiols, S-nitrosylated proteins and the nitrosyl-metal complexes can carry NO over long distances. In this perspective, the mechanism of DAF regulation by NO gives further insight in to a number of pathophysiological conditions involving DAF. To our knowledge, data presented here provide for the first time a new insight on the mechanism of NO mediated down-regulation of DAF both at the transcriptional and post-transciptional levels. In addition, a brief exposure to NO can delocalize and internalize DAF protein from the cell surface lipid rafts, limiting the availabilty of DAF for bacterial binding [29]. Thus, NO appears to be an important host defense mechanism against E. coli infection by an immediate delocalization of pre-existing DAF from the cells surface and a long term down-regulation of DAF expression.
The regulation of DAF promoter activity by various factors has been studied by different groups. As mentioned earlier all of these studies described up-regulation of DAF promoter by various factors. The promoter region present within the first 206 nucleotides upstream of transcription start point containing binding sites for the transcription factors such as Sp1 and CRE is the main positive regulator of the DAF gene [13;30]. Results of our transient transfection studies in Ishikawa cells using a series of DAF gene upstrem deletion constructs are consistent with these previous reports. The deletion constructs from − 602 to − 191 reduced the promoter activity ~2–3 fold and further deletion down to − 108 reduced the promoter activity more than 90%. There are conflicting reports on the negative regulatory elements on DAF gene upstream sequence. Ewulonu et al [30] reported a negative regulator in the region − 355 to − 286 whereas Thomas et al [13] did not find any negative regulatory elements. However, with the human DAF promoter, in Ishikawa cells we found a negative regulator in the region − 437 to − 277 which is consistent to that reported by Ewulonu et al.
We mapped the NO response region on the DAF gene upstream sequence to − 437 to − 277 region. This region in the murine DAF gene contains one binding site each for transcription factors AP2 and NFκB [31]. However, in human DAF gene this region contains a binding site for transcription factor Sp1. Infact, we found 11 sites with high similarity to the consensus Sp1 binding site within − 300 region. Nitric oxide inhibited the binding of Sp1 to binding site in the − 437 to − 277 region but not to the site in the − 191 to − 108 region where the core promoter was found to be located. Regulation of gene expression by Sp1 through multiple binding sites is a known phenomenon [32;33]. Discrepancy in DNA binding activity of Sp1 protein due to its post-translational modification is commonly observed and is attributed to the variations in Sp1 binding site sequences and flanking sequences [34]. The data presented here suggest that negative regulatory element and the NO-response region are present in the same − 437 to − 277 region and NO reduces binding of Sp1 to its consensus site in this region. Therefore, it is also possible that Sp1 may inhibit the repressor protein binding in this region and by reducing Sp1 binding NO is enhancing the repressor binding and thus downregulating the DAF expression. NO can also disrupt the DNA binding of Sp1, which is a zinc finger protein, by releasing zinc from thiol groups [35]. Therefore, NO down-regulation of DAF may also involve effects on the binding of other zinc-finger transcription factors including MZF-1 and GATA2 for which binding sites are located flanking the Sp1 site in the NO-response region. These scenarios would suggest that Sp1 is only a part of a larger NO response complex.
About 3275, corresponding to ~ 16% of human protein coding genes have been reported to have at least one WUAUUUAUW type of consensus sequence at their 3′-UTR [36]. The 3′-UTR of DAF gene has a class II ARE which is characterized by two or more overlapping sequences of the type UUAUUUA (U/A)(U/A). The class II AREs are predominantly found in rapidly inducible mRNAs such as mRNAs of cytokines whereas class I AREs which are characterized by dispersed copies of AUUUA motif are predominantly found in the mRNAs of transcription factors and cell cycle regulatory proteins [24]. The actual physiological significance of ARE sequence in DAF mRNA is enigmatic. However, because of the similarity in AREs between cytokine and DAF the regulation of ARE in DAF appears to be closely related to the activation of cytokines that elicit complement. The factors that control AREs in cytokines and DAF are not known. Our results presented here suggest that NO is a regulator of DAF-ARE. When there is a bacterial insult, ARE mediated modulation of mRNA provides a tool to rapidly decrease the DAF level and thus limits the availability of receptor for bacterial binding. A rapid response to reduce the DAF level is apparent because the adhesion of E. coli to endometrial epithelial cells occurs immediately after the contact and reaches saturation in about 60 minutes [37;38]. In addition to the ARE region, we also observed a decrease in the reporter activity with the deletion of upstream flanking sequence of ARE (plasmid Δ0.72) in DAF gene 3′-UTR. This region contains CU rich sequences similar to the types (C/U) CCA, and CCC(U/A) which are part of the DICE machinery. Several trans-acting factors bind to DICE and stabilize mRNA by repressing translation [39;40]. NO can modulate the binding of DICE and trans-factors, resulting in the translational repression of many transcripts [39–41]. In this context regulation of DAF mRNA turnover by NO is a complex phenomenon involving several sequence elements on its 3′-UTR.
Studies have shown that HuR protein binds to ARE at the 3′-UTR of many mRNAs and stabilizes them [24]. Our studies using RNA immuno-precipitation assay revealed a decrease in the association between DAF-ARE and HuR protein upon NO treatment. This decrease was not due to decreases in either DAF mRNA or HuR protein levels with 4 h of exposure to DTNO. However, HuR protein levels were also down-regulated by prolonged exposure (24h) to NO. Therefore, the down-regulation of HuR protein may further contribute to the destabilization of DAF mRNA upon extended NO production. The down-regulation of HuR by NO has been previously reported [42]. NO can either enhance or inhibit the binding of HuR to different mRNAs and alters the stability of mRNAs [43]. This discrepancy in binding of HuR to mRNA could be due to different post-translational modifications induced by NO. Although, NO can activate several protein kinases through cGMP dependent and independent pathways, we did not observe any phosphorylation of HuR in this study. However, phosphorylation of HuR by Chk1 and Chk2 has been reported and implicated in cytoplasmic accumulation of HuR [44]. On the other hand, studies have shown that NO can induce direct S-nitrosylation of thiol groups in various RNA binding proteins and therefore, can alter their binding to RNA [45;46]. Therefore, S-nitrosylation of HuR protein in Ishikawa cells and reduction in rHuR binding to DAF-ARE upon in vitro NO treatment suggest that S-nitrosylation of HuR is the probable mechanism of its reduced binding to DAF mRNA.
In summary, we have shown that DAF gene is down-regulated by NO through the regulation of its promoter activity and mRNA stability. Transcription factor Sp1 and RNA binding protein HuR are instrumental in mediating the inhibition effect of NO on DAF gene expression. Although the present study utilized endometrial cell model to address the role of NO in the regulation of DAF in the milieu of uterine infection and other pregnancy complications, such phenomenon may exist in modulating other DAF associated functions in other systems.
Materials and methods
Cell culture
Ishikawa cells are a human endometrial cell line derived from a well differentiated endometrial adenocarcinoma that has been shown to mimic endometrial epithelial cells [39;47]. Ishikawa cells were routinely cultured in Eagle’s minimum essential medium (MEM) containing 2 mM L-glutamine, 100 µg of penicillin/streptomycin/ml and 10 mM HEPES (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FBS) (Gemini Bioproducts, West Sarcamento, CA, USA) at 37 °C in humidified 5% CO2 atmosphere. When the cells were exposed to NO donor or NOS substrate or NOS inhibitor, MEM devoid of L-arginine and phenol red (Atlanta biological, Lawrenceville, GA, USA) but supplemented with 5% FBS was used.
Plasmids
HeLa cell genomic DNA was used to PCR-amplify 0.734 kb of DAF promoter using the forward primer 5′–AATTGGTACCATGACTCCCACCCGAACAAG–3′, and reverse primer 5′–CCCCCAAGCTTGGCGCGCCGG–3′. The PCR amplified promoter fragment was cloned in pβgal-basic β-galactosidase vector (Clontech, Mountain View, CA, USA) using KpnI and HindIII restriction sites. The resultant plasmid was designated as pβgalb-0.734 kb (−734 to −1, +1 is A of ATG site). Several 5′-deletions were generated in the 0.734-kb DAF promoter. The nucleotide sequence of the PCR forward primers for generation of deletion plasmids were as follows: 5′-AATTGGTACCGCTGGCCTTTGACAGACCTC–3′ for 0.602 kb, 5′-AATTGGTACCGGCCTGCGATACTTGAGCAC-3′ for 0.437 kb, 5′-AATTGGTACCCCTTCCCCTCCCCACTCTC-3′ for 0.277 kb, 5′-AATTGGTACCCCCCTACTCCACCCGTCTTG-3′ for 0.191 kb, and 5′-AATTGGTACCGGTATTGCGGAGCCACGAG-3′ for 0.108 kb. The same reverse primer 5′–CCCCCAAGCTTGGCGCGCCGG–3’ was used to generate all deletion plasmids. The DAF 3’-UTR was analyzed for the presence of adenosine-uracil rich elements (ARE) using the programme AREsite (http://rna.tbi.univie.ac.at/AREsite) [36]. The human DAF 3′-UTR sequence was obtained from NCBI accession number, NM_000574. He La cell cDNA was used to PCR-amplify 1.35 kb of DAF 3′-UTR using the forward primer 5′-CGCCGGTAAATTCGTAACCATGGGCTTGCTGACTTAG-3′ and reverse primer 5′-CACCGGCGAAGAAATACACATTAAAGTCTTTACAGTG-3′. The PCR amplified 3′-UTR fragment was cloned downstream to βgal gene in pβgal-control β-galactosidase vector (Clontech) using SgrAI restriction sites. The resultant plasmid was designated as pβgalc-1.35 kb. Three 5′-deletions were generated in the 1.35 kb 3′-UTR and cloned to pβgal-control β-galactosidase vector using EcoRI and BamHI restriction sites. The resultant plasmids were designated as Δ0.35, Δ0.72 and Δ0.99. The Δ0.35 and Δ0.99 were generated using the forward primers 5′-GAATTCCACAAGATCTGTAATGTTATTTCC-3′ and 5′-GAATTCCTACAAGCAGTTCAGAATGCC-3′ respectively. The same reverse primer 5′-TCGACGGATCCCTAGAGGATCTG-3′ was used to generate these two deletion plasmids. Before generating Δ0.72, a “T” was introduced to create an EcoR1 site (GAATTC) downstream of SgrAI by site directed mutagenesis. The forward primer 5′-GATTATATATTATTTCTGAATTCGAGATGTCCATAG-3′ and the reverse primer 5′-TATGGACATCTCGAATTCAGAAATATATAATC-3′ were used to generate Δ0.72 deletion plasmid.
Transient transfection and reporter assay
Ishikawa cells were grown in 6-well cell culture plates. When they achieved 70–80% confluency, cells were co-transfected with 1 µg each of βgal reporter plasmids and luciferase reporter plasmid pGL3 (Promega, Madison, WI, USA) using Lipofectamine 2000 reagent (Invitrogen). Transfected and treated cells were scraped using rubber policeman with 75 µl of reporter lysis buffer (Promega). The β-galactosidase activities in the cell lysates were measured using Luminescent β-gal assay kit (Clontech). The firefly luciferase activities in the cell lysates were measured using Luciferase assay system (Promega).
Transcription factor ELISA
Biotin tagged duplex DNA fragments were synthesized by Integrated DNA Technologies (Coralville, IA, USA). The binding assay for transcription factors was performed as follows. Reacti-Bind NeutrAvidin coated 96-well plates (Thermo Scientific, Rockford, IL, USA) were blocked using wash buffer (25 mM Tris, 0.15 M sodium chloride, pH 7.2, 0.05% Tween-20) containing 0.01% acetylated BSA (Promega). Biotinylated DNA duplexes (0.1 pmole) were immobilized on to the neutravidin coated wells by incubating in wash buffer for 2 h at room temperature. Ishikawa cells were exposed to nitric oxide donor, DTNO (1 mM) for 4 h and nuclear fractions were prepared using NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific) according to the manufacturer’s instructions. Nuclear fraction equivalent to 1.75 µg of protein was added to the immobilized DNA and incubated for 30 min at room temperature. The wells were washed three times for 5 min each using wash buffer and incubated with rabbit anti-Sp1 (Millipore, Billerica, MA, USA) or anti-CREB (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti NFΚB (Santa Cruz Biotechnology) antibodies in wash buffer for 1.5 h at room temperature. After three washes of 5 min each using wash buffer, goat anti rabbit IgG conjugated to horse radish peroxidase (1: 1000 dilution, 200 µl) (Southern Biotech, Birmingham, AL, USA) was added and incubated for 2 h at room temperature. Wells were washed 4 times for 5 min each; 100 µl of 1-step Turbo TMB-ELISA substrate (Thermo Scientific) was added and incubated for 30 min at room temperature. The reaction was stopped by adding 100 µl of 1 M H2SO4 and the absorbance was read at 450nm.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed using a kit from Millopore according to the procedure provided by the manufacturer. Briefly, Ishikawa cells were treated with DTNO (1mM) for 4 h and control and treated cells were fixed in 1% formaldehyde for 15 min. Cells were scraped, pelleted by centrifugation and re-suspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1). DNA was sheared using a sonicator (QSonica, Newtown, CT, USA) with four 15-s pulses at setting 3. Sheared chromatin was immunoprecipitated with anti-Sp1 antibody or control IgG by incubating overnight at 4 °C. The cross-links were reversed by heating at 65 °C for 4 h, and the chromatin was deproteinated with 10 µg/ml proteinase K. The relative binding of Sp1 to DAF promoter regions were quantitated by qRT-PCR using the following set of primers: forward 5′-ACTCAAGCGCGGGGATGCTC-3′, reverse 5′-TGCGATGACCTGCCTTCTAG-3′ for − 438 to − 399 region and forward 5′-GACCGCACCTCTGACCACAA-3′, reverse 5′-CCGGAGCGAGTTGCAGTAAG-3′ for − 166 to −127 region.
RNA isolation and quantitative reverse transcription-PCR (RT-qPCR)
Total RNA was isolated from the cells using RNeasy kit (Qiagen, Valencia, CA, USA) according to the procedure recommended by the manufacturer. RNA extraction was followed by DNAase 1 (Qiagen) treatment to remove DNA contamination. Total RNA of 1 µg was reverse transcribed into complementary DNA using avian myeloblastosis virus reverse transcriptiase (Promega) and random oliginucleotide hexamers (Invitogen). Quantitative real time reverse transcriptase PCR was done with CFX96 system and SYBR green master mix (Bio-Rad, Hercules, CA, USA). A comparative cycle of threshold fluorescence (CT) method was used with housekeeping gene as an internal control. The CT value for the housekeeping gene was subtracted from the CT value of the target gene to obtain a ΔCT. The ΔCT of the calibrator (the mean ΔCT of the control cells) was then subtracted from each sample to give a ΔΔCT value. This was then inserted into 2T−ΔΔC to give a final expression relative to the calibrator.
Determination of DAF mRNA half-life
Ishikawa cells grown in 6-well cell culture plates were treated with 1 µg/ml Actinomycin D for 4 h to arrest the transcription. Cells were then exposed to 1 mM DTNO or spent DTNO (1mM DTNO kept at 37 °C for 48 h) or the substrate for NOS enzyme L-arginine (0.3mM) or L-arginine combined with NOS inhibitor L-NAME (3mM) for 0, 4, and 8 h. The relative amount of DAF mRNA at each time point with respect to the housekeeping gene glyceraldahyde-3-phosphate dehydrogenase (GAPDH) was determined by RT-qPCR. The primer pairs for DAF were obtained from SA Biosciences (Frederick, MD, USA). The forward primer 5′- GGTCTCCTCTGACTTCAACA- 3′ and reverse primer 5′- AGCCAAATTCGTTGTCATAC -3′ was used for the qPCR of GAPDH. The relative amount of DAF mRNA remained at each time period was calculated taking relative amount of DAF mRNA at time 0 as 100%. A decay curve was obtained for the two treatments and control by plotting % remaining DAF mRNA versus time. From the nonlinear single exponential regression curves decay constants were determined using GraphPad Prism 3.0 programme (GraphPad Inc, San Diego, CA, USA). The mRNA half-life was then determined using the formula t1/2 = ln2/kdecay, where t1/2 is DAF mRNA half life and kdecay is decay constant.
RNA Immuno-precipitation (RIP) Assay
RNA immuno-precipitation assay is used to assess the association between RNA binding proteins and mRNA [48]. The RNA immuno-precipitation assay was performed using Ribocluster Profiler RIP-Assay kit (MBL, Woburn, MA, USA) according to the manufacturer’s instructions. Briefly, Ishikawa cells grown in 15 cm dishes were treated with 1 mM DTNO for 4 h. Control cells and DTNO exposed cells were scraped with nuclease free PBS, pelleted by centrifuging at 300×g for 5 min at 4 °C and then lysed using lysis buffer at 4 °C on ice for 10 min. Cell lysates were centrifuged at 12,000×g for 5 min at 4 °C and the supernatants were collected. The lysates were then pre-cleared by incubating with 50% slurry of agarose G beads (Pierce, Rockford, IL, USA) in wash buffer for 1 h at 4 °C with constant shaking. The RIP-certified anti HuR antibody or normal rabbit IgG (MBL) were immobilized on agarose G beads by incubating with 50% slurry of agarose G beads in wash buffer for 30 min at 4 °C with constant shaking. Precleared cell lysates were incubated with HuR antibody or IgG coated agarose G beads in separate tubes for 3 h at 4 °C with constant shaking followed by centrifugation at 2,000×g for 1 min at 4 °C. The supernatants were carefully removed and the precipitates were washed four times with wash buffer. The total mRNA from agaroseG-HuR antibody-HuR complex or agarose G- IgG complex was isolated using RNA isolation reagents provided with the RIP assay kit according to the manufacturer’s instructions. The relative amount of DAF mRNA was determined by RT-qPCR. The relative amount of HuR mRNA was determined by RT-qPCR using previously described primers [49]. GAPDH mRNA was used as internal control.
Preparation of recombinant HuR
Plasmid pTYB11-HuRwt, containing the complete open reading frame of human HuR subcloned downstream of an intein/chitin-binding domain tag, was described by Fialcowitz-White [50]. Recombinant HuR was expressed from this vector in E. coli ER2566 cells. Briefly, cells were grown shaking at 37 °C in SOB medium containing MgCl2 (10 mM) and ampicillin (50 µg/ml) to OD600 ≈ 0.6 – 0.8. Expression of recombinant HuR was induced with isopropyl 1-thio-β-D-galactopyranoside (1 mM) in a 25 °C shaker for 5 h. After induction, cultures were centrifuged at 3300×g for 20 min. Cell pellets were resuspended in ice-cold buffer A (500 mM NaCl, 1 mM EDTA in 20 mM phosphate buffer, pH 7.0) with 20 µM phenylmethanosulfonyl fluoride and disrupted by sonication. The lysate was clarified by centrifugation at 20,000×g for 20 min at 4 °C, then loaded onto a chitin column (New England Biolabs, Ipswich, MA, USA) equilibrated with buffer A. After lysate was loaded, the column was washed with 10 column volumes of buffer A followed by 3 column volumes of buffer A containing 50 mM dithiotheritol (DTT), then capped and stored at 4 °C for 40 h to permit DTT-induced self-cleavage of the intein tag. The released tagless-HuR protein was eluted from the column in buffer A. To concentrate the protein and to remove DTT, the eluent was loaded onto an Amicon Ultra-4 concentrator (10 kDa molecular weight cutoff) and repeatedly concentrated in buffer A. Concentrated protein was then centrifuged at 20,000×g for 15 minutes to remove any particulates. Purified HuR was quantified against a titration of bovine serum albumin by Coomassie Blue-stained SDS-polyacrylamide gel electrophoresis.
DAF 3′-UTR mRNA-rHuR binding assay
Biotin tagged short mRNA sequence, 5′-biotin-UAAUAUUUAUUUAUAUUUAUUUAUGACAG-3′ containing 3′- ARE of DAF mRNA was synthesized by Integrated DNA Technologies. The binding assay was performed as follows. Reacti-Bind NeutrAvidin coated 96-well plates (Thermo Scientific) were blocked using wash buffer (25 mM Tris, 0.15 M sodium chloride, pH 7.2, 0.05% Tween-20) containing 0.01% acetylated BSA (Promega). Different amounts of biotin labeled DAF mRNA (0.01, 0.02, 0.04, 0.06, 0.08, 0.1 and 0.12 pmoles) were immobilized on to avidin plates by incubating in wash buffer for 2 h at room temperature. The recombinant HuR (rHuR) was exposed to 1 mM DTNO in binding buffer (Tris HCl, pH 8.0 containing 50 mM KCl, 2 mM DTT, 0.5 mM EDTA, 0.1 µg/µl acetylated BSA and 1 µg/µl heparin) for 30 min at room temperature. The DTNO exposed rHuR was purified by passing through Zeba spin desalting column (Thermo Scientific). The non-exposed (control) or DTNO exposed rHuR (8.7 nM, 20 µl) was incubated with immobilized DAF mRNA in binding buffer for 30 min at room temperature. The wells were washed three times for 5 min each using wash buffer and then incubated with rabbit anti HuR antibody (1: 1000 dilution, 200 µl) (MBL) for 2 h at room temperature. After three washes of 5 min each using wash buffer, goat anti rabbit IgG conjugated to horse radish peroxidase (1: 1000 dilution, 200 µl) (Southern Biotech) was added and incubated for 2 h at room temperature. Wells were washed 4 times for 5 min each; 100 µl of 1-step Turbo TMB-ELISA substrate (Thermo Scientific) was added and incubated for 30 min at room temperature. The reaction was stopped by adding 100 µl of 1 M H2SO4 and the absorbance was read at 450nm.
Biotin switch assay
The biotin switch assay was performed according to the published procedure [51]. Briefly, the protein concentrations in cell lysates were adjusted to 0.8 µg/µl using HEN buffer (Hepes-NaOH, 250 mM, pH 7.0 containing 1 mM EDTA and 0.1 mM neocuproine). Then 4 volumes of blocking buffer {HEN and SDS (25% w/v in H2O) in 9:1 ratio containing 20 mM MMTS} was added and incubated at 50 °C for 20 min with frequent vortexing. Ten volumes of pre-chilled acetone was added and incubated at −20 °C for 20 min. The contents were centrifuged at 2000×g for 10 min at 4 °C and the pellets were resuspended in 0.1 ml of HENS buffer (HEN buffer containing 1% SDS) per mg of protein in the starting sample. Then 1:3 volume of labeling solution (4 mM biotin-HPDP) and 1:50 volume of ascorbate solution (50 mM) were added. After incubation for 1 h at 25 °C, 2 volumes of pre-chilled acetone was added and incubated at −20 °C for 20 min. The contents were again centrifuged at 2000×g for 10 min at 4 °C. The pellets were resuspended in 0.1 ml of HENS buffer per mg of protein in the starting sample and the biotinylated proteins were purified by precipitation using neutravidin as follows. The resuspended pellets were incubated with 200 µl of 50% slurry of neutravidin agarose beads for 1 h at room temperature with constant rotation. The contents were centrifuged at 500×g for 1 min, the supernatants named as unbound were removed carefully. The pellets were washed once using 200 µl PBS, centrifuged again at 500×g for 1 min. Supernatants were carefully discarded and to the pellets named as eluate, 20 µl SDS-PAGE buffer was added, boiled at 100 °C for 10 min, centrifuged briefly and the proteins in the supernatants were separated by electrophoresis. The presence of HuR protein and β-actin (positive control for S-nitrosylated protein) was investigated by western blotting.
Western blotting
Protein concentrations were determined using Pierce BCA protein assay kit (Thermo Scientific), against BSA standard. Proteins (5 µg) were separated by polyacrylamide gel electrophoresis. To separate proteins in the immuno-precipitated samples, the sample buffer was added to the precipitated agarose G beads, boiled for 10 min at 100 °C in a dry bath and then centrifuged at 10,000×g for 5 min. Equal volumes (20 µl) of supernatants were loaded on to the polyacrylamide gel. After the electrophoresis, proteins were elctro-transferred to PVDF membranes. Membranes were blocked with TBST (20 mM Tris, 500 mM NaCl, 1% Tween-20, pH 7.5) containing 5% fat free milk for 1 h at room temperature and incubated with the primary antibodies overnight at 4 °C. The antibodies against β-actin and β-tubulin were purchased from Cell signaling Technology (Danvers, MA USA, USA). Antibody against HuR which was used for immuno-precipitation and western blot was purchased from Promega. Antibody against phosphorylated proteins was purchased from Invitrogen. Blots were then washed with TBST and incubated with horseradish-peroxidase conjugated secondary antibodies for 1 h at room temperature. The bound antibody was visualized on a blue sensitive autoradiography film using supersignal west pico chemiluminescence substrate (GE healthcare, Little Chalfont, UK, USA) according to the instructions provided by the manufacturer.
Statistical analysis
GraphPad Prism (version 3) software was used for statistical analysis. Comparisons between the groups were made by analysis of variance for multiple groups or by t test for two groups. Statistical significance (p < .05) between the groups was determined by Bonferroni’s multiple comparison method.
Acknowledgement
Financial support from the National Institute of Health (NIH) through grants R01HL102866, R01HL58144 and R01HD057013 to Dr. Chandra Yallmpalli and grant R01CA102482 to Dr. Gerald M. Wilson is greatly appreciated.
The abbreviations used are
- DAF
Decay Accelerating Factor
- NO
Nitric oxide
- 3′-UTR
3′-Untranslated region
- ARE
Adenosine-uracil rich element
- DTNO
diethylenetriamine NONOate
- SNAP
L-NAME (NG-nitro-L-argininemethyl ester)
- MMTS
methyl methanethiosulfonate
- biotin-HPDP
N-[6-(biotinamido)hexyl]-3′-(2′-pyridyl dithio)propionamide.
References
- 1.Gibbs RS, Romero R, Hillier SL, Eschenbach DA, Sweet RL. A review of premature birth and subclinical infection. Am. J. Obstet. Gynecol. 1992;166:1515–1528. doi: 10.1016/0002-9378(92)91628-n. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch E, Saotome I, Hirsh D. A model of intrauterine infection and preterm delivery in mice. Am. J. Obstet. Gynecol. 1995;172:1598–1603. doi: 10.1016/0002-9378(95)90503-0. [DOI] [PubMed] [Google Scholar]
- 3.Krohn MA, Hillier SL, Nugent RP, Cotch MF, Carey JC, Gibbs RS, Eschenbach DA. The genital flora of women with intraamniotic infection. Vaginal Infection and Prematurity Study Group. J. Infect. Dis. 1995;171:1475–1480. doi: 10.1093/infdis/171.6.1475. [DOI] [PubMed] [Google Scholar]
- 4.Nowicki B, Fang L, Singhal J, Nowicki S, Yallampalli C. Lethal outcome of uterine infection in pregnant but not in nonpregnant rats and increased death rate with inhibition of nitric oxide. Am. J. Reprod. Immunol. 1997;38:309–312. doi: 10.1111/j.1600-0897.1997.tb00521.x. [DOI] [PubMed] [Google Scholar]
- 5.Nowicki B, Singhal J, Fang L, Nowicki S, Yallampalli C. Inverse relationship between severity of experimental pyelonephritis and nitric oxide production in C3H/HeJ mice. Infect. Immun. 1999;67:2421–2427. doi: 10.1128/iai.67.5.2421-2427.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang L, Nowicki BJ, Dong YL, Yallampalli C. Localized increase in nitric oxide production and the expression of nitric oxide synthase isoforms in rat uterus with experimental intrauterine infection. Am. J. Obstet. Gynecol. 1999;181:601–609. doi: 10.1016/s0002-9378(99)70499-0. [DOI] [PubMed] [Google Scholar]
- 7.Hart A, Pham T, Nowicki S, Whorton EB, Jr, Martens MG, Anderson GD, Nowicki BJ. Gestational pyelonephritis--associated Escherichia coli isolates represent a nonrandom, closely related population. Am. J. Obstet. Gynecol. 1996;174:983–989. doi: 10.1016/s0002-9378(96)70337-x. [DOI] [PubMed] [Google Scholar]
- 8.Hart A, Nowicki BJ, Reisner B, Pawelczyk E, Goluszko P, Urvil P, Anderson G, Nowicki S. Ampicillin-resistant Escherichia coli in gestational pyelonephritis: increased occurrence and association with the colonization factor Dr adhesin. J Infect. Dis. 2001;183:1526–1529. doi: 10.1086/320196. [DOI] [PubMed] [Google Scholar]
- 9.Goluszko P, Popov V, Selvarangan R, Nowicki S, Pham T, Nowicki BJ. Dr fimbriae operon of uropathogenic Escherichia coli mediate microtubule-dependent invasion to the HeLa epithelial cell line. J. Infect. Dis. 1997;176:158–167. doi: 10.1086/514018. [DOI] [PubMed] [Google Scholar]
- 10.Nowicki B, Hart A, Coyne KE, Lublin DM, Nowicki S. Short consensus repeat-3 domain of recombinant decay-accelerating factor is recognized by Escherichia coli recombinant Dr adhesin in a model of a cell-cell interaction. J. Exp. Med. 1993;178:2115–2121. doi: 10.1084/jem.178.6.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvarangan R, Goluszko P, Popov V, Singhal J, Pham T, Lublin DM, Nowicki S, Nowicki B. Role of decay-accelerating factor domains and anchorage in internalization of Dr-fimbriated Escherichia coli. Infect. Immun. 2000;68:1391–1399. doi: 10.1128/iai.68.3.1391-1399.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang L, Nowicki BJ, Urvil P, Goluszko P, Nowicki S, Young SL, Yallampalli C. Epithelial invasion by Escherichia coli bearing Dr fimbriae is controlled by nitric oxide-regulated expression of CD55. Infect. Immun. 2004;72:2907–2914. doi: 10.1128/IAI.72.5.2907-2914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas DJ, Lublin DM. Identification of 5'-flanking regions affecting the expression of the human decay accelerating factor gene and their role in tissue-specific expression. J Immunol. 1993;150:151–160. [PubMed] [Google Scholar]
- 14.Kaul A, Nagamani M, Nowicki B. Decreased expression of endometrial decay accelerating factor (DAF), a complement regulatory protein, in patients with luteal phase defect. Am J Reprod. Immunol. 1995;34:236–240. doi: 10.1111/j.1600-0897.1995.tb00947.x. [DOI] [PubMed] [Google Scholar]
- 15.Parker CJ. Molecular basis of paroxysmal nocturnal hemoglobinuria. Stem Cells. 1996;14:396–411. doi: 10.1002/stem.140396. [DOI] [PubMed] [Google Scholar]
- 16.Murray KP, Mathure S, Kaul R, Khan S, Carson LF, Twiggs LB, Martens MG, Kaul A. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59- in benign and malignant endometrial tissue. Gynecol. Oncol. 2000;76:176–182. doi: 10.1006/gyno.1999.5614. [DOI] [PubMed] [Google Scholar]
- 17.Niehans GA, Cherwitz DL, Staley NA, Knapp DJ, Dalmasso AP. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin) Am J Pathol. 1996;149:129–142. [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmad SR, Lidington EA, Ohta R, Okada N, Robson MG, Davies KA, Leitges M, Harris CL, Haskard DO, Mason JC. Decay-accelerating factor induction by tumour necrosis factor-alpha, through a phosphatidylinositol-3 kinase and protein kinase C-dependent pathway, protects murine vascular endothelial cells against complement deposition. Immunology. 2003;110:258–268. doi: 10.1046/j.1365-2567.2003.01733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andoh A, Fujiyama Y, Sumiyoshi K, Sakumoto H, Okabe H, Bamba T. Tumour necrosis factor-alpha up-regulates decay-accelerating factor gene expression in human intestinal epithelial cells. Immunology. 1997;90:358–363. doi: 10.1111/j.1365-2567.1997.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasu J, Mizuno M, Uesu T, Takeuchi K, Inaba T, Ohya S, Kawada M, Shimo K, Okada H, Fujita T, Tsuji T. Cytokine-stimulated release of decay-accelerating factor (DAF;CD55) from HT-29 human intestinal epithelial cells. Clin. Exp. Immunol. 1998;113:379–385. doi: 10.1046/j.1365-2249.1998.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young SL, Lessey BA, Fritz MA, Meyer WR, Murray MJ, Speckman PL, Nowicki BJ. In vivo and in vitro evidence suggest that HB-EGF regulates endometrial expression of human decay-accelerating factor. J. Clin. Endocrinol. Metab. 2002;87:1368–1375. doi: 10.1210/jcem.87.3.8350. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, Wang W, Wesley RA, Danner RL. A Sp1 binding site of the tumor necrosis factor alpha promoter functions as a nitric oxide response element. J. Biol. Chem. 1999;274:33190–33193. doi: 10.1074/jbc.274.47.33190. [DOI] [PubMed] [Google Scholar]
- 23.Lee KA, Masson N. Transcriptional regulation by CREB and its relatives. Biochim. Biophys. Acta. 1993;1174:221–233. doi: 10.1016/0167-4781(93)90191-f. [DOI] [PubMed] [Google Scholar]
- 24.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lafarga V, Cuadrado A, Lopez dS I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell Biol. 2009;29:4341–4351. doi: 10.1128/MCB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andoh A, Shimada M, Araki Y, Fujiyama Y, Bamba T. Sodium butyrate enhances complement-mediated cell injury via down-regulation of decay-accelerating factor expression in colonic cancer cells. Cancer Immunol. Immunother. 2002;50:663–672. doi: 10.1007/s00262-001-0239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cummings JH. Short chain fatty acids in the human colon. Gut. 1981;22:763–779. doi: 10.1136/gut.22.9.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniel P, Brazier M, Cerutti I, Pieri F, Tardivel I, Desmet G, Baillet J, Chany C. Pharmacokinetic study of butyric acid administered in vivo as sodium and arginine butyrate salts. Clin. Chim. Acta. 1989;181:255–263. doi: 10.1016/0009-8981(89)90231-3. [DOI] [PubMed] [Google Scholar]
- 29.Banadakoppa M, Goluszko P, Liebenthal D, Yallampalli C. Nitric Oxide Induces Segregation of Decay Accelerating Factor (DAF or CD55) from the Membrane Lipid- Rafts and its Internalization in Human Endometrial Cells. Cell Biol. Int. 2012 doi: 10.1042/CBI20110586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewulonu UK, Ravi L, Medof ME. Characterization of the decay-accelerating factor gene promoter region. Proc. Natl. Acad. SciUSA. 1991;88:4675–4679. doi: 10.1073/pnas.88.11.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cauvi DM, Cauvi G, Pollard KM. Constitutive expression of murine decay-accelerating factor 1 is controlled by the transcription factor Sp1. J. Immunol. 2006;177:3837–3847. doi: 10.4049/jimmunol.177.6.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De LA, Sacchetta P, Nieddu M, Di IC, Favaloro B. Important roles of multiple Sp1 binding sites and epigenetic modifications in the regulation of the methionine sulfoxide reductase B1 (MsrB1) promoter. BMC. Mol. Biol. 2007;8:39. doi: 10.1186/1471-2199-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji C, Casinghino S, McCarthy TL, Centrella M. Multiple and essential Sp1 binding sites in the promoter for transforming growth factor-beta type I receptor. J. Biol. Chem. 1997;272:21260–21267. doi: 10.1074/jbc.272.34.21260. [DOI] [PubMed] [Google Scholar]
- 34.Chu S, Ferro TJ. Sp1: regulation of gene expression by phosphorylation. Gene. 2005;348:1–11. doi: 10.1016/j.gene.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Berendji D, Kolb-Bachofen V, Meyer KL, Grapenthin O, Weber H, Wahn V, Kroncke KD. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Lett. 1997;405:37–41. doi: 10.1016/s0014-5793(97)00150-6. [DOI] [PubMed] [Google Scholar]
- 36.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39:D66–D69. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishikawa Y. Adherence of Escherichia coli in pathogenesis of endometritis and effects of estradiol examined by scanning electron microscopy. Infect. Immun. 1985;47:318–321. doi: 10.1128/iai.47.1.318-321.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishikawa Y, Baba T. In vitro adherence of Escherichia coli to endometrial epithelial cells of rats and influence of estradiol. Infect. Immun. 1985;50:506–509. doi: 10.1128/iai.50.2.506-509.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holcik M, Liebhaber SA. Four highly stable eukaryotic mRNAs assemble 3' untranslated region RNA-protein complexes sharing cis and trans components. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2410–2414. doi: 10.1073/pnas.94.6.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3' end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 41.Wang S, Zhang J, Theel S, Barb JJ, Munson PJ, Danner RL. Nitric oxide activation of Erk1/2 regulates the stability and translation of mRNA transcripts containing CU-rich elements. Nucleic Acids Res. 2006;34:3044–3056. doi: 10.1093/nar/gkl386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akool e, Kleinert H, Hamada FM, Abdelwahab MH, Forstermann U, Pfeilschifter J, Eberhardt W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol. Cell Biol. 2003;23:4901–4916. doi: 10.1128/MCB.23.14.4901-4916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuwano Y, Rabinovic A, Srikantan S, Gorospe M, Demple B. Analysis of nitric oxide-stabilized mRNAs in human fibroblasts reveals HuR-dependent heme oxygenase-1 upregulation. Mol. Cell Biol. 2009;29:2622–2635. doi: 10.1128/MCB.01495-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HH, Abdelmohsen K, Gorospe M. Regulation of HuR by DNA Damage Response Kinases. J. Nucleic Acids. 2010;2010 doi: 10.4061/2010/981487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim S, Ponka P. Role of nitric oxide in cellular iron metabolism. Biometals. 2003;16:125–135. doi: 10.1023/a:1020788603046. [DOI] [PubMed] [Google Scholar]
- 46.Liu W, Rosenberg GA, Liu KJ. AUF-1 mediates inhibition by nitric oxide of lipopolysaccharide-induced matrix metalloproteinase-9 expression in cultured astrocytes. J. Neurosci. Res. 2006;84:360–369. doi: 10.1002/jnr.20895. [DOI] [PubMed] [Google Scholar]
- 47.Nishida M, Kasahara K, Kaneko M, Iwasaki H, Hayashi K. Establishment of a new human endometrial adenocarcinoma cell line, Ishikawa cells, containing estrogen and progesterone receptors] Nippon Sanka Fujinka Gakkai Zasshi. 1985;37:1103–1111. [PubMed] [Google Scholar]
- 48.Kuwano Y, Pullmann R, Jr, Marasa BS, Abdelmohsen K, Lee EK, Yang X, Martindale JL, Zhan M, Gorospe M. NF90 selectively represses the translation of target mRNAs bearing an AU-rich signature motif. Nucleic Acids Res. 2010;38:225–238. doi: 10.1093/nar/gkp861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan YX, Chen H, Kilberg MS. Interaction of RNA-binding proteins HuR and AUF1 with the human ATF3 mRNA 3'-untranslated region regulates its amino acid limitation-induced stabilization. J. Biol. Chem. 2005;280:34609–34616. doi: 10.1074/jbc.M507802200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fialcowitz-White EJ, Brewer BY, Ballin JD, Willis CD, Toth EA, Wilson GM. Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J. Biol. Chem. 2007;282:20948–20959. doi: 10.1074/jbc.M701751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaffrey SR, Snyder SH. The biotin switch method for the detection of Snitrosylated proteins. Sci. STKE. 2001;2001:l1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]