

Summary
Tissue factor (TF) is a transmembrane glycoprotein and an essential component of factor VIIa-TF enzymatic complex that triggers activation of the coagulation cascade. Formation of TF-FVIIa complexes on cell surfaces not only trigger the coagulation cascade but also transduce cell signaling via activation of protease-activated receptors. Tissue factor is expressed constitutively on cell surfaces of a variety of extravascular cell types, including fibroblasts and pericytes in and surrounding blood vessel walls and epithelial cells but generally absent on cells that come in contact with blood directly. However, TF expression could be induced in some blood cells, such as monocytes and endothelial cells, following an injury or pathological stimuli. Tissue factor is essential for hemostasis, but aberrant expression of TF leads to thrombosis. Therefore, a proper regulation of TF activity is critical for the maintenance of hemostatic balance and health in general. TF-FVIIa coagulant activity at the cell surface is influenced not only by TF protein expression levels but also independently by a variety of mechanisms, including alterations in membrane phospholipid composition and cholesterol content, thiol-dependent modifications of TF allosteric disulfide bond, and other post-translational modifications of TF. In this article, we critically review key literature on mechanisms by which TF coagulant activity is regulated at the cell surface in the absence of changes in TF protein levels with specific emphasis on recently published data and provide the authors’ perspective on the subject.
Keywords: cholesterol, cryptic, decryption, encryption, factor VIIa, glycosylation, phospholipids, protein disulfide isomerase, tissue factor
Introduction
Tissue factor (TF), a plasma membrane anchored glycoprotein, is the primary initiator of coagulation in both physiological and pathological conditions. TF functions as the allosteric cofactor for plasma clotting protease factor VIIa (FVIIa) and TF-FVIIa complex initiates the coagulation cascade by activating clotting proteins factors IX and X through limited proteolysis. Tissue factor is expressed constitutively on cell surfaces of many extravascular cells, including fibroblasts and pericytes in and surrounding blood vessel walls and epithelial cells [1,2]. Although cells that come in contact with blood do not typically express TF, some of them, e.g., monocytes and endothelial cells, do express TF in disease conditions [3,4]. At present, it is not entirely clear how the coagulant activity of TF on cell surfaces is altered in the absence of changes in TF antigen levels or TF-FVIIa complex formation. It is also unclear whether TF expressed at the cell surface could undergo post-translational modifications and how they affect TF function at the cell surface. In this article, we critically review existing literature on regulation of TF activity on cell surfaces with specific emphasis on recently published data and provide the authors’ perspective on this subject. Here, we limit our discussion to how alterations to the cell surface membrane, particularly exposure of anionic phospholipids to the outer leaflet of plasma membrane, and potential post-translational modifications of TF could alter TF coagulant activity at the cell surface. For detailed information on regulation of cell-associated TF by other mechanisms, including release of TF microparticles from cell surfaces, the readers are referred to recent review articles on these areas [5–7].
Not all TF present on cell surfaces participates in activating the coagulation
It was reported in 1975 by Maynard et al. that TF on cells exists in a latent form, and the procoagulant activity of TF on intact cells could be increased by many folds by treating the cells with trypsin or other proteases [8]. Restricted formation of the TF-FVIIa complex on cell surfaces could not be the reason for TF latency as numerous studies performed later showed that TF expressed on a variety of cell types was capable of binding to FVII or FVIIa [9–14]. Later, Bach and Rifkin showed that intact bovine fibroblasts, pericytes and kidney cells manifested significantly less TF activity compared to their disrupted counter parts, supporting the hypothesis that TF activity on unperturbed cells is not fully expressed [15]. Correlation of FVIIa binding to the cell surface TF and expression of cell surface TF-FVIIa procoagulant activity in fibroblasts showed a linear relationship between FVIIa binding to TF and TF-FVIIa procoagulant activity at low levels of FVIIa binding but the relationship became nonstoichiometric with higher amounts of FVIIa bound to TF [14], indicating not all TF-FVIIa complexes formed at the cell surface are capable of activating factor X. Le et al. [11], using an ovarian carcinoma cell line as a model system, found that TF-FVIIa coagulant activity was fully manifested on intact monolayers after only 1 min of incubation of cells with FVIIa whereas 30 to 60 min were required for FVIIa to bind all available TF sites on the cell surface. This and other experiments reported in this publication provided clear evidence for the existence of two different populations of TF on cell surfaces, a minor population of TF (~ <20%) that can rapidly bind to FVIIa and account for all TF procoagulant activity on intact cells, and a major population of TF that could bind to FVIIa but the subsequent TF-FVIIa complexes formed fail to activate factor X. These two populations of TF were termed as “functional” and “non-functional” TF, respectively, in the earlier publication [11] but they are now commonly referred to as “coagulant active” and “cryptic” TF. It is not entirely clear whether cryptic TF is completely inert in supporting the coagulation or contains low procoagulant activity. Recent studies from the authors’ laboratories indicate that cryptic TF is close to completely inactive in activating factor X (less than 5% of the activity that was found with active TF; unpublished data of the authors, 2012). It is important to note here that despite any potential differences between the two forms of TF in their ability to interact with FVIIa, both the coagulant active and cryptic TF form stable high-affinity associations with FVII and FVIIa at 10 nM, a concentration equivalent to the plasma concentration of FVII (see rev [16,17]). Therefore, it is important to be cautious in comparing TF mutants that exhibit low procoagulant activity because of the severe defect in FVIIa binding (Kd > 100 nM) with cryptic TF in elucidating mechanisms by which TF procoagulant activity on cell surfaces is regulated.
The encrypted TF at the cell surface could be converted to become procoagulantly active TF in many ways, from destroying plasma membrane integrity by freezing and thawing and treating with non-ionic detergents to cell activation by Ca2+ ionophore treatment or pathophysiological stimuli, such as LPS, thrombin, cytokines or any apoptotic stimulus. These treatments may induce different levels of TF procoagulant activity at the cell surface and probably underlying mechanisms responsible for increased TF activity may vary, but the common theme appears to be the loss of phospholipid asymmetry.
Phospholipid asymmetry: its role in regulation of TF activity on cell surfaces
Membrane phospholipid asymmetry is considered to be a general property of biological membranes. The outer leaflet of the plasma membrane is abundant with phosphatidylcholine (PC) and sphingomyelin (SM) and contains only traces of phosphatidylserine (PS), whereas the inner leaflet is enriched with PS, phosphatidylethanolamine (PE), phosphoinositide and phosphatidic acid. PS asymmetry in the cell membrane is maintained with an expenditure of energy in the form of ATP hydrolysis and involves the concerted action of three classes of aminophospholipid translocases – flippases (catalyzes the inward transport of PS), floppases (catalyzes the outward flow of PS) and scramblases (catalyzes the bidirectional movement of lipids) [18]. The inhibition or activation of one or more of these enzymes by responses elaborated in a variety of diseases, such as infection, hypertension, atherosclerosis and diabetes or agents that induce Ca2+ influx will result in loss of membrane phospholipid asymmetry, which leads to increased exposure of negatively charged PS to the outer leaflet of cell surface membrane [19,20].
It is well known from TF reconstitution studies with lipids that TF procoagulant activity requires its association with phospholipids [21,22], and the presence of anionic phospholipids, such as PS, in the phospholipid mixture greatly accelerates TF procoagulant activity [15,23–27]. Studies performed with purified TF reconstituted into liposomes showed that TF-FVIIa requires high levels of PS (~30% of total phospholipids) for optimal procoagulant activity [26]. However, when PE is incorporated into liposomes, it dramatically lowers the PS requirement for optimal proteolytic activation of FX by the TF-VIIa complex [26]. In fact, recent studies showed that any phospholipid other than with a choline head group can augment TF-FVIIa coagulant activity supported by low levels of PS [28]. These studies clearly suggest that phospholipid composition in the cell membrane may regulate cell surface TF-FVIIa coagulant activity. It is safe to conclude that the limited availability of PS at outer cell surface membrane due to phospholipid asymmetry is primarily responsible for restricting TF coagulant activity at the cell surface. In addition, it is also possible that high SM content on the outer leaflet of cell surface membrane could further downregulate TF-FVIIa coagulant activity at the cell surface as TF reconstituted in phospholipid vesicles containing SM were found to have a lower procoagulant activity compared to TF reconstituted with PC alone [26](unpublished data of the authors).
The importance of phospholipid asymmetry in regulating cell surface TF activity is quite evident since any stimulus or process that can disrupt PS asymmetry and increase the appearance of PS on the cell surface would invariably increase TF activity at the cell surface. A number of studies showed that treatment of a variety of cell types with Ca2+ ionophore, an agent that rapidly increases cytosolic Ca2+ and disrupts PS asymmetry through Ca2+ influx-mediated inhibition of flippase activity and activation of scramblase, markedly increases TF activity at the cell surface [15,29–32]. Similarly, sulfhydryl reactive compounds such as N-ethylmaleimide (NEM), which increases PS efflux by inhibiting flippase, was also shown to increase TF activity at the cell surface [33]. Biologically relevant processes, such as cell-activating events, complement activation and apoptosis that result in PS exposure also led to increased TF activity [34–36]. Treatment of cells with nonionic detergents or physical disruption of cells such as freezing-thawing and sonication markedly enhances TF activity [37–39]. More importantly, blocking the activity of PS exposed at the cell surface by annexin V or lactahedrin inhibited the increase of TF procoagulant activity [29,31,32,40–42]. Inhibition of increased anionic phospholipids and not basal PS levels was shown to be responsible for increased TF activity associated with decryption [43]. However, few reports in the literature indicate that TF activity at the cell surface could be increased independent of PS as annexin V is not fully capable of blocking the enhanced TF coagulant activity associated with TF decryption [29,31]. However, it is important to note here that the phospholipid binding property of annexin V is complex [44]. Annexin V binding to PS depends on the microenvironment in which PS is located and is influenced by steric hindrance of other membrane associated proteins at the cell surface [45–47].
Despite strong implication that PS regulates TF activity at the cell surface, it is difficult to examine the specific contribution of externalized PS in the development of TF activity at the cell surface as none of the studies quantitatively measured PS content at the cell surface in relation to TF activity. Annexin V binding to cells and/or the development of prothrombinase activity were used commonly to evaluate the extent of PS exposure at the cell surface [30,33,41,45,47–49], but both of these measurements could be influenced by microenvironment of the membrane and changes other than PS at the cell surface [45,46,50,51]. More importantly, the concentration of PS in the close vicinity of the TF molecule and not the same amount of PS equally distributed through out the cell surface membrane is important for the expression of TF procoagulant activity at the cell surface. Moreover, based on limited concentrations of PS in cell surface membranes (including PS levels in the inner leaflet of the membrane), it is doubtful whether PS concentration could ever reach to a level capable of optimally supporting TF-FVIIa coagulant activity at the cell surface. Therefore, other factors that augment the activity of low level of PS could play an important role in regulating PS-dependent TF-FVIIa coagulant activity.
Potential mechanisms by which PS regulates TF-FVIIa coagulant activity at the cell surface
Most of our understanding of how phospholipids regulate TF-FVIIa coagulant activity was derived from studies carried out in purified systems using TF relipidated in a predefined lipid content, mostly in PC, PS and PE [24,26–28,52–56], and lately by molecular modeling [28,57]. Findings of these studies have been reviewed recently by Morrissey and colleagues [58,59] and will not be discussed in detail here unless they are relevant for understanding how PS could regulate TF-FVIIa activity at the cell surface. Although studies in artificial membranes with defined lipid content are useful for understanding the potential effects of phospholipids on TF activity, they may not accurately reflect the behavior of complex cell membrane surfaces.
It is widely accepted that the ability of PS to bind vitamin-K dependent clotting proteins via their γ–carboxyglutamic acid-containing domain (Gla domain), which effectively increases the local concentrations of these proteins on the lipid surface, greatly accelerates the rate of catalysis of the clotting reactions. Therefore, it is possible that PS exposure at the cell surface would increase TF-FVIIa coagulant activity by increasing FX binding to the cell surface that could laterally diffuse on the surface to come in contact with membrane anchored TF-FVIIa. In this scenario, the major effect of PS in TF-FVIIa activation of FX comes largely from a decrease in apparent Km for FX [26,55]. Consistent with such a possibility, the increased exposure of PS at the cell surface following Ca2+-ionophore stimulation was shown to lower the apparent Km for FX [15,30]. However, a more noteworthy finding of these studies is that the PS effect on Vmax is more pronounced than the reduction in apparent Km, indicating that the PS effect on TF-FVIIa activation of FX is largely independent of FX binding to PS. In agreement with this notion, exposure of PS at the outer leaflet of cell surface membrane in fibroblasts following NEM treatment was found to enhance TF-FVIIa activation of FX by an increase in Vmax without an accompanying decrease in the apparent Km [33]. Further, blockade of FX binding to the cell surface with a 10-fold molar excess of competing substrate prothrombin fragment 1 failed to attenuate the increased TF-FVIIa activation of FX [33].
It is not entirely clear at present how the exposure of PS at the cell surface increases TF procoagulant activity. In the presence of FVIIa concentrations close to the FVII concentration in plasma (i.e., 10 nM), formation of TF-FVIIa complexes on cell surfaces is not limiting since at this concentration almost all TF sites on cell surfaces are occupied with FVIIa (see [16,17]). Furthermore, our recent studies showed that PS had no effect on FVIIa binding to TF embedded in lipid membrane [60]. If so, exposure of PS on the cell surface is likely to enhance TF-FVIIa activation of FX by transforming the inactive TF-FVIIa complexes to coagulant active TF-FVIIa complexes. In this model, a direct interaction between PS and encrypted TF or TF-FVIIa would result in structural changes in TF or TF-FVIIa complex that would expose the macromolecular substrate binding site to FX and FIX on TF-FVIIa (Fig. 1). It has been suggested that possible electrostatic interactions between PS polar head groups and Lys165/Lys166 in TF could change the quaternary structure of TF by altering the orientation of the extracellular domain relative to the membrane surface, which may facilitate the precise alignment of the TF-FVIIa active site to the scissile bonds of the membrane bound FIX and FX [61,62].
Figure 1.
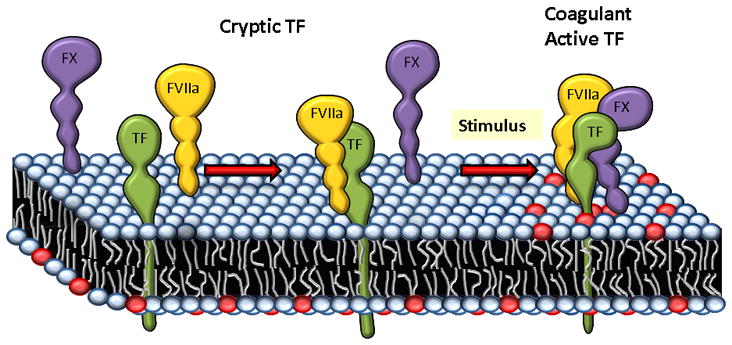
A schematic representation of how exposure of phosphatidylserine (PS) to the outer leaflet of plasma membrane may increase TF coagulant activity at the cell surface. In cells expressing TF, FVIIa binds TF, but majority of TF-FVIIa complexes on the cell surface unable to interact with substrate factor X, therefore they remain coagulant inactive. Upon a stimulus, PS (depicted in red color circles) from the inner leaflet of the plasma membrane would translocate to the outer leaflet. TF or TF-FVIIa complexes directly interact with PS head groups and then undergo conformational changes to recognize the substrate factor X. Factor X binding to PS via its Gla domain may also play a role for efficient activation of factor X by TF-FVIIa complexes on the cell surface.
Recent studies of molecular dynamic interaction of sTF and FVIIa in solution and on the surface of PS membranes revealed that isolated FVIIa undergoes large structural fluctuations whereas sTF is structurally stable and upon complex formation sTF restricts the motion of FVIIa significantly [57]. More importantly, sTF was shown to directly interact with PS through several residues located at the bottom of TF C-terminal domain, including Lys165 and Lys166. Although the conformation of sTF appears to be relatively unchanged on the surface of the membrane, its orientation against the membrane appears to change significantly. Interestingly, some of the TF residues that come in direct contact with PS are in the region that has been proposed to directly interact with FX [63–65]. Therefore, it is likely that direct interaction of PS head group with specific residues in TF exosite may alter the conformation of these residues. This along with a change in orientation of TF-FVIIa complex following its interaction with the membrane may be responsible for promoting TF-FVIIa complex to interact with substrates FX and FIX, thus transforming cryptic TF-FVIIa complexes into coagulant active TF-FVIIa complexes. FVIIa (in TF-FVIIa complex) binding to the exposed PS on the cell surface via its Gla domain may also play a role in such precise alignment of TF-FVIIa with the substrate.
Finally, it may be important to note here that at present the discussion on how PS could modulate TF-FVIIa activity at the cell surface is limited to the coagulant function and not the cell signaling function of TF-FVIIa. Although it has been suggested that cryptic and not the coagulant active TF is capable of inducing cell signaling [66], we are not aware of any study that actually examined whether and how PS exposure at the cell surface modulates TF-FVIIa signaling function. However, correlation of FVIIa binding to TF and the subsequent development of TF-FVIIa signaling function showed a linear relationship between the amount of FVIIa bound to TF and TF-FVIIa signal quantum, indicating all TF-FVIIa complexes formed on the cell surface are capable of inducing cell signaling [67,68].
Role of thiol pathways in regulating TF activity at cell surfaces
As discussed above, availability of anionic phospholipids at the cell surface, which are normally sequestered in the inner leaflet of the plasma membrane, plays a critical role in regulating TF activity at the cell surface. It has been shown that PS externalization is under the control of thiol-dependent inactivation of the PS translocase that moves aminophospholipids from the outer to the inner leaflet [69]. Thiolation also significantly lowers the threshold for Ca2+ required to activate outward PS transport by the scramblase and may directly activate a stress response that leads to a surge in Ca2+ into the cytosol [69]. Therefore, in principle, thiol pathways may play important roles in regulation of TF activity at cell surfaces and such mechanisms could be physiologically operative by regulated oxidation of specific protein sulfhydryls or by their conjugation with glutathione. Consistent with the possibility that thiol pathways may regulate TF activity at the cell surface, treatment of fibroblasts with sulfhydryl reagents (NEM) increased TF activity at the cell surface, and this was accompanied by increased PS exposure at the cell surface [33]. Recently, Popescu et al. [48] showed that inhibition of protein disulfide isomerase (PDI), an oxidoreductase and a protein known to be part of a thiol-exchange pathway, markedly increased the cell surface TF procoagulant activity by increasing the exposure of PS at the cell surface. These investigators showed that PDI regulates PS exposure by affecting the activities of both the flippase and floppase [48] (Fig. 2).
Figure 2.
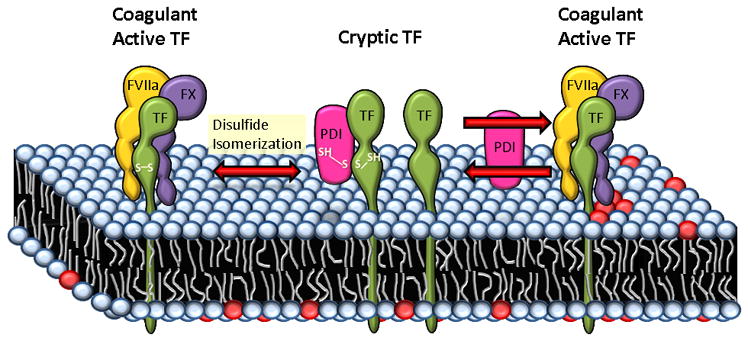
Proposed mechanisms of PDI-mediated regulation of TF coagulant activity at the cell surface. As per one of the proposed models, Cys186-Cys209 disulfide bond of TF is critical for its coagulant activity and post-translational modifications of this allosteric disuflide bond such as reduction, S-nitrosylation and glutathionation keep TF in a cryptic state. PDI can regulate the coagulant activity of TF through formation/breaking or reshuffling of this disulfide bond by its oxido-reductase or isomerase activity. PDI could also regulate TF coagulant activity through S-nitrosylation and/or glutathionation of thiols in TF. In a second model, PDI through its oxido-reductase activity modulates PS dynamics at the cell surface. PDI reductase activity helps in maintaining a low exposure of PS at the cell surface by affecting both flippase and floppase activities. Inhibition of PDI at the cell surface increases PS (shown as red color circles) exposure, which facilitates the conversion of cryptic TF to the coagulant active form. Both PDI-mediated disulfide bond switch and PDI-induced changes in the lipid environment may occur simultaneously and their net effect may dictate TF activity status at the cell surface.
Recent studies have also suggested that thiol-disulfide exchange reactions could regulate TF procoagulant activity at the cell surface independent of PS exposure by modification of the allosteric Cys186-Cys209 disulfide bond of TF [31,66] (Fig. 2). The rationale for this is based on following observations/assumptions - mutagenesis of Cys186 and Cys209 in the TF C-terminal module severely impaired factor X activation, a favorable redox potential of Cys186-Cys209 disulfide bond and close proximity between reduced Cys186 and Cys209 thiols, and solvent-exposed Cys186-Cys209 stabilizes TF-terminal fibronectin III module that is the contact site for FVIIa Gla-domain and one of the exosites for docking of macromolecular substrate [31,66,70]. Although a number of studies, including our own, clearly documented that mutagenesis of Cys186 and/or Cys209 in TF severely impaired TF-dependent FX activation [32,66,71,72], these data should not be construed as evidence that thiol pathways regulate TF procoagulant activity at the cell surface by switching on and off the Cys186-Cys209 disulfide bond in TF at the cell surface. The published studies clearly showed that mutagenesis of Cys186 and/or Cys209 severely impaired TF protein synthesis and/or processing such that only a small fraction of mutant TF was present at the cell surface relative to wild-type TF when the cells were transfected similarly with either the disulfide mutants or wild-type TF [32,66,71]. Furthermore, the affinity of FVIIa binding to the mutant was decreased by 10 to 50-fold compared to wild-type TF [32,71], suggesting that the Cys186-Cys209 disulfide bond is required for TF to maintain FVIIa binding conformation. The severe impairment in FVIIa binding could explain why TF mutants lacking the Cys186-Cys209 disulfide bond exhibited very low procoagulant activity even at FVIIa concentrations that are sufficient to saturate both active and cryptic TF populations (10 nM of FVIIa) but showed substantially higher, in some cases close to normal coagulant activity, when very high concentrations of FVIIa were used to overcome the binding impairment (Table 1). Recently, it had been reported that murine TF disulfide mutants (conserved at position Cys190 and Cys213 in murine) are expressed normally on the cell surface and TFC190/213A mutant exerted marginal procoagulant activity even at high concentrations of FVIIa (~15% of wild-type TF). Interestingly, TFC190A mutant expressed as much 40% of the procoagulant activity of wild-type TF even at 1 nM concentration of FVIIa [73]. It had been suggested that dimerization of TF as the resultant of the mutation is responsible for this TF mutant retaining substantial amount of coagulant activity [73], which runs contrary to earlier observations that indicate TF dimerization either reduces [61] or had no measurable effect [74] on TF coagulant activity (see later for more discussion on this).
Table 1.
Procoagulant activity of TF mutants lacking Cys186-Cys209 disulfide bond in different studies
| TF | FXa generated, nM/min | Ref | |
|---|---|---|---|
| FVIIa, 10 nM | FVIIa, 100–250 nM | ||
| TF wt | 45.0 (100%)1 | 30.0 (100%) | Rehemtulla et al.[71] |
| TFC186/S/C209S | 7.5 (17%) | 12.0 (40%) | |
| TF wt | 1.3 (100%)2 | Ahamed et al.[66] | |
| TFC209A | 0.1 (8%) | ||
| TF wt | 9.8 (100%)3 | 9.6 (100%) | Kothari et al.[32] |
| TFC186S/C209S | 3.9 (40%) | 8.1 (84%) | |
| TF wt | 0.45 (100%)4 | Ruf and Versteeg [86] | |
| TFC209A | 0.06 (13%) | ||
| TF wt | 3.2 (100%)5 | Kothari et al.[43] | |
| TFC209S | 0.9 (13%) | ||
| TFwt | 5.0 (100%)6 | 6.0 (100%) | Van den Hengel et al.[72] |
| TFC209A | 0.2 (4%) | 1.0 (17%) | |
| TFC209S | 1.3 (26%) | 3.0 (50%) | |
| TFC186S/C209S | 0.5 (10%) | 1.0 (17%) | |
FX, 2 μM, CHO cells;
concentrations of FX and FVIIa were not clearly specified, HUVEC;
FX, 1 μM, HUVEC;
FX, 100 nM, HUVEC;
FX, 175 nM, HUVEC;
FX, 100 nM, BHK cells.
In addition to mutagenesis data, several lines of other evidence have been provided to implicate that thiol pathways play an important role in regulation of TF activity at the cell surface by targeting the Cys186-Cys209 disulfide bond, and this evidence has been discussed in recent review articles [16,75–77]. In a nut shell, the evidence includes - (1) treatment of myeloid cells with thiol-oxidizing agent, HgCl2, or dithiol cross linkers increases cell surface TF activity by many fold [31,78]; (2) Blocking free thiols significantly impaired TF activation on cells [31,66]; (3) PDI associates with cell surface TF and knockdown of PDI increases TF procoagulant activity by 2-fold in keratinocytes [66]; (4) inactivation of TF coagulant activity upon nitrosylation and blockade of this inhibition by vicinal thiol blocker [66]; (5) ATP activation of P2X7 receptor increased availability of solvent-accessible extracellular thiols, increased TF activity at the cell surface and released TF-bearing microparticles in macrophages [79]; and (6) blocking PDI activity by specific mAb or by the inhibitor bacitracin inhibited thrombus formation in various murine thrombosis models [79–81]. Despite the above cited evidence, the importance of thiol pathways in regulating TF activity at the cell surface, particularly by targeting Cys186-Cys209 disulfide bond, appears to be circumstantial at present as many loopholes exist in the above evidence and data from other cell model systems failed to support the above hypothesis. They include - (1) analysis of human TF purified from brain or transfected mammalian cells showed that all four cysteines in the extracellular domain participate in forming two disulfide bonds and no free sulfhydryl groups exist in the TF protein [82–84]; (2) there was no experimental data demonstrating that TF on cell surfaces is present in a reduced form with respect to Cys186-Cys209 disulfide bond, and the oxidation with HgCl2 or PDI-mediated thiol exchange reaction reforms the disulfide bridge; (3) Cys186 and Cys209 may not be available for PDI when TF is bound to FVII/FVIIa [17] (this does not rule out the possibility that PDI acts on the Cys186-Cys209 disulfide bond when TF is not occupied by FVII or FVIIa); (4) HgCl2 treatment of various cell types was shown to increase PS on the outer leaflet of the plasma membrane [15,29,32,41,61,85]; (5) no demonstrable association of PDI and TF at the cell surface and knock-down of PDI has no effect on TF procoagulant activity in cancer cells [41]; (6) ATP activation of P2X7 receptor increased free thiols but also increased the TF procoagulant activity on macrophages (there is no information on whether the increased free thiols come from TF, PDI or some other proteins); (7) data from in vivo murine thrombosis models at best show that PDI contributes to local thrombin generation following the vascular injury, but they do not show that PDI activates TF by forming the Cys186-Cys209 disulfide bond and such change is responsible for thrombus formation.
It has been suggested that differences in cell types may be responsible for the conflicting data on potential modifications of allosteric Cys186-Cys209 disulfide bond by PDI-mediated thiol exchange reactions and its role in regulating TF procoagulant activity on cell surfaces [75,78,86]. It was assumed that cancer cells, fibroblasts or certain overexpression systems constitutively express active TF whereas TF on macrophages and monocytic cell lines was largely non-procoagulant on the cell surface, and thus it had been proposed that one should carefully choose right experimental systems to study TF activation [75,78]. However, there is no experimental evidence or rationale for the above assumption. Original studies, which identified that a majority of TF present on the cell surface is coagulant inactive, was obtained using an ovarian cancer cell model system [11]. Further, our ongoing studies revealed that high levels of TF procoagulant activity expressed in cancer cells simply reflect high levels of TF antigen present on these cells and not that these cells express mostly active TF (unpublished data of Kothari, Pendurthi and Rao, April 2012). Despite the noted controversy and valid concerns on the validity of PDI-mediated thiol pathways regulating TF activity on cell surfaces by modifying the Cys186-Cys209 disulfide bond, one should not negate the possibility of thiol pathways regulating TF activity on the cell surface by other mechanisms and the contribution of PDI to thrombus formation in vivo.
Self-association/disassociation of tissue factor
Chemical cross-linking of TF on intact cells revealed that a small fraction of TF on unperturbed cells exists as dimers and multimers, suggesting that TF may be self-associated on cell surfaces [61,87]. Both the cytoplasmic and transmembrane domains appear to be necessary for optimal self-association of TF [87]. Stimulation of HL60 cells with Ca2+ ionophore, which markedly increased TF activity at the cell surface, reduced the self-association of TF [61]. Based on this and other supporting data [61], it was suggested that TF on the surface of unperturbed cells exists as dimers, and the dimeric form of TF lacks the procoagulant activity, and calcium influx into cytosol triggers a calmodulin-dependent process, which results in dissociation of inactive TF dimers to procoagulant TF monomers [61]. According to this model, the substrate binding site is not readily accessible when TF become dimerized as the FX/FIX docking site on TF is at the interface of homodimers and the dissociation of dimeric TF into monomeric form would expose the substrate binding site on TF [62] (Fig. 3). However, Donate et al. found that the dimerized form of TF has the same factor X-activating efficiency as the monomeric form [74]. The pros and cons of the hypothesis that self-association and dissociation of TF on cell surfaces could influence the coagulant activity of TF on the cell surface was discussed in detail in an earlier review [62], and also summarized in a recent review [16].
Figure 3.
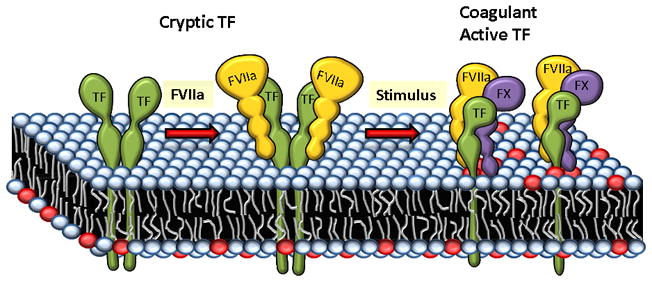
Transition of cryptic TF to coagulant active form involves transformation of dimeric TF to monomeric form. Dimeric TF lacks the coagulant activity as it can only bind to FVIIa but not to the substrate factor X. A stimulus that is capable of exposing PS (depicted in red color circles) at the cell surface also leads to transformation of dimeric TF to monomeric form that is capable of interacting with the substrate (the figure is a modified version of an earlier depiction by Key and Bach [133]).
Tissue factor glycosylation
It is well recognized that glycosylation plays an important role in cell surface receptor functions [88]. TF contains four potential N-linked glycosylation sites, three of them are in the extracellular domain at Asn11, Asn124, Asn137 and one in the cytoplasmic domain at Asn261. Studies of Paborsky et al. [83] revealed the presence of carbohydrates at all three glycosylation sites of the extracellular domain. At present, the effect of glycosylation on TF coagulant activity is not entirely clear as opposing results were obtained in different studies [83,89–94]. In very early studies, Pitlick et al. [89] found that concanavalin A, a member of the lectin family of carbohydrate binding proteins that preferentially bind to glucosyl and mannosyl residues [95], reversibly inhibited TF procoagulant activity. Further supporting the importance of carbohydrates for TF procoagulant activity on cell surfaces, tunicamycin, the inhibitor of N-linked glycosyl reaction, was shown to inhibit surface TF procoagulant activity in LPS-stimulated murine macrophages [93]. Bona et al. found that tunicamycin treatment decreased the TF procoagulant activity on cells by inhibiting the transport of TF to the cell surface [94]. However, based on similar procoagulant activity of rTF purified from eukaryotic expression system (human kidney 293 cells) or in E.coli, it had been concluded that glycosylation is not essential for TF procoagulant activity [83,96]. The observation that glycosylation site mutants of soluble rTF expressed in yeast exhibit a similar procoagulant activity as of rTF produced in E.coli and CHO cells further supported the conclusion that TF glycosylation does not influence TF procoagulant activity [90].
However, a recent careful and systematic comparison of the activity of TF purified from placenta and rTF derived from E.coli or insect expression systems revealed that natural placental TF was more catalytically active than other forms of TF [91]. Furthermore, deglycosylation of placental TF resulted in a significant decrease in TF coagulant activity [91]. Mass spectrophotometric analysis revealed that rTF1-243 derived from E.coli expression system had no carbohydrates attached to the backbone of the protein as expected, and placental TF was more heavily modified than rTF1-263 from insect cells [91]. Although all three potential glycosylation sites in the extracellular domain of both rTF1-263 and placental TF have carbohydrate attachments, the extent of glycosylation and carbohydrate composition was different between the two proteins, as well as between each glycosylated site within the protein [91,97]. Taken together, these data indicate that the presence of carbohydrates and the heterogeneity in the carbohydrate composition would significantly influence TF procoagulant activity.
In contrast to the above data, recent studies examining the role of carbohydrates on TF function by analyzing the procoagulant activity of wild-type TF and TF mutants lacking one or more N-linked glycans expressed in CHO or human endothelial cells showed no significant differences in cell surface TF activity among them [92]. At present the reason for the discrepancy between the data obtained in cell systems using TF mutagenesis approach [92] and analysis of purified placental TF [91] on the importance of glycosylation for TF procoagulant activity is unknown. It is possible that TF embedded in the cell membrane may behave differently from that of TF incorporated into liposomes in terms of the requirement of carbohydrate for its optimal coagulant activity. Alternatively, the carbohydrate composition of rTF expressed even in mammalian cells could be different from that of carbohydrates on natural placental TF and these differences could influence the interaction of TF with FVIIa and FX differently. In fact, there is some indirect evidence that glycosylation of TF from various species may vary and human TF was heterogeneous with respect to its carbohydrate moiety [98]. If rTF expressed in mammalian cells contain different carbohydrates from that of natural TF from placenta, then it would not only explain conflicting data in the literature on the importance of glycosylation to TF function but also raise an interesting possibility that TF activity could be modulated differentially in various cell types and diseases, particularly in cancer as altered glycosylation is a prominent and universal feature of cancer cells [99].
Tissue factor phosphorylation and palmitoylation
Among various posttranslational modifications of proteins, phosphorylation is the most important modification because protein phosphorylation plays a cardinal role in regulating many cellular processes in eukaryotes [100–102]. Protein phosphorylation is carried out by protein kinases, which transfer phosphate to the hydroxyl groups of the side chains of three amino acids – serine, threonine and tyrosine. Both the amino acid sequence motif surrounding the serine/threonine/tyrosine residues and the three-dimensional structure of the substrate proteins contribute to the phosphorylation specificity [103]. TF protein contains two phosphorylation sites (Ser253 and Ser258), both of them in the cytoplasmic domain. Ser253 was shown to be phosphorylated by PKCa-mediated mechanism [104,105]. Subsequent to the phosphorylation of Ser253, a proline-directed kinase phosphorylates Ser258 [105–107]. Phosphorylation of Ser258 was shown to be enhanced in vivo when palmitoylation of TF cytoplasmic domain is inhibited [105]. Studies of Ahamed and Ruf showed that TF-dependent PAR2 signaling leads to TF phosphorylation [106]. Although a number of studies reported that TF phosphorylation influences cell signaling, migration and angiogenesis [108–110], we are not aware of any report that clearly demonstrate that TF phosphorylation regulates TF procoagulant activity on cell surfaces. This is consistent with the data that the cytoplasmic domain of TF is not required for TF procoagulant activity on cell surfaces [111] or for its de-encryption [112,113]. Studies demonstrating that various PKC activators and inhibitors influence TF activity (see rev [6,114]) reflect the importance of PKC-mediated phosphorylation for TF gene transcription and protein expression rather than TF phosphorylation per se influencing the coagulant activity of TF. However, it had been reported recently that TF phosphorylation regulates TF incorporation into microparticles and its release [115].
Cys residues located near transmembrane regions are often sites for reversible attachment of the fatty acid palmitate [116–118]. Palmitoylation has been found to influence a wide spectrum of structural and functional features of proteins, including membrane binding and targeting [116,117]. Although transmembrane proteins do not require palmitoylation for membrane association, palmitoylation does affect the structure and function of integral membrane proteins by facilitating both protein-protein and protein-lipid interactions, and by targeting them to cholesterol and sphingolipid-rich microdomains and caveolae [116,117].
Studies of Bach and coworkers demonstrated the acylation of the cysteine in the cytoplasmic region of TF (Cys245) by both palmitic acid and steric acids [82]. They also showed that deacylation of TF with hydroxylamine resulted in spontaneous generation of disulfide-linked TF dimers, suggesting that TF dimers, a minor component found in most TF preparations could be the artifact produced by deacylation of Cys245 and subsequent interchain disulfide bond formation. However, this may also reflect the possibility that Cys245 in a minor fraction of TF was not acylated and thus it is free to form disulfide linkage with other proteins. The later possibility is consistent with the observation that traces of TF dimers and multimers are present in living cells [61,87]. There is no evidence at present that palmitoylation regulates TF activity at the cell surface, at least directly. It is possible that palmitoylation may regulate TF procoagulant activity indirectly by maintaining TF in a monomeric form, targeting TF into caveolae/lipid rafts or regulating TF phosphorylation. These mechanisms have been discussed in detail in other sections of this review. However, it is important to note that the link between the above regulatory processes and TF palmitoylation is not clearly established.
Caveolae, lipid rafts and cholesterol: Modulation of tissue factor procoagulant activity
Lipids in membranes do not always mix uniformly and often can cluster to form microdomains. Interaction of cholesterol with sphingolipids in the plasma membrane form liquid-ordered phase microdomains that are resistant to detergent solubilization and freely float in the plasma membrane, which are often referred to as lipid rafts [119,120]. Lipid rafts in association with caveolin forms flask shaped invaginations in the membrane, caveolae [121]. Caveolae/lipid rafts are known to play a critical role in facilitating protein-protein and protein-lipid interactions, and influence numerous cellular events in membrane traffic and signal transduction [121–124]. Ultrastructural localization of TF in a variety of cell types showed that a fraction of TF in these cells is associated with lipid rafts/caveolae [125–128]. It has been suggested that lipid raft/caveolae-associated TF represents the encrypted form, which can be activated rapidly by disruption of these structures [125,129,130]. It was shown in ECV304 cells that TF translocates to caveolae following ternary complex formation with FVIIa, TFPI, FXa and the TF translocated to caveolae exhibited a lower procoagulant activity even in the presence of anti-TFPI antibodies, suggesting that glycosphingolipid-enriched microdomains may inhibit TF directly [129]. In other cell types, TF may be associated with caveolae and lipid rafts independent of TFPI [127,128,131]. Dietzen et al. [130] reported that although TF is not stably associated with lipid rafts in HEK293 cells, disruption of lipid rafts by treating the cells with methyl β-cyclodextrin (mβCD), which removes cholesterol from the membrane, increased TF activity at the cell surface. These investigators have suggested that lipid rafts are required for the maintenance of cellular TF in an encrypted state probably by limiting the transbilayer transport of PS. However, similar experiments in other cell types yielded opposite results. Both in fibroblasts and cancer cells, disruption of caveolae/lipid rafts by mβCD treatment was shown to decrease TF procoagulant activity at the cell surface [128,131]. Interestingly, disruption of caveolae by treating the cells with filipin was found to increase TF procoagulant activity [131] whereas caveolae disruption by silencing caveloin-1 had no effect on TF procoagulant activity [128]. These variable results indicate that caveolae per se may not regulate procoagulant activity of TF at the cell surface, but the methodology used for caveolae disruption might be influencing the activity of TF independent of its association with caveolae. For example, the decrease in TF procoagulant activity where caveolae was disrupted with mβCD treatment may be the result of reduced cholesterol content in the plasma membrane. Increased TF procoagulant activity where caveolae was disrupted with filipin treatment could have been the result of the increased concentration of cholesterol in membrane patches as filipin sequesters the membrane cholesterol by forming complex with it [132]. The observation that depletion of membrane cholesterol reduces the cell surface TF activity and increasing the cholesterol concentration in the membrane by loading cells with cholesterol increases the cell surface TF activity in fibroblasts and cancer cells [128,131] fits with the concept that membrane cholesterol regulates TF activity. Cholesterol modulation of TF activity on cell surfaces appears to be independent of PS. FVIIa binding studies suggest that membrane cholesterol may modulate TF affinity to FVIIa [131]. At present whether cholesterol modulates TF procoagulant activity by direct interaction between cholesterol and TF or as a critical component necessary to form lipid rafts is not entirely clear.
Concluding remarks
Most of the evidence in the literature point out that exposure of anionic phospholipids, such as PS, on the outer leaflet of plasma membrane is the key regulator of TF procoagulant activity at the cell surface. However, a role for PS does not automatically exclude the involvement of other mechanisms in regulating TF activity on cell surfaces. Recent data that suggest thiol- and PDI-dependent pathways regulate TF procoagulant activity on cell surfaces through modification of Cys186-Cys209 disulfide bond are interesting but direct evidence for this is still lacking. There is no real disagreement on that the Cys186-Cys209 disulfide bond is critical for TF processing to the cell surface and to the proper conformation of TF to bind FVIIa, and PDI- and thiol-pathways, which influence various cellular functions and proteins, regulate TF procoagulant activity and contribute to TF-dependent thrombosis in vivo. However, connecting these two processes and to conclude that PDI-mediated switching on/off of the Cys186-Cys209 disulfide bond plays a major regulatory role in TF-dependent hemostasis and thrombosis requires more robust and convincing evidence. There is some evidence that other post-translational modifications of TF, particularly glycosylation, may regulate TF coagulant activity but future studies need to address potential differences between natural TF and rTF in their carbohydrate composition, and whether TF relipidated in phospholipid vesicles would behave the same as TF embedded in the cell membrane to draw meaningful conclusions.
Acknowledgments
This work was supported by National Institutes of Health grants HL58869, HL65500 and HL1074830. The authors acknowledge Janet Arras’s help in preparing figures.
Footnotes
Disclosure and conflict of interests
None
References
- 1.Fleck RA, Rao LVM, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59:421–37. doi: 10.1016/0049-3848(90)90148-6. [DOI] [PubMed] [Google Scholar]
- 2.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues: Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–97. [PMC free article] [PubMed] [Google Scholar]
- 3.Contrino J, Hair G, Kreutzer DL, Rickles FR. In situ detection of tissue factor in vascular endothelial cells: Correlation with the malignant phenotype of human breast disease. Nature Medicine. 1996;2:209–15. doi: 10.1038/nm0296-209. [DOI] [PubMed] [Google Scholar]
- 4.Osterud B, Flaegstad T. Increased thromboplastin activity in monocytes of patients with meningococcal infection: Related to an unfavorable prognosis. Thromb Haemost. 1983;49:5–7. [PubMed] [Google Scholar]
- 5.Owens AP, III, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–97. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butenas S, Amblo-Krudysz J, Mann KG. Posttranslational modifications of tissue factor. Front Biosci (Elite Ed) 2012;4:381–91. doi: 10.2741/385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osterud B, Bjorklid E. Tissue factor in blood cells and endothelial cells. Front Biosci (Elite Ed) 2012;4:289–99. doi: 10.2741/e376. [DOI] [PubMed] [Google Scholar]
- 8.Maynard JR, Heckman CA, Pitlick FA, Nemerson Y. Association of tissue factor activity with the surface of cultured cells. J Clin Invest N Y. 1975;55:814–22. doi: 10.1172/JCI107992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broze GJ., Jr Binding of human factor VII and VIIa to monocytes. J Clin Invest N Y. 1982;70:526–35. doi: 10.1172/JCI110644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fair DS, MacDonald MJ. Cooperative interaction between factor VII and cell surface-expressed tissue factor. J Biol Chem. 1987;262:11692–8. [PubMed] [Google Scholar]
- 11.Le DT, Rapaport SI, Rao LVM. Relations between factor VIIa binding and expression of factor VIIa/tissue factor catalytic activity on cell surfaces. J Biol Chem. 1992;267:15447–54. [PubMed] [Google Scholar]
- 12.Sakai T, Lund-Hansen T, Paborsky L, Pedersen AH, Kisiel W. Binding of human factors VII and VIIa to a human bladder carcinoma cell line (J82) implications for the initiation of the extrinsic pathway of blood coagulation. J Biol Chem. 1989;264:9980–8. [PubMed] [Google Scholar]
- 13.Walsh JD, Geczy CL. Discordant Expression of Tissue Factor Antigen and Procoagulant Activity on Human Monocytes Activated with LPS and Low Dose Cycloheximide. Thromb Haemost. 1991;66:552–8. [PubMed] [Google Scholar]
- 14.Ploplis VA, Edgington TS, Fair DS. Initiation of the extrinsic pathway of coagulation. Association of factor VIIa with a cell line expressing tissue factor. J Biol Chem. 1987;262:9503–8. [PubMed] [Google Scholar]
- 15.Bach R, Rifkin DB. Expression of tissue factor procoagulant activity: regulation by cytosolic calcium. Proc Natl Acad Sci U S A. 1990;87:6995–9. doi: 10.1073/pnas.87.18.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rao LVM, Kothari H, Pendurthi UR. Tissue factor: Mechanisms of decryption. Front Biosci. 2012;E4:1513–27. doi: 10.2741/477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bach RR, Monroe D. What is wrong with the allosteric disulfide bond hypothesis? Arterioscler Thromb Vasc Biol. 2009;29:1997–8. doi: 10.1161/ATVBAHA.109.194985. [DOI] [PubMed] [Google Scholar]
- 18.Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res. 2003;44:233–42. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Zwaal RF, Bevers EM, Comfurius P, Rosing J, Tilly RH, Verhallen PF. Loss of membrane phospholipid asymmetry during activation of blood platelets and sickled red cells; mechanisms and physiological significance. Mol Cell Biochem. 1989;91:23–31. doi: 10.1007/BF00228075. [DOI] [PubMed] [Google Scholar]
- 20.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–88. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 21.Chargaff E. Remarks on the role of lipids in blood coagulation. Arch Sci Physiol. 1948;2:269–71. [Google Scholar]
- 22.Nemerson Y. The phospholipid requirement of tissue factor in blood coagulation. J Clin Invest. 1968;47:72–80. doi: 10.1172/JCI105716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjorklid E, Storm E. Purification and Some Properties of the Protein Component of Tissue Thromboplastin from Human Brain. Biochem J. 1977;165:89–96. doi: 10.1042/bj1650089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnaswamy S, Field KA, Edgington TS, Morrissey JH, Mann KG. Role of the membrane surface in the activation of human coagulation factor X. J Biol Chem. 1992;267:26110–20. [PubMed] [Google Scholar]
- 25.Rapaport SI, Rao LVM. The tissue factor pathway: How it has become a “prima ballerina”. Thromb Haemost. 1995;74:7–17. [PubMed] [Google Scholar]
- 26.Neuenschwander PF, Bionco-Fisher E, Rezaie AR, Morrissey JH. Phosphatidylethanolamine augments factor VIIa-tissue factor activity: Enhancement of sensitivity to phosphatidylserine. Biochem. 1995;34:13988–93. doi: 10.1021/bi00043a004. [DOI] [PubMed] [Google Scholar]
- 27.Shaw AW, Pureza VS, Sligar SG, Morrissey JH. The local phospholipid environment modulates the activation of blood clotting. J Biol Chem. 2007;282:6556–63. doi: 10.1074/jbc.M607973200. [DOI] [PubMed] [Google Scholar]
- 28.Tavoosi N, Davis-Harrison RL, Pogorelov TV, Ohkubo YZ, Arcario MJ, Clay MC, Rienstra CM, Tajkhorshid E, Morrissey JH. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286:23247–53. doi: 10.1074/jbc.M111.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolberg AS, Monroe DM, Roberts HR, Hoffmann MR. Tissue factor de-encryption:ionophore treatment induces changes in tissue factor activity by phosphatidylserine-dependent and -independent mechanisms. Blood Coag Fibrinol. 1999;10:201–10. [PubMed] [Google Scholar]
- 30.Kunzelmann-Marche C, Satta N, Toti F, Zhang Y, Nawroth PP, Morrissey JH, Freyssinet J-M. The influence exerted by a restricted phospholipid microenvironment on the expression of tissue factor activity at the cell plasma membrane surface. Thromb Haemost. 2000;83:282–9. [PubMed] [Google Scholar]
- 31.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochem. 2006;45:12020–8. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- 32.Kothari H, Nayak RC, Rao LV, Pendurthi UR. Cystine186-cystine 209 disulfide bond is not essential for the procoagulant activity of tissue factor or for its de-encryption. Blood. 2010;115:4273–83. doi: 10.1182/blood-2009-09-241356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le DT, Rapaport SI, Rao LVM. Studies of the mechanism for enhanced cell surface factor VIIa/tissue factor activation of factor X in fibroblast monolayers after their exposure to N-ethylmalemide. Thromb Haemost. 1994;72:848–55. [PubMed] [Google Scholar]
- 34.Henriksson CE, Klingenberg O, Ovstebo R, Joo GB, Westvik AB, Kierulf P. Discrepancy between tissue factor activity and tissue factor expression in endotoxin-induced monocytes is associated with apoptosis and necrosis. Thromb Haemost. 2005;94:1236–44. doi: 10.1160/TH05-07-0463. [DOI] [PubMed] [Google Scholar]
- 35.Carson SD, Johnson DR. Consecutive Enzyme Cascades: Complement Activation at the Cell Surface Triggers Increased Tissue Factor Activity. Blood. 1990;76:361–7. [PubMed] [Google Scholar]
- 36.Greeno EW, Bach R, Moldow CF. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Laboratory investigation. 1996;75:281–9. [PubMed] [Google Scholar]
- 37.Carson SD. Manifestation of cryptic fibroblast tissue factor occurs at detergent concentrations which dissolve the plasma membrane. Blood Coag Fibrin. 1996;7:303–13. doi: 10.1097/00001721-199604000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Carson SD, Kuszynski CA, Pirruccello SJ. Fibroblasts restrict tissue factor from vesicles which form in response to low concentrations of detergent. Blood Coag Fibrin. 1996;7:314–24. doi: 10.1097/00001721-199604000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Rao LVM, Robinson T, Hoang AD. Factor VIIa/tissue factor-catalyzed activation of factors IX and X on a cell surface and in suspension: A kinetic study. Thromb Haemost. 1992;67:654–9. [PubMed] [Google Scholar]
- 40.Fu Y, Zhou J, Li H, Cao F, Su Y, Fan S, Li Y, Wang S, Li L, Gilbert GE, Shi J. Daunorubicin induces procoagulant activity of cultured endothelial cells through phosphatidylserine exposure and microparticles release. Thromb Haemost. 2010;104:1235–41. doi: 10.1160/TH10-02-0102. [DOI] [PubMed] [Google Scholar]
- 41.Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110:3900–8. doi: 10.1182/blood-2007-07-101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J, Shi J, Hou J, Cao F, Zhang Y, Rasmussen JT, Heegaard CW, Gilbert GE. Phosphatidylserine exposure and procoagulant activity in acute promyelocytic leukemia. J Thromb Haemost. 2010;8:773–82. doi: 10.1111/j.1538-7836.2010.03763.x. [DOI] [PubMed] [Google Scholar]
- 43.Kothari H, Rao LVM, Pendurthi UR. Cys186-Cys209 disulfide-mutated tissue factor does not equal cryptic tissue factor: no impairment in decryption of disulfide mutated tissue factor. Blood. 2010;116:502–3. [Google Scholar]
- 44.van Heerde WL, De Groot PG, Reutelingsperger CPM. The complexity of the phospholipid binding protein annexin v. Thromb Haemost. 1995;73:172–9. [PubMed] [Google Scholar]
- 45.Ravanat C, Archipoff G, Beretz A, Freund G, Cazenave JP, Freyssinet JM. Use of annexin-V to demonstrate the role of phosphatidylserine exposure in the maintenance of haemostatic balance by endothelial cells. Biochem J. 1992;282:7–13. doi: 10.1042/bj2820007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuart MC, Reutelingsperger CP, Frederik PM. Binding of annexin V to bilayers with various phospholipid compositions using glass beads in a flow cytometer. Cytometry. 1998;33:414–9. [PubMed] [Google Scholar]
- 47.Rao LVM, Tait JF, Hoang AD. Binding of annexin V to a human ovarian carcinoma cell line (OC-2008). Contrasting effects on cell surface factor VIIa/tissue factor activity and prothrombinase activity. Thromb Res. 1992;67:517–31. doi: 10.1016/0049-3848(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 48.Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116:993–1001. doi: 10.1182/blood-2009-10-249607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dachary-Prigent J, Freyssinet J, Pasquet J, Carron J, Nurder AT. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: A flow cytometry study showing a role for free sulfhydryl groups. Blood. 1993;81:2554–65. [PubMed] [Google Scholar]
- 50.Smeets EF, Comfurius P, Bevers EM, Zwaal RF. Contribution of different phospholipid classes to the prothrombin converting capacity of sonicated lipid vesicles. Thromb Res. 1996;81:419–26. doi: 10.1016/0049-3848(96)00014-x. [DOI] [PubMed] [Google Scholar]
- 51.Tait JF, Gibson D, Fujikawa K. Phospholipid binding properties of human placental anticoagulant protein-I, a member of the lipocortin family. J Biol Chem. 1989;264:7944–9. [PubMed] [Google Scholar]
- 52.Bach R, Gentry R, Nemerson Y. Factor VII binding to tissue factor in reconstituted phospholipid vesicles: induction of cooperativity by phosphatidylserine. Biochem. 1986;25:4007–20. doi: 10.1021/bi00362a005. [DOI] [PubMed] [Google Scholar]
- 53.Forman SD, Nemerson Y. Membrane-dependent coagulation reaction is independent of the concentration of phospholipid-bound substrate: fluid phase factor X regulates the extrinsic system. Proc Natl Acad Sci U S A. 1986;83:4675–9. doi: 10.1073/pnas.83.13.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hathcock JJ, Rusinova E, Gentry RD, Andree H, Nemerson Y. Phospholipid Regulates the Activation of Factor X by Tissue Factor/Factor VIIa (TF/VIIa) via Substrate and Product Interactions. Biochem. 2005;44:8187–97. doi: 10.1021/bi050338b. [DOI] [PubMed] [Google Scholar]
- 55.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991;266:2158–66. [PubMed] [Google Scholar]
- 56.Bom VJJ, Bertina RM. The contributions of Ca2+, phospholipids and tissue-factor apoprotein to the activation of human blood-coagulation factor X by activated factor VII. Biochem J. 1990;265:327–36. doi: 10.1042/bj2650327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohkubo YZ, Morrissey JH, Tajkhorshid E. Dynamical view of membrane binding and complex formation of human factor VIIa and tissue factor. J Thromb Haemost. 2010;8:1044–53. doi: 10.1111/j.1538-7836.2010.03826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrissey JH, Tajkhorshid E, Rienstra CM. Nanoscale studies of protein-membrane interactions in blood clotting. J Thromb Haemost. 2011;9 (Suppl 1):162–7. doi: 10.1111/j.1538-7836.2011.04300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morrissey JH, Tajkhorshid E, Sligar SG, Rienstra CM. Tissue factor/factor VIIa complex: role of the membrane surface. Thromb Res. 2012;129 (Suppl 2):S8–10. doi: 10.1016/j.thromres.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sen P, Neuenschwander PF, Pendurthi UR, Rao LV. Analysis of factor VIIa binding to relipidated tissue factor by surface plasmon resonance. Blood Coagul Fibrinolysis. 2010;21:376–9. doi: 10.1097/MBC.0b013e328333b084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bach RR, Moldow CF. Mechanism of tissue factor activation on HL-60 cells. Blood. 1997;89:3270–6. [PubMed] [Google Scholar]
- 62.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–61. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 63.Huang Q, Neuenschwander PF, Rezaie AR, Morrissey JH. Substrate recognition by tissue factor-factor VIIa. J Biol Chem. 1996;271:21752–7. doi: 10.1074/jbc.271.36.21752. [DOI] [PubMed] [Google Scholar]
- 64.Kirchhofer D, Lipari MT, Moran P, Eigenbrot C, Kelley RF. The tissue factor region that interacts with substrates factor IX and Factor X. Biochem. 2000;39:7380–7. doi: 10.1021/bi000182+. [DOI] [PubMed] [Google Scholar]
- 65.Rao LVM, Ruf W. Tissue factor residues Lys165 and Lys166 are essential for rapid formation of quaternary complex of tissue factor.VIIa with Xa. tissue factor pathway inhibitor. Biochem. 1995;34:10867–71. doi: 10.1021/bi00034a020. [DOI] [PubMed] [Google Scholar]
- 66.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci U S A. 2006;103:13932–7. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pendurthi UR, Allen KE, Ezban M, Rao LVM. Factor VIIa and thrombin induce the expression of Cyr61 and connective tissue growth factor, extracellular matrix signaling proteins that could act as possible downstream mediators in factor VII. tissue factor-induced signal transduction. J Biol Chem. 2000;275:14632–41. doi: 10.1074/jbc.275.19.14632. [DOI] [PubMed] [Google Scholar]
- 68.Petersen LC, Albrektsen T, Hjorto GM, Kjalke M, Bjorn SE, Sorensen BB. Factor VIIa/tissue factor-dependent gene regulation and pro-coagulant activity: effect of factor VIIa concentration. Thromb Haemost. 2007;98:909–11. [PubMed] [Google Scholar]
- 69.Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–64. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- 70.Liang HP, Brophy TM, Hogg PJ. Redox properties of the tissue factor Cys186-Cys209 disulfide bond. Biochem J. 2011;437:455–60. doi: 10.1042/BJ20110718. [DOI] [PubMed] [Google Scholar]
- 71.Rehemtulla A, Ruf W, Edgington TS. The integrity of the cysteine 186-cysteine 209 bond of the second disulfide loop of tissue factor is required for binding of factor VII. J Biol Chem. 1991;266:10294–9. [PubMed] [Google Scholar]
- 72.van den Hengel LG, Kocaturk B, Reitsma PH, Ruf W, Versteeg HH. Complete abolishment of coagulant activity in monomeric disulfide-deficient tissue factor. Blood. 2011;118:3446–8. doi: 10.1182/blood-2011-06-364612. [DOI] [PubMed] [Google Scholar]
- 73.van den Hengel LG, Osanto S, Reitsma PH, Versteeg HH. Murine tissue factor coagulant activity is critically dependent on the presence of an intact allosteric disulfide bond. Haematologica. 2012 doi: 10.3324/haematol.2012.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Donate F, Kelly CR, Ruf W, Edgington TS. Dimerization of tissue factor supports solution-phase autoactivation of factor VII without influencing proteolytic activation of factor X. Biochem. 2000;39:11467–76. doi: 10.1021/bi000986p. [DOI] [PubMed] [Google Scholar]
- 75.Versteeg HH, Ruf W. Thiol pathways in the regulation of tissue factor prothrombotic activity. Curr Opin Hematol. 2011;18:343–8. doi: 10.1097/MOH.0b013e32834981de. [DOI] [PubMed] [Google Scholar]
- 76.Rao LV, Kothari H, Pendurthi UR. Tissue factor encryption and decryption: facts and controversies. Thromb Res. 2012;129 (Suppl 2):S13–S17. doi: 10.1016/j.thromres.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruf W. Role of thiol pathways in TF procoagulant regulation. Thromb Res. 2012;129 (Suppl 2):S11–S12. doi: 10.1016/j.thromres.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liang HPH, Hogg PJ. Critical importance of the cell system when studying tissue factor de-encryption. Blood. 2008;112:912–3. doi: 10.1182/blood-2008-05-158766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Furlan-Freguia C, Marchese P, Gruber A, Ruggeri ZM, Ruf W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J Clin Invest. 2011;121:2932–44. doi: 10.1172/JCI46129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–22. doi: 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–31. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bach R, Konigsberg WH, Nemerson Y. Human tissue factor contains thioester-linked palmitate and sterate on the cytoplasmic half-cystine. Biochem. 1988;14:4227–31. doi: 10.1021/bi00412a004. [DOI] [PubMed] [Google Scholar]
- 83.Paborsky LR, Harris RJ. Post-translational modifications of recombinant human tissue factor. Thromb Res. 1990;60:367–76. doi: 10.1016/0049-3848(90)90219-3. [DOI] [PubMed] [Google Scholar]
- 84.Krudysz-Amblo J, Jenny RJ, Knigy T, Matthews D, Mann KG, Butenas S. Allosteric cysteine oxidation does not play a role in tissue factor decryption. Blood. 2011 [Google Scholar]
- 85.Kaneko H, Kakkar VV, Scully MF. Mercury compounds induce a rapid increase in procoagulant activity of monocyte-like U937 cells. Br J Haematol. 1994;87:87–93. doi: 10.1111/j.1365-2141.1994.tb04875.x. [DOI] [PubMed] [Google Scholar]
- 86.Ruf W, Versteeg HH. Tissue factor mutated at the allosteric Cys186-Cys209 disulfide bond is severely impaired in decrypted procoagulant activity. Blood. 2010;116:500–1. doi: 10.1182/blood-2010-04-281287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roy S, Paborsky LR, Vehar GA. Self-association of tissue factor as revealed by chemical crosslinking. J Biol Chem. 1991;266:4665–8. [PubMed] [Google Scholar]
- 88.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–67. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 89.Pitlick FA. Concanavalin A inhibits tissue factor coagulant activity. J Clin Invest. 1975;55:175–9. doi: 10.1172/JCI107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stone MJ, Ruf W, Miles DJ, Edgington TS, Wright PE. Recombinant soluble human tissue factor secreted by Saccharomyces cerevisiae and refolded from E. coli inclusion bodies: glycosylation of mutants, activity and physical characterization. Biochem J. 1995;310:605–14. doi: 10.1042/bj3100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krudysz-Amblo J, Jennings ME, Mann KG, Butenas S. Carbohydrates and activity of natural and recombinant tissue factor. J Biol Chem. 2010;285:3371–82. doi: 10.1074/jbc.M109.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kothari H, Rao LV, Pendurthi UR. Glycosylation of tissue factor is not essential for its transport or functions. J Thromb Haemost. 2011;9:1511–20. doi: 10.1111/j.1538-7836.2011.04332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shands JW., Jr Macrophage factor X activator formation: metabolic requirements for synthesis of components. Blood. 1985;65:169–75. [PubMed] [Google Scholar]
- 94.Bona R, Lee E, Rickles F. Tissue factor apoprotein: intracellular transport and expression in shed membrane vesicles. Thromb Res. 1987;48:487–500. doi: 10.1016/0049-3848(87)90405-1. [DOI] [PubMed] [Google Scholar]
- 95.Goldstein IJ, Hollerman CE, Smith EE. Protein-carbohydrate interaction. II. Inhibition studies on the interaction of concanavalin A with polysaccharides. Biochem. 1965;4:876–83. doi: 10.1021/bi00881a013. [DOI] [PubMed] [Google Scholar]
- 96.Paborsky LR, Tate KM, Harris RJ, Yansura DG, Band L, McCray G, Gorman CM, O’Brien DP, Chang JY, Swartz JR, Fung VP, Thomas JN, Vehar GA. Purification of Recombinant Human Tissue Factor. Biochem J. 1989;28:8072–7. doi: 10.1021/bi00446a016. [DOI] [PubMed] [Google Scholar]
- 97.Krudysz-Amblo J, Jennings ME, Matthews DE, Mann KG, Butenas S. Differences in the fractional abundances of carbohydrates of natural and recombinant human tissue factor. Biochim Biophys Acta. 2011;1810:398–405. doi: 10.1016/j.bbagen.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van den Besselaar AMHP, Bertina RM. Interaction of thromboplastin apoprotein of different tissues with concanavalin A- Evidence for heterogeneous glycosylation of the human apoprotein. Thromb Haemost. 1984;52:192–5. [PubMed] [Google Scholar]
- 99.Varki A, Kannagi R, Toole BP. Essentials of glycobiology. 2. New York: CSH Press; 2009. Glycosylation changes in cancer; pp. 617–32. [Google Scholar]
- 100.Cohen P. The regulation of protein function by multisite phosphorylation -a 25 year update. T I B S. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 101.Pawson T, Scott JD. Protein phosphorylation in signaling--50 years and counting. Trends Biochem Sci. 2005;30:286–90. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 102.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70:375–87. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 103.Via A, Diella F, Gibson TJ, Helmer-Citterich M. From sequence to structural analysis in protein phosphorylation motifs. Front Biosci. 2011;16:1261–75. doi: 10.2741/3787. [DOI] [PubMed] [Google Scholar]
- 104.Zioncheck TF, Roy S, Vehar GA. The cytoplasmic domain of tissue factor is phosphorylated by a protein kinase C-dependent mechanism. J Biol Chem. 1992;267:3561–4. [PubMed] [Google Scholar]
- 105.Dorfleutner A, Ruf W. Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood. 2003;102:3998–4005. doi: 10.1182/blood-2003-04-1149. [DOI] [PubMed] [Google Scholar]
- 106.Ahamed J, Ruf W. Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J Biol Chem. 2004;279:23038–44. doi: 10.1074/jbc.M401376200. [DOI] [PubMed] [Google Scholar]
- 107.Mody RS, Carson SD. Tissue factor cytoplasmic domain peptide is multiply phosphorylated in vitro. Biochem. 1997;36:7869–75. doi: 10.1021/bi9701235. [DOI] [PubMed] [Google Scholar]
- 108.Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller BM, Friedlander M, Ruf W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004;10:502–9. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 109.Dorfleutner A, Hintermann E, Tarui T, Takada Y, Ruf W. Cross-talk of integrin alpha3beta1 and tissue factor in cell migration. Mol Biol Cell. 2004;15:4416–25. doi: 10.1091/mbc.E03-09-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ryden L, Grabau D, Schaffner F, Jonsson PE, Ruf W, Belting M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int J Cancer. 2010;126:2330–40. doi: 10.1002/ijc.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sorensen BB, Freskgard P-O, Nielsen LS, Rao LVM, Ezban M, Petersen LC. Factor VIIa-induced p44/42 mitogen-activated kinase activation requires the proteolytic activity of factor VIIa and is independent of the tissue factor cytoplasmic domain. J Biol Chem. 1999;274:21349–54. doi: 10.1074/jbc.274.30.21349. [DOI] [PubMed] [Google Scholar]
- 112.Wolberg AS, Kon RH, Monroe DM, Ezban M, Roberts HR, Hoffman M. Deencryption of cellular tissue factor is independent of its cytoplasmic domain. Biochem Biophys Res Commun. 2000;272:332–6. doi: 10.1006/bbrc.2000.2783. [DOI] [PubMed] [Google Scholar]
- 113.Carson SD, Bromberg ME. Tissue factor encryption/de-encryption is not altered in the absence of the cytoplasmic domain. Thromb Haemost. 2000;84:657–63. [PubMed] [Google Scholar]
- 114.Egorina EM, Sovershaev MA, Osterud B. Regulation of tissue factor procoagulant activity by post-translational modifications. Thromb Res. 2008;122:831–7. doi: 10.1016/j.thromres.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 115.Collier ME, Ettelaie C. Regulation of the incorporation of tissue factor into microparticles by serine phosphorylation of the cytoplasmic domain of tissue factor. J Biol Chem. 2011;286:11977–84. doi: 10.1074/jbc.M110.195214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Milligan G, Parenti M, Magee AI. The dynamic role of palmitoylation in signal transduction. Trends Biochem Sci. 1995;20:181–7. doi: 10.1016/s0968-0004(00)89004-0. [DOI] [PubMed] [Google Scholar]
- 117.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 118.Mumby SM. Reversible palmitoylation of signaling proteins. Curr Opin Cell Biol. 1997;9:148–54. doi: 10.1016/s0955-0674(97)80056-7. [DOI] [PubMed] [Google Scholar]
- 119.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 120.Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, Okamoto T, Lisanti MP. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289–304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schlegel A, Lisanti MP. The caveolin triad: caveolae biogenesis, cholesterol trafficking, and signal transduction. Cytokine Growth Factor Rev. 2001;12:41–51. doi: 10.1016/s1359-6101(00)00022-8. [DOI] [PubMed] [Google Scholar]
- 122.Anderson RGW. Caveolae: where incoming and outgoing messengers meet. Proc Natl Acad Sci USA. 1993;90:10909–13. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 124.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. The journal of clinical investigation. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mulder AB, Smit JW, Bom VJJ, Blom NR, Ruiters MHJ, Halie R, van der Meer J. Association of smooth muscle cell tissue factor with caveolae. Blood. 1996;88:1306–13. [PubMed] [Google Scholar]
- 126.Mulder AB, Smit JW, Bom VJJ, Blom NR, Halie MR, van der Meer J. Association of endothelial tissue factor and thrombomodulin with caveolae. Blood. 1996;88:3667–3670M. [PubMed] [Google Scholar]
- 127.Mandal SK, Pendurthi UR, Rao LVM. Cellular localization and trafficking of tissue factor. Blood. 2006;107:4746–53. doi: 10.1182/blood-2005-11-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Awasthi V, Mandal SK, Papanna V, Rao LV, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol. 2007;27:1447–55. doi: 10.1161/ATVBAHA.107.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sevinsky JR, Rao LVM, Ruf W. Ligand induced protease receptor translocation into caveolae: A mechanism regulating cell surface proteolysis of the tissue factor dependent coagulation pathway. J Cell Biol. 1996;133:293–304. doi: 10.1083/jcb.133.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dietzen DJ, Page KL, Tetzloff TA. Lipid rafts are necessary for tonic inhibition of cellular tissue factor procoagulant activity. Blood. 2004;103:3038–44. doi: 10.1182/blood-2003-07-2399. [DOI] [PubMed] [Google Scholar]
- 131.Mandal SK, Iakhiaev A, Pendurthi UR, Rao LV. Acute cholesterol depletion impairs functional expression of tissue factor in fibroblasts: modulation of tissue factor activity by membrane cholesterol. Blood. 2005;105:153–60. doi: 10.1182/blood-2004-03-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141:905–15. doi: 10.1083/jcb.141.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Key NS, Bach RR. Tissue factor as a therapeutic target. Thromb Haemost. 2001;85:375–6. [PubMed] [Google Scholar]