

Abstract
Proteins with JAB1/MPN/MOV34 metalloenzyme (JAMM/MPN+) domains are widespread among all domains of life, yet poorly understood. Here we report the purification and characterization of an archaeal JAMM/MPN+ domain protein (HvJAMM1) from Haloferax volcanii that cleaves ubiquitin-like small archaeal modifier proteins (SAMP1/2) from protein conjugates. HvJAMM1 cleaved SAMP1/2 conjugates generated in H. volcanii as well as isopeptide-and linear-linked SAMP1-MoaE in purified form. Cleavage of linear linked SAMP1-MoaE was dependent on the presence of the SAMP domain and the C-terminal VSGG motif of this domain. While HvJAMM1 was inhibited by size exclusion chromatography and metal chelators, its activity could be restored by addition of excess ZnCl2. HvJAMM1 residues (Glu31, His88, His90, Ser98 and Asp101) that were conserved with the JAMM/MPN+ active-site motif were required for enzyme activity. Together, these results provide the first example of a JAMM/MPN+ zinc metalloprotease that independently catalyzes the cleavage of ubiquitin-like (isopeptide and linear) bonds from target proteins. In archaea, HvJAMM1 likely regulates sampylation and the pools of ‘free’ SAMP available for protein modification. HvJAMM1-type proteins are thought to release the SAMPs from proteins modified post-translationally as well as those synthesized as domain fusions.
Keywords: archaea, protease, ubiquitin, proteasome, post-translational modification
INTRODUCTION
Ubiquitylation is a post-translational mechanism that is important to DNA repair, protein quality control, cell survival and cell death in eukaryotes (Kerscher et al., 2006). The process of ubiquitylation involves the covalent attachment of ubiquitin (Ub) to protein targets through a series of reactions mediated by Ub-activating (E1), Ub-conjugating (E2) and Ub-ligase (E3) enzymes (Hochstrasser, 2009). Typically, the C-terminal carboxyl group of Ub is ligated to a lysine side chain of a substrate protein forming a covalent amide (isopeptide) bond. Following ligation of a single moiety of Ub to the protein target, further molecules of Ub can be added to the attached Ub forming diverse poly-Ub chains (Xu et al., 2009).
Deubiquitylating enzymes (DUBs) are important regulators of the levels of free (unconjugated) Ub and Ub-modified proteins (Amerik and Hochstrasser, 2004; Katz et al., 2010). DUBs remove molecules linked to the C-terminal carboxyl of the last residue of Ub (Gly76) and, thus, encompass a broad group of enzymes that hydrolyze diverse (ester, peptide and isopeptide) bonds. DUBs that cleave isopeptide bonds often catalyze the disassembly and editing of the Ub moieties attached to protein substrates. DUBs that hydrolyze peptide bonds can release Ub from inactive precursors that are translated from Ub gene fusions or can cleave Ub moieties that are attached post-translationally to the Nα-amino group of a target protein. DUBs can also reactivate C-terminal modified forms of Ub that are generated non-specifically by small nucleophiles in the cell.
DUBs are a broad group of proteases divided into five subfamilies including the JAB1/MPN/MOV34 (JAMM/MPN+ or JAMM) metalloproteases, the Ub C-terminal hydrolases (UCHLs), the Ub-specific proteases (UBPs), ovarian tumor proteases (OTUs) and the Josephin domain proteases (JDs) (Nijman et al., 2005). Ub-like proteases (ULPs) that remove Ub-related proteins (such as SUMO, Nedd8 and Smt3) from target proteins have also been described (Li and Hochstrasser, 2000). The majority of these enzymes use a nucleophilic cysteine thiol in their active site for hydrolysis and, thus, are cysteine-type proteases (Komander, 2010). In contrast, the less common DUBs of the JAMM subfamily use a catalytic metal ion for activity (Komander, 2010).
Archaea are predicted to encode metalloproteases of the JAMM subfamily but not cysteine-type DUBs. Members of the JAMM subfamily are widespread in all domains of life, yet the biological function of these proteins is poorly understood. The X-ray crystal structure of an archaeal JAMM protein (the Archaeoglobus fulgidus AfJAMM) has provided a framework for modeling the active sites of the JAMM isopeptidase subunits of 26S proteasomes (Rpn11/Poh1) and COP9 signalosomes (Csn5/Jab1) in eukaryotes (Tran et al., 2003; Ambroggio et al., 2004). However, an activity for an archaeal JAMM protein has not been reported.
Archaea have a mechanism of post-translational modification, termed sampylation, that shares many biochemical features with the ubiquitylation system of eukaryotes (Maupin-Furlow, 2012). In the archaeon Haloferax volcanii, the Ub-like proteins SAMP1 and SAMP2 are covalently attached to proteins by a mechanism that requires the Ub-activating E1 enzyme homolog termed UbaA (Humbard et al., 2010; Miranda et al., 2011). Much like Ub, SAMP2 is attached through isopeptide bonds to lysine residues of target proteins (Humbard et al., 2010). To reverse and/or regulate this process, proteases are predicted to be synthesized and cleave the SAMPs from SAMP-modified proteins (Humbard et al., 2010).
Here we report an H. volcanii JAMM domain protein (HvJAMM1) that functions as a zinc-dependent metalloprotease in the release of the Ub-like SAMPs from isopeptide-and linear-linked protein substrates. While HvJAMM1 has relatively broad spectrum of activity in removing SAMP1/2 from diverse proteins, this protease is unable to hydrolyze unmodified proteins otherwise hydrolyzed by proteinase K. HvJAMM1 is the first example of a JAMM domain protein that removes Ub-like proteins from diverse protein targets independent of a multisubunit complex and, thus, adds an important insight into this group of poorly characterized enzymes.
RESULTS
Two major groups of archaeal JAMM domain proteins
While archaea do not encode apparent homologs of cysteine-type isopeptidases (of the MEROPS database peptidase clans CA or CE), archaea are predicted to synthesize proteases of the Mov34-MPN-PAD-1 superfamily (cl13996) with JAB1/MPN/MOV34 metalloenzyme (JAMM) domains (MEROPS M67 family). Here we used hierarchical clustering to understand the relationships of the amino acid sequences of the ‘JAMM’ domain proteins of archaea to eukaryotic and bacterial homologs with known or putative functions (Fig. 1). The JAMM domain proteins that function as DUBs or isopeptidases in eukaryotes included in the analysis were Rpn11/Poh1 (Verma et al., 2002; Yao and Cohen, 2002), Csn5/Jab1 (Cope et al., 2002), AMSH and AMSH-LP (McCullough et al., 2004; Sato et al., 2008; Davies et al., 2011), 2A-DUB/MYSM1/KIAA1915 (Zhu et al., 2007), and Brcc36 (Sobhian et al., 2007; Cooper et al., 2009; Patterson-Fortin et al., 2010; Cooper et al., 2010; Feng et al., 2010). The cluster analysis also included the bacterial JAMM domain proteins Mycobacterium tuberculosis Mec+ (Rv1334) shown to catalyze the hydrolysis of cysteine from CysO-cysteine adduct formed by cysteine synthase M (CysM) (Burns et al., 2005), Pseudomonas fluorescens QbsD thought to remove the cysteine-phenylalanine residues from the C-terminus of the sulfur carrier QbsE (Godert et al., 2007), and Ttc1133 associated with the recently described TtuB-system of protein modification in Thermus thermophilus (Shigi, 2012). Note that the bacterial TtuB, CysO and QbsE are related to Ub and SAMP1/2 in their overall β-grasp structural fold (Burroughs et al., 2007).
Figure 1.
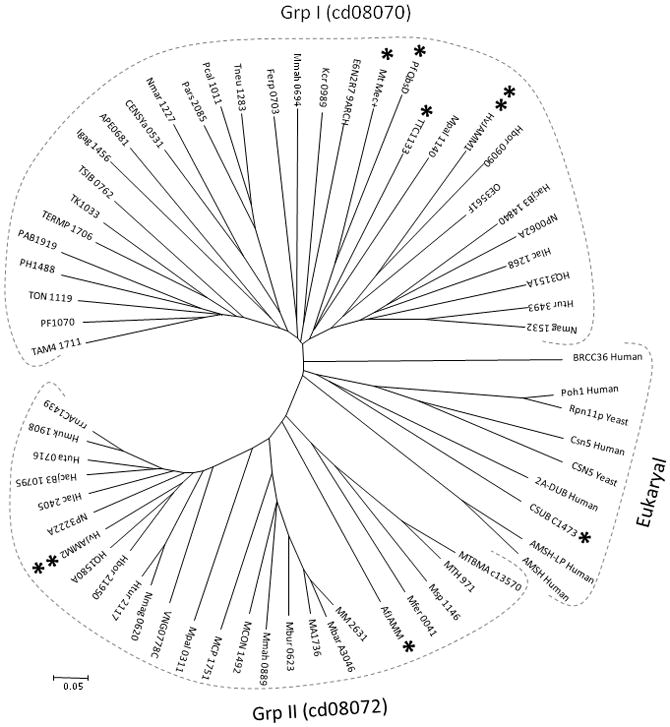
Dendrogram of archaeal JAMM domain proteins of the Mov34-MPN-PAD-1 superfamily (cl13996) compared to select homologs from eukaryotes and bacteria. Highlighted are HvJAMM1/2 (**), AfJAMM (*) and CSUB_C1473 (*) from archaea as well as Mec+, QbsD and TTC1133 (*) from bacteria, which are discussed in text. Abbreviation, organism source, and NCBI GenBank GI sequence identification numbers for each protein sequence are listed in Suppl. Table S3.
Archaeal JAMM domains were found to cluster into two major groups including group I (cd08070 family) and group II (cd08072 family) (Fig. 1). The one exception was the JAMM domain of CSUB_C1473 (Rpn11I), predicted by metagenomics to be encoded by Candidatus Caldiarchaeum subterraneum of the proposed crenarchaeal lineage ‘Aigarchaeota’ (Nunoura et al., 2011). Rpn11I clustered intimately with the JAMM domains of eukaryotic isopeptidases and DUBs (Fig. 1), as was previously observed by Nunoura et al. (2011). The JAMM domains of the bacterial Ttc1133, QbsD and Mec+ clustered to group I (Fig. 1). The archaeal AfJAMM that has been characterized structurally but not enzymatically (Tran et al., 2003; Ambroggio et al., 2004) clustered to group II (Fig. 1). Most halophilic archaea, such as H. volcanii, harbored JAMM domain homologs that clustered to both group I and II (Fig. 1). Based on gene neighborhood and co-occurrence patterns (Szklarczyk et al., 2011), JAMM domains of group I were associated with cysteine synthase (CysM) and E1-like enzymes (e.g., MoeB, ThiF), and those of group II were linked with archaeal B-type ATP-dependent Lon proteases (LonB) and Ub-like SAMP1 (Suppl. Fig. S1). The association of archaeal JAMM domains to sampylation and energy-dependent protein degradation based on gene neighborhood and co-occurrence patterns, combined with the structural relationship of archaeal JAMM domains to eukaryotic deubiquitylases and Ub-like isopeptidases, suggested that archaeal JAMM proteins may cleave SAMP conjugates.
HvJAMM1/2 purified from recombinant E. coli
To further characterize representative archaeal JAMM domain proteins of the Mov34-MPN-PAD-1 superfamily, H. volcanii HVO_2505 (HvJAMM1) of group I and HVO_1016 (HvJAMM2) of group II were selected for expression from pET-based plasmid vectors with N-terminal poly-histidine tags (His6-) in Escherichia coli. HvJAMM1 and HvJAMM2 were the only two JAMM domain (and DUB) homologs predicted for H. volcanii, and both proteins had conserved amino acid residues of the JAMM motif (EXnH[S/T]HX7SX2D, where X represents any residue) (Suppl. Fig. S2). HvJAMM2 included a C-terminal extension of 21 amino acid residues not present in HvJAMM1 or AfJAMM (Suppl. Fig. S2).HvJAMM1 and HvJAMM2 were readily purified from recombinant E. coli to apparent homogeneity by HisTrap HP Ni2+-affinity chromatography and size exclusion chromatography (SEC) (Suppl. Fig. S3). Based on SEC, HvJAMM1 purified as a 22.4-kDamonomer and HvJAMM2 purified as a mixture of 24.7-kDa monomeric and 50-kDa dimeric forms (compared to the theoretical molecular masses of 16.9/19.8 kDa for the His-tagged protein sequences of HvJAMM1/2, respectively). In previous studies, the related AfJAMM behaved as a monomer during purification by SEC and crystallized in an asymmetric unit as a dimer (Tran et al., 2003; Ambroggio et al., 2004). While an ~10-kDa discrepancy was found between the molecular mass of HvJAMM1 estimated by SDS-PAGE (26 kDa) compared to its theoretical molecular mass (16.9 kDa), most haloarchaeal proteins (like HvJAMM1) feature a negative surface charge and are active and stable in high salt (Mevarech et al., 2000). These unusual properties of HvJAMM1 may account for its slow migration in SDS-PAGE gels. Further analysis of HvJAMM1, by MALDI-TOF and ESI-MS, revealed observed average masses of 16,771.40 to 16,771.63 Da that were consistent with the theoretical average mass of 16,771.17 Da for this protein (Table 1).
Table 1.
Metal content and molecular masses of HvJAMM1 wild-type (WT) and variant proteins.
| HvJAMM1 | Metal content (mol/mol protein)1 | Average Mass (Da)2 | Mass Spec | ||
|---|---|---|---|---|---|
| Zinc | Nickel | Theoretical | Observed | ||
| WT | 0.84 (0.12) | 0.24 (0.018) | 16,771.17 | 16,771.40 | ESI-TOF |
| 16,771.63 | MALDI-TOF | ||||
| E31D | n.d. | n.d. | 16,757.14 | 16,757.21 | ESI-TOF |
| H88N | 0.01 | 0.07 | 16,748.13 | 16,749.74 | MALDI-TOF |
| H90Q | 0.04 | 0.04 | 16,762.16 | 16,762.88 | MALDI-TOF |
| D94N | n.d. | n.d. | 16,770.19 | 16,770.03 | ESI-TOF |
| S98A | 0.37 | 0.13 | 16,775.17 | 16,775.46 | MALDI-TOF |
| D101E | 1.1 | 0.34 | 16,785.20 | 16,785.75 | MALDI-TOF |
| C115S | n.d. | n.d. | 16,755.11 | 16,755.11 | ESI-TOF |
Metal content determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) of HisTrap HTP purified proteins. Metal content of HvJAMM1 purified by size exclusion chromatography in parenthesis.
Average mass in Da with theoretical mass based on the HvJAMM1 (HVO_2505) polypeptide sequence with addition of an N-terminal poly-histidine linker (His6-tag) (MGSSHHHHHHSSGLVPRGSH-) and amino acid modifications as indicated. Observed masses are based on MALDI-TOF MS and ESI-MS as indicated.
Abbreviations: WT, wild-type; n.d., not determined.
HvJAMM1 purified in an active form
A variety of purification methods, substrates and assay conditions were used to characterize the HvJAMM1/2 proteins. Substrates included diglycine-Amc, Ub-Amc, unlabeled proteins (i.e., hemoglobin, cytochrome c, carbonic anhydrase, creatinine phosphokinase, β-amylase and bovine serum albumin) and SAMP1/2 conjugates. HvJAMM2 did not hydrolyze these substrates under any conditions tested (see methods for details). In contrast, HvJAMM1 was activate after dilution of the cell lysate of recombinant E. coli into buffer supplemented with high salt (2M NaCl), but not low salt (150 mM NaCl) (see below for details on the types of enzyme activities detected). Therefore, the buffers used for the purification and assay of HvJAMM1 were routinely supplemented with molar concentrations of salt similarly to conditions used for the stability and activity of other haloarchaeal proteins (Connaris et al., 1999; Mevarech et al., 2000). Whether or not HvJAMM2 was synthesized and/or refolded into an active form remains to be determined, since the substrates used for assay may not have been optimal or physiological.
HvJAMM1 is a desampylase
HvJAMM1 was found to catalyze the metal-dependent cleavage of SAMP1/2 conjugates (Fig. 2). To detect this activity, SAMP1/2 proteins were synthesized with an N-terminal Flag-tag in H. volcanii. The Ub-like protein conjugates that were formed were boiled to remove the native hydrolytic activity of the cells and used as substrates for assay of HvJAMM1. Products were separated by reducing SDS-PAGE, and the conjugated and free forms of SAMP1/2 were detected by α-Flag immunoblot (Fig. 2). With this approach, HvJAMM1 was found to reduce the levels of SAMP1/2 conjugates and moderately increase the levels of free (unconjugated) SAMP1/2 in the cell lysate (Fig. 2). Addition of the metal chelator EDTA inhibited this reaction, suggesting a metal ion is required for HvJAMM1 activity (Fig. 2). Fluorescent staining for total protein by Sypro Ruby demonstrated that the majority of the proteins in the cell lysate were not degraded by HvJAMM1 (Fig. 2). Thus, HvJAMM1 did function as a general protease, but instead was relatively specific for cleaving SAMP1/2 conjugates.
Figure 2.
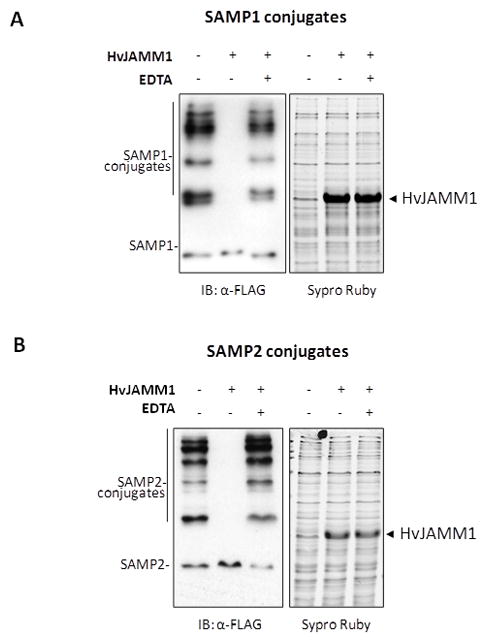
HvJAMM1 is a Zn2+-dependent JAMM metalloprotease. Desampylation activity of HvJAMM1 was monitored using Flag-SAMP1 S85K (A) and Flag-SAMP2 (B) conjugates as described in Experimental Procedures. Similar results were observed for Flag-SAMP1 conjugates. EDTA was included as a negative control to disrupt HvJAMM1 activity based on the conservation of residues in the HvJAMM1 amino acid sequence that are likely to coordinate a catalytic zinc ion. Total protein was stained with Sypro Ruby to confirm equal loading and qualitatively assess whether or not HvJAMM1 is a non-specific protease that hydrolyzes the cellular proteome.
Further analysis supported the conclusion that HvJAMM1 is not a general protease or peptidase. In particular, HvJAMM1 was unable to hydrolyze bovine serum albumin, hemoglobin, creatine phosphokinase, carbonic anhydrase, β-amylase or cytochrome c (Suppl. Fig. S4). Long-term incubation with HvJAMM1 had little if any impact on the level or length of these proteins (based on Coomassie Blue staining after separation by SDS-PAGE) (Suppl. Fig. S4). In contrast, the unmodified proteins were readily hydrolyzed by proteinase K, a broad spectrum subtilisin-type protease (Suppl. Fig. S4). In further defining the substrate specificity of HvJAMM1, this enzyme was found to be inactive in hydrolyzing the amide bond that links aminomethylcoumarin (AMC) to the C-terminus of monomeric Ub (Ub-AMC) or diglycine (GG-AMC) (Suppl. Fig. S5). The scissile bond of AMC-linked substrates differs in structure from true peptide bonds, which may account for these findings. As a positive control, Ub-AMC was found to be readily hydrolyzed by the eukaryotic cysteine-type isopeptidase T (Ub specific peptidase 5, USP5) in similar assay (Suppl. Fig. S5). Neither USP5 nor HvJAMM1 hydrolyzed GG-AMC. Together, these results reveal HvJAMM1 is not a general protease but is instead a metal-dependent protease that is relatively specific for the cleavage of SAMP1/2 from the Ub-like conjugates formed in H. volcanii.
HvJAMM1 activity optima
To determine optimal conditions for in vitro assay, HvJAMM1 activity was monitored at various enzyme concentrations, assay times, temperatures, salt concentrations, and pH conditions (Suppl. Fig. S6). Activity was readily detected within 1 h of assay at enzyme concentrations of 1.25–50 μM using 0.2 mg/ml substrate (boiled cell lysate with SAMP1/2 conjugates) (Suppl. Fig. S6 a, b). HvJAMM1 displayed optimal activity at temperatures of 40–50 °C, NaCl concentrations of 0.7–2 M and buffers of neutral to basic pH (pH 7–10) (Suppl. Fig. S6 c–e). HvJAMM1 was active over a wide range of temperature (20–60 °C). However, the enzyme was not active at 70 °C and displayed little to no activity at low concentrations of salt (150 mM NaCl) or low pH (pH 6.5 and below) (Suppl. Fig. S6c–e). Like HvJAMM1, the hydrolytic activities of Mec+ and AMSH can be detected within 1 h of assay (Burns et al., 2005; Davies et al., 2011), but AMSH and AMSH-LP can require assay times of 18–20 h (McCullough et al., 2004; Sato et al., 2008). Compared to other JAMM domain proteins, HvJAMM1 was similarly active at neutral to basic pH, but was active over a wider range of temperatures (20–60 vs. 20–37 °C) and salt concentrations (0.7–2 vs. 0–0.5 M NaCl) (McCullough et al., 2004; Burns et al., 2005; Cooper et al., 2009; Cooper et al., 2010). The ‘extreme’ conditions, in which HvJAMM1 is active, are consistent with the growth optima of 45 °C and 1.7–2.5 M NaCl for H. volcanii (Mullakhanbhai and Larsen, 1975; Robinson et al., 2005).
SAMP1 is linked to MoaE by isopeptide bonds
To further examine the substrate preference of HvJAMM1 and to determine whether SAMP1 forms isopeptide bonds with its protein targets, SAMP1-MoaE conjugates were purified from H. volcanii and used as substrates for HvJAMM1 activity assays. While we previously demonstrated by MS/MS that SAMP2 is attached to proteins by isopeptide bonds (Humbard et al., 2010), the covalent bond joining SAMP1 to its protein targets was not known. Here we co-expressed SAMP1 and MoaE from separate genes and used tandem affinity purification (TAP) to isolate SAMP1-MoaE conjugates (where, SAMP1 was expressed with an N-terminal Flag-tag and MoaE was expressed with a C-terminal StrepII-tag in H. volcanii). A single protein band was detected in the purified fractions, which was specific for StrepII and Flag and was resistant to boiling in reducing SDS-PAGE buffer (Fig. 3). Thus, SAMP1-MoaE conjugates were readily purified from H. volcanii.
Figure 3.
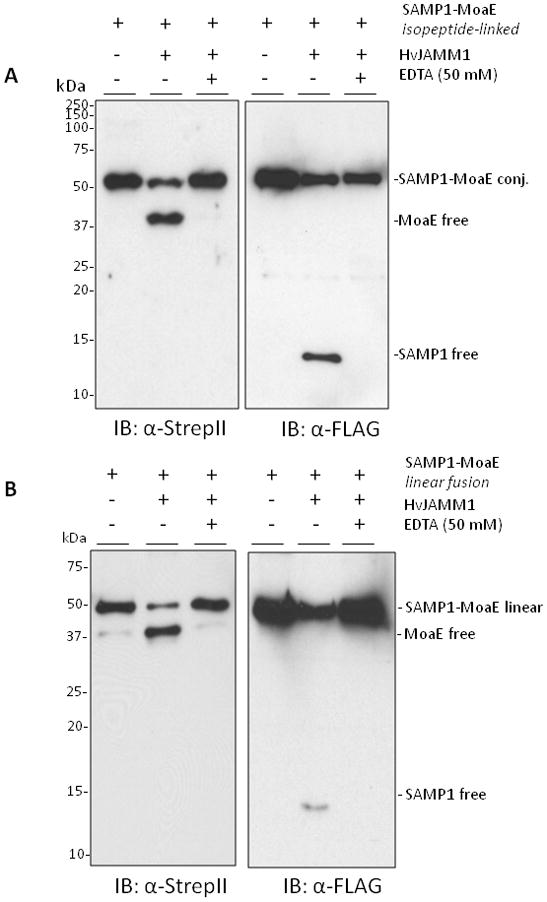
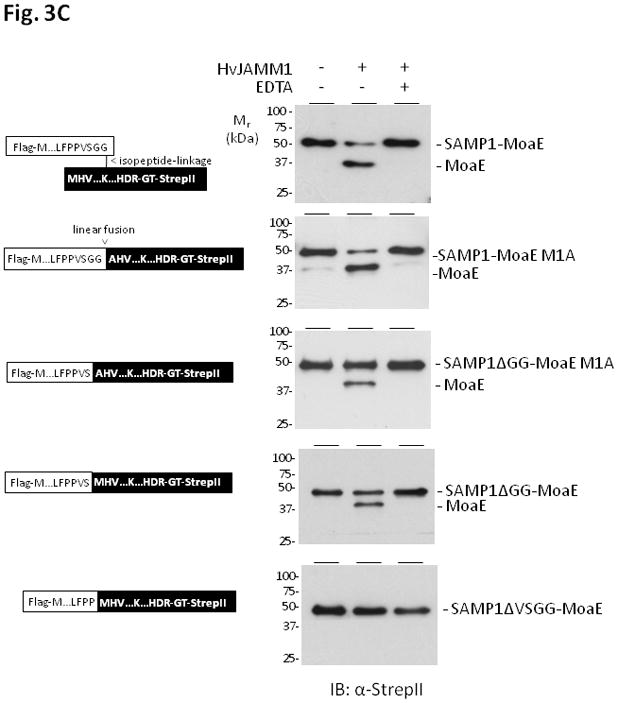
HvJAMM1releases SAMP1 from isopeptide (A) and linear (B and C) linkages to MoaE. Desampylation activity was monitored by α-Flag and α-StrepII immunoblot where the Flag-tag is incorporated at the N-terminus of SAMP1 and the StrepII tag is incorporated at the C-terminus of MoaE. Site-directed modifications were incorporated into the linear peptide bond linking SAMP1 to MoaE, and EDTA was included to inactivate HvJAMM1 as indicated. Protein bands specific for the free and conjugated forms of SAMP1 and MoaE migrated slower in SDS-PAGE gels than their theoretical molecular masses (i.e., 39 kDa for FLAG-SAMP1-MoaE-StrepII, 29 kDa for MoaE-StrepII, 12 for FLAG-SAMP1). This slow migration is similar to HvJAMM1 (this study), H. volcanii 20S proteasomes (Wilson et al., 1999) and other proteins such as eukaryotic Ub and Ub-like proteins (Van der Veen and Ploegh, 2012).
To determine the site(s) of modification and type of covalent bond linking SAMP1 to MoaE, a MS-based proteomic approach was used. SAMP1 S85K-MoaE conjugates were generated and purified from H. volcanii. The S85K modification was introduced near the C-terminal diglycine motif of SAMP1 to generate a -GlyGly (114.1 Da) footprint on lysine residues of the target protein after trypsin digest. Trypsin cleaves peptide bonds at the carboxyl side of the amino acids lysine or arginine, but not lysine residues that are modified by Ub-like isopeptide bonds. Unmodified SAMP1 would leave a -NGEAAALGEATAAGDELALFPPVSGG footprint, which was too bulky to map by MS/MS. Incorporation of the SAMP1 S85K variant, thus, facilitated mapping the sites of modification. Signature tryptic peptides generated from SAMP1S85K-MoaE conjugates were fragmented by MS/MS to map the -GlyGly (114.1 Da) footprints indicative of SAMP1 modification (Suppl. Fig. S7).
Based on the MS/MS profile of the signature peptides generated from the SAMP1 S85K-MoaE conjugates, isopeptide bonds (typical of Ub) were detected between the α-carboxyl group of the C-terminal Gly87 of SAMP1 and the ε-amino groups of two lysine residues of MoaE (Lys240 and Lys247) (Suppl. Fig. S7). Tryptic peptides specific for SAMP2, polySAMP1 chains, or doubly-modified MoaE were not detected. However, a peptide (LFADLAEVAGSR) indicative of unmodified Lys4 of SAMP1 was detected. Lys4 is the only other lysine residue present in SAMP1, besides the Lys85 introduced for MS/MS footprinting. Thus, MoaE appears to be modified by a single moiety of SAMP1 at two different sites (Lys240 and Lys247) through isopeptide linkages. These results also demonstrate that isopeptide conjugates can be purified for use as defined substrates in HvJAMM1 activity assays.
Interestingly, the two lysine residues (Lys240 and Lys247) of H. volcanii MoaE that were modified by SAMP1 are analogous to the lysine residues implicated in the catalytic activity of the large subunit of the E. coli molybdopterin synthase (MoaE Lys119 and Lys126, respectively) (Rudolph et al., 2003). Thus, samp1ylation of MoaE may serve to reduce/inhibit synthesis of molybdopterin, a sulfur-containing intermediate in molybdenum cofactor (MoCo) biosynthesis.
HvJAMM1 cleaves isopeptide-linked SAMP1-MoaE
HvJAMM1 was found to cleave isopeptide-linked SAMP1-MoaE (Fig. 3a). To detect this activity, HvJAMM1 was incubated with the isopeptide linked Flag-SAMP1-MoaE-StrepII conjugates purified from H. volcanii. The reaction mixtures were separated by reducing SDS-PAGE, and product formation was monitored by α-StrepII and α-FLAG immunoblot. With this approach, HvJAMM1 was found to desampylate MoaE and generate free SAMP1. HvJAMM1 required a metal ion for this activity (based on its inactivation by EDTA) (Fig. 3a). The free forms of SAMP1 and MoaE generated after ‘desampylation’ of MoaE were not readily hydrolyzed by HvJAMM1 (Fig. 3a). Thus, HvJAMM1 appears to be a metalloprotease that can cleave isopeptide-linked SAMP conjugates. In particular, HvJAMM1 may reactivate molybdopterin synthase by cleaving SAMP1 from the catalytic lysine residues of MoaE.
HvJAMM1 cleaves linear-linked SAMP1-MoaE
HvJAMM1 was also found to cleave linear fusions of SAMP1 to MoaE. For this analysis, the 3′ end of the gene encoding Flag-SAMP1 was genetically fused to the 5′ end of the gene encoding MoaE-StrepII to synthesize a single linear polypeptide. The linear fusion was synthesized in H. volcanii ΔubaA, purified by tandem affinity chromatography, and used as a substrate for assay with HvJAMM1. Similarly to the isopeptide linked SAMP1-MoaE, the linear fusion of SAMP1 to MoaE was cleaved by HvJAMM1 to generate ‘free’ forms of SAMP1 and MoaE (Fig. 3b). To assess whether or not the amino acid sequence linking SAMP1 to MoaE impacted the ability of HvJAMM1 to cleave this linear protein fusion, the amino acid residues in this region were systematically altered. In particular, the diglycine (GG) and valine and serine residues immediately N-terminal to this motif (within the SAMP1 domain) were deleted (SAMP1ΔGG-and SAMP1ΔGGVS-MoaE, respectively). Interestingly, while deletion of the diglycine motif of the linear fusion of SAMP1 to MoaE reduced cleavage by HvJAMM1, it did not abolish this activity. Significant cleavage of the ΔGG variants was detected (Fig. 3c). In contrast, deletion of the VSGG motif rendered the SAMP1-MoaE fusion inaccessible to cleavage by HvJAMM1 (Fig. 3c). Based on these results, HvJAMM1 is a relatively broad spectrum JAMM-type metalloprotease. HvJAMM1 can cleave proteins attached to SAMP1 by linear an disopeptide bonds. Furthermore, the C-terminal diglycine motif of SAMP1 is not required for HvJAMM1 mediated-cleavage of linear protein fusions.
HvJAMM1 appears essential
To determine the role of HvJAMM1 in H. volcanii, the encoding gene (HVO_2505, jamm1) was targeted for in-frame deletion using the pop-in/pop-out method of homologous recombination (Allers et al., 2004). This method has been developed for H. volcanii and is used routinely by our lab to generate deletion strains (Zhou et al., 2008; Sherwood et al., 2009; Humbard et al., 2010; Rawls et al., 2010; Miranda et al., 2011)(HM1109 of this study, Suppl. Table S1). For deletion of jamm1, H. volcanii ΔpyrE2 strains (where pyrE2 encodes orotate phosphoribosyl transferase needed for uracil biosynthesis) were transformed to uracil prototrophy using pJAM1769 (Suppl. Table S1), a pTA131-derived plasmid that carries a pyrE2 marker and ~500 bp of DNA flanking the 5′ and 3′ ends of jamm1. Integration of the plasmid (pop-in) onto the H. volcanii genome was confirmed by PCR using primers that annealed ~700 bp 5′ and 3′ of jamm1 (not on the plasmid used for integration) (Suppl. Fig. S8). After integrant outgrowth, strains were counter-selected for uracil auxotrophy by plating for resistance to 5-fluoroorotic acid (5-FOA). Strains that underwent intrachromosomal crossover and loss of the pyrE2 marker were identified based on their inability to convert 5-FOA to the toxic analog 5-fluorouracil. Typically, this type of crossover results in a mixture of isolates with the target gene restored to wild type or deleted. However, in this study, only strains with wild-type jamm1 were identified after screening 460 isolates (Suppl. Fig. S8). This result provides compelling, albeit preliminary, evidence for the essentiality of jamm1.
HvJAMM1 reduces the levels of SAMP1/2 conjugates in vivo
To further probe the function of HvJAMM1 in H. volcanii, active and inactive (E31D) forms of the enzyme were synthesized with Flag-SAMP1/2 in HM1096 (a strain with a wild-type copy of jamm1 on the chromosome) (Fig. 4). The inactive variant (HvJAMM1 E31D) was generated by site-directed mutagenesis based on analogy to conserved residues of the JAMM motif. The levels of the SAMP1/2 conjugates were monitored by α-Flag immunoblot in cells grown to stationary phase. With this approach, the levels of SAMP1/2 conjugates were found to significantly decrease by in trans expression of the gene encoding the active form of HvJAMM1 but not the inactive variant (HvJAMM1 E31D) (Fig. 4). This decrease was reproducibly stimulated by a temperature upshift to 50 °C compared to maintaining the cells at 42 °C for 12 h prior to harvest (Fig. 4). Thus, HvJAMM1 mediates a decrease in the level of SAMP1/2 modified proteins in the cell, and this activity appears to be stimulated by heat shock.
Figure 4.
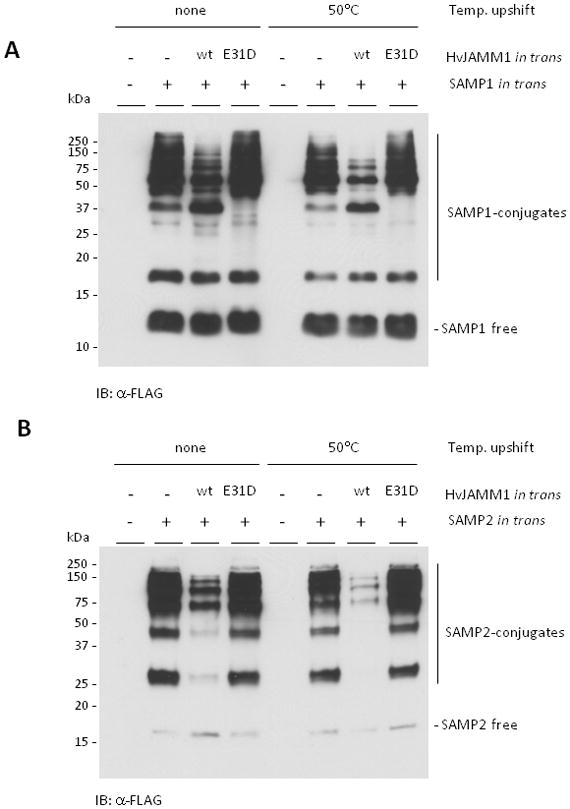
In trans expression of HvJAMM1stimulates the desampylation of SAMP1 (A) and SAMP2 (B) conjugates and is stimulated by temperature upshift of stationary phase cells to 50 °C. H. volcanii HM1096 (Δsamp1 Δsamp2 Δhvo_2177) was used as parent with plasmids pJAM202c (empty vector), pJAM947 (Flag-SAMP1), pJAM1810 (Flag-SAMP1 and HvJAMM1), pJAM1811 (Flag-SAMP1 and HvJAMM1 E31D), pJAM949 (Flag-SAMP2), pJAM1812 (Flag-SAMP2 and HvJAMM1), pJAM1814 (Flag-SAMP2 and HvJAMM1 E31D). HvJAMM1 wild type (wt) and inactive variant (E31D) are indicated. Strains were grown in ATCC974 medium at 42 °C for 60 h and shifted to 50 °C or maintained at 42 °C for 12 h. Cell pellets were boiled in reducing SDS-PAGE lysis buffer. Proteins were separated by SDS-PAGE and analyzed by α-Flag immunoblot.
SAMP1 derived from linear SAMP1-MoaE is functional in H. volcanii
By synthesis of linear linked SAMP1-MoaE in appropriate H. volcanii deletion strains, we addressed whether or not SAMP1 with a C-terminal protein extension was functional in protein modification. Since the C-terminal glycine of SAMP1 is required to form isopeptide bonds with target proteins (Suppl. Fig. S7) (Humbard et al., 2010), protein modification by SAMP1 would be an indicator of the in vivo cleavage of SAMP1 from linear SAMP1-MoaE. Consistent with this possibility, linear linked SAMP1-MoaE was functional in the formation of SAMP1 modified proteins only in the presence of wild-type ubaA (where ubaA encodes a homolog of non-canonical ubiquitin activating E1 enzymes) (Suppl. Fig. S9a,b). H. volcanii strains expressing linear SAMP1-MoaE generated samp1ylated proteins that were distinct from the original SAMP1-MoaE fusion and were not present in an ubaA deletion strain (Suppl. Fig. S9a,b).
We also examined whether or not linear linked SAMP1-MoaE was functional in sulfur transfer. SAMP1 and MoaE are required for dimethylsulfoxide (DMSO) reductase activity in H. volcanii, presumably because SAMP1 and MoaE are needed to form the MoCo-active site of DMSO reductase (Miranda et al., 2011). Residues analogous to the diglycine motif of SAMP1 form a C-terminal thiocarboxylate (MoaD-COSH) that is required for sulfur transfer in MoCo biosynthesis in E. coli (Schmitz et al., 2007). Thus, growth with DMSO as a terminal electron acceptor was used to monitor SAMP1-MoaE cleavage, since the C-terminus of SAMP1 most likely needs to be free and available for MoCo biosynthesis in H. volcanii. Expression of linear linked SAMP1-MoaE was found to restore the ability of an H. volcanii moaE samp1 deletion to respire on DMSO, with the diglycine motif of SAMP1-MoaE required for this complementation (Suppl. Fig S9c).
Together, these results reveal that linear linked SAMP1-MoaE can function in protein modification and MoCo biosynthesis when produced in H. volcanii. Since the C-terminus of SAMP1 appears essential for both of these processes, we propose that H. volcanii can cleave the linear linked SAMP1-MoaE. HvJAMM1 is the most likely candidate for this cleavage activity based on its in vitro activity profile.
Tertiary structure of HvJAMM1 can be modeled to JAMM metalloenzymes
To further understand the function of HvJAMM1, its tertiary structure was modeled with a catalytic zinc ion using the atomic coordinates of the X-ray crystal structures of AfJAMM as template (Fig. 5). The active site of HvJAMM1 was superimposed onto the structures of AfJAMM (PDB: 1R5X) (Ambroggio et al., 2004) and human AMSH (PDB: 3RZU)(Davies et al., 2011). Thus, HvJAMM1 was predicted to bind a catalytic zinc ion in a tetrahedral coordination sphere provided by the sidechains of His88, His90 and Asp101 and a nucleophilic water molecule. The sidechain of HvJAMM1 Glu31 was predicted to provide hydrogen bonding to the water molecule that coordinates the catalytic zinc, similarly to AfJAMM Glu22 (Ambroggio et al., 2004), human AMSH-LP Glu392 (Sato et al., 2008) and human AMSH Glu280 (Davies et al., 2011). Likewise, HvJAMM1 Ser98 was envisaged to function as an intermediate stabilizing residue and affect catalysis in a manner similar to the reaction mechanism proposed for AMSH-LP and in analogy to thermolysin (Sato et al., 2008). A cysteine residue (Cys115) that modeled near the active site of HvJAMM1 was not predicted to be required for catalytic activity based on its configuration (Fig. 5).
Figure 5.
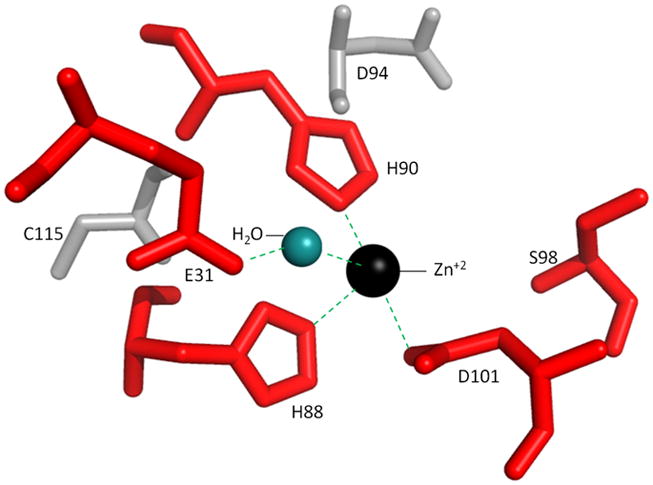
Active site region of HvJAMM1 modeled using AfJAMM as template. The catalytic zinc ion and water (not to scale) are included to assist in understanding the mechanism of HvJAMM1-mediated desampylation. E31, H88, H90, S98 and D101 (red) are conserved active site residues predicted to be required for HvJAMM1 activity.
HvJAMM1 is inhibited by NEM and metal chelators
To initiate an understanding of catalytic mechanism, known protease inhibitors were tested for their influence on HvJAMM1 activity. Chemical compounds included phenylmethanesulfonylfluoride (PMSF; which can bind specifically the active site serine residue of serine proteases), N-ethylmaleimide (NEM; an irreversible modifier that alkylates cysteine residues), and the chelators 1,10-phenalthroline (phen) and N, N, N′, N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) (used widely in biological systems to chelate heavy metals, particularly Zn2+)(Salvesen and Nagase, 2001). With this approach, HvJAMM1 was found to be relatively insensitive to PMSF, but was inhibited at low molar ratios of inhibitor to enzyme by NEM (at 20:1), phen (at 50:1) and TPEN (at 5:1) (Fig. 6a, Suppl. Fig. S10). Addition of excess Zn2+ to TPEN-treated HvJAMM1 restored its activity, while addition of Fe2+, Cu2+ and Ni2+ did not reactivate the enzyme (Fig. 6a). SEC resulted in release of zinc and inactivation of HvJAMM1 (Table 1). Like TPEN-treatment, activity of the SEC-purified HvJAMM1 was restored by addition of excess Zn2+ (but not the other divalent metals). This finding is comparable to the JAMM metalloprotease Rpn11, which is inactivated by TPEN and restored by the addition of ZnCl2 (Guterman and Glickman, 2004). The zinc metalloproteases thermolysin, carboxypeptidase A and Csn5 are also inactivated by metal chelators but cannot be restored by the addition of excess zinc (Holland et al., 1995; Gomez-Ortiz et al., 1997; Cope et al., 2002). The mechanistic reason for NEM inhibition of HvJAMM1 is not fully understood, since HvJAMM1 is not predicted to be a cysteine protease. However, NEM may sterically inhibit the activity of HvJAMM1 by addition of a bulky group to the cysteine residue (C115) near its active site (Fig. 5). While not common, JAMM isopeptidases are reported to be inhibited by NEM including AMSH (McCullough et al., 2004), BRISC-associated Brcc36, and proteasomal Poh1 (Cooper et al., 2009). In contrast to HvJAMM1 and AMSH, NEM inhibition of the Brcc36-and Poh1-complexes is only observed under certain buffer conditions (unprotonated Tris, pH 8). NEM is proposed to alkylate a cysteine residue of Brcc36 and Poh1 that allows unprotonated Tris to displace the nucleophilic water and inactivate the enzyme (Cooper et al., 2009).
Figure 6.
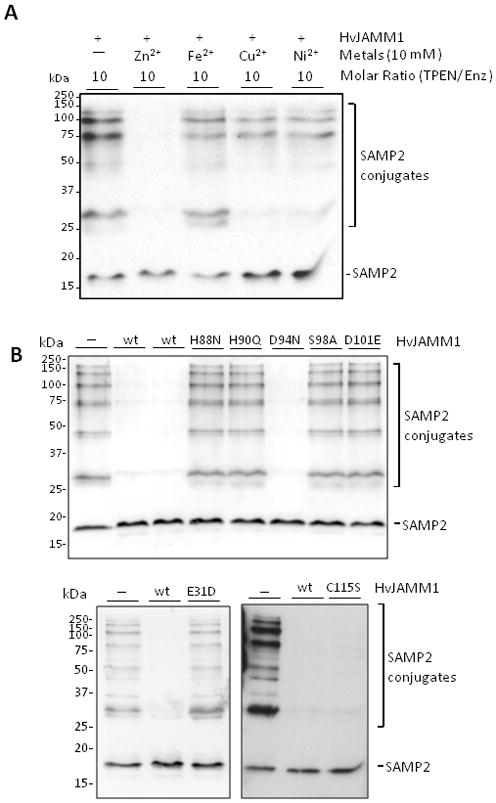
Divalent Zn2+ (A) and conserved active site residues (E31, H88, H90, S98 and D101) (B) are crucial for HvJAMM1 activity. In contrast, HvJAMM1 C115S and D94N variants retain desampylating activity. Reactions were performed using SAMP2 conjugates as a substrate under standard assay conditions (as described in Experimental Procedures). The metal chelator TPEN, divalent metals and HvJAMM1 wild-type and site-directed variant proteins were included in assays as indicated.
HvJAMM1 requires conserved JAMM motif residues for activity
In addition to chemical inhibition, site-directed mutagenesis was used to probe the HvJAMM1 active site. HvJAMM1 Glu31, His88, His90, Ser98 and Asp101 were targeted for mutagenesis based on their predicted requirement for enzyme activity (see 3D modeling above). HvJAMM1 Asp94 and Cys115 were also modified, as negative controls, based on their close proximity to the HvJAMM1 active site, yet likely non-essential role in catalysis.
HvJAMM1 protein variants were purified and their structural properties were compared to the unmodified enzyme by reducing SDS-PAGE, size exclusion chromatography (SEC), mass spectrometry (MALDI-TOF and ESI-MS), metal content (ICP-AES) and circular dichroism (CD)(Table 1, Suppl. Fig. S11). With exception of metal content, the site-directed changes did not alter the overall structural properties of HvJAMM1. Similarly to the unmodified form, the HvJAMM1 variants purified as monomers by SEC and had observed average masses (determined by MALDI-TOF and ESI-MS) consistent with their theoretical average masses (Table 1). While altering the acidic residue (Asp) at position 94 to a polar residue (Asn) increased the rate of migration of HvJAMM1 by SDS-PAGE (Suppl. Fig. S10), like the other variants, the observed mass of the E94N variant was similar to its theoretical mass (Table 1). Significant overlap in the CD spectra was also observed for HvJAMM1 wild-type and variant proteins (in the absence and presence of excess ZnCl2) providing support for similarity in overall structural-fold of these proteins (Suppl. Fig. S10). Metal content analysis revealed that wild-type HvJAMM1 coordinated a Zn2+ atom, while HvJAMM1 H90Q, H88N and S98A variants were significantly reduced in Zn2+ content. Among the variants, H90Q and H88N had the most pronounced effect, reducing the mol/mol of protein content from nearly 1 in wild type to less than 0.05 in the variant proteins (Table 1). The D101E mutation did not significantly alter the Zn2+ content of HvJAMM1 (Table 1).
The HvJAMM1 site-directed variants were also examined for their ability to hydrolyze SAMP2 conjugates (Fig. 6b). Amino acid residues Glu31, His88, His90, Ser98 and Asp101 were found to be important for HvJAMM1 activity, while Asp94 and Cys115 were not required (Fig. 6b). While the D101E form of HvJAMM1 retained Zn2+, this variant was inactive in cleavage of SAMP2 conjugates. An analogous modification in human AMSH (E280A) abolishes deubiquitylating activity, while having little to no impact on retention of zinc (Davies et al., 2011). Based on the atomic structure of AMSH E280A compared to wild-type, an alternative non-active site residue (Asp309) appears to swing around into the active site and assist in coordinating the zinc in an unproductive conformation (Davies et al., 2011). Thus, HvJAMM1 D101E may coordinate zinc in a catalytically inactive state through an alternative amino acid side chain.
DISCUSSION
HvJAMM1 provides the first example of a JAMM enzyme that independently cleaves Ub-like proteins from diverse protein targets. Only a few other JAMM/MPN+ metalloproteases are known to act independent of a multisubunit complex, but these latter enzymes have narrow substrate specificity (e.g., Mec+ hydrolyzes cysteine from CysO, human AMSH/AMSH-LPhydrolyzesK63-linked diUb chains). JAMM isopeptidases that cleave Ub-like proteins from protein targets typically require association in multisubunit complexes for activity. For example, Csn5/Jab1 requires association in the COP9 signalosome (CSN) to catalyze the removal of the Ub-like Nedd8/Rub1 from the cullin subunit of E3 Ub ligases of the cullin-RING ligase (CRL) family (Copeet al., 2002). Likewise, Rpn11/Poh1 must be associated in ATP-hydrolyzing 19S regulatory particles to detect its DUB activity, suggesting that its substrates must be proceeding on the pathway of proteolysis by 26S proteasomes (Yao and Cohen, 2002; Finley, 2009).
Here we propose a catalytic mechanism for HvJAMM1-mediated cleavage of SAMP1/2 conjugates based on the results of this study (Fig. 7) and in analogy to AMSH and AMSH LP homologs. In this model, a catalytic Zn2+ is tetrahedrally coordinated by the side chains of HvJAMM1 His88, His90 and Asp101 and a water molecule. The side chain of HvJAMM1 Glu31 functions by general base mechanism to polarize the nucleophilic water molecule (coordinated to zinc) that attacks the carbonyl carbon of the scissile bond during catalysis. A negatively charged tetrahedral intermediate forms that is stabilized by hydrogen bonding to the side chain of Ser98. The water molecule coordinated to the side chain of Glu31 completes the acylation step by donating protons to the nitrogen of the isopeptide bond undergoing hydrolysis. After SAMP1/2 release, a hydrogen bond forms between Glu31 and the protein target that releases the ‘desampylated’ product upon collapse.
Figure 7.
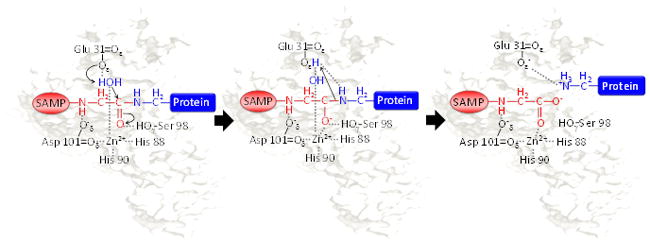
Proposed catalytic mechanism of HvJAMM1. Schematic representation of the catalytic mechanism proposed for HvJAMM1 in cleaving the isopeptide bond of SAMP-modified proteins. Highlighted are SAMP (blue) conjugated to the target protein (pink) and the nucleophilic water (red) that is coordinated to the catalytic Zn2+.
Interestingly, HvJAMM1 was found to cleave two distinct Ubl proteins (SAMP1/2) from a diversity of proteins including those that were linear and isopeptide-linked. This type of broad substrate specificity has not been previously reported for a JAMM metalloprotease and may explain why the encoding gene appears essential in H. volcanii. SAMP1/2 are linked through isopeptide bonds to protein targets by a mechanism that requires UbaA, a non-canonical Ub activating E1 enzyme homolog (Humbard et al., 2010; Miranda et al., 2011)(this study). Thus, the cleavage of isopeptide-linked SAMP conjugates by HvJAMM1 may provide a means for the cell to reverse the action of UbaA and, thus, regulate sampylation, recycle the SAMPs, and maintain cellular homeostasis. The broad substrate specificity of HvJAMM1 also suggests that this protease could reactivate SAMPs that are otherwise inactivated at their C-termini by small molecules (e.g., nucleophilic attack of adenylated and thioester forms of the SAMPs by glutathione and polyamines). Why HvJAMM1 cleaves SAMPs linear linked to protein domains such as MoaE is not clear. However, HvJAMM1 does require an Ub-like domain (SAMP) fusion for this linear cleavage activity.
Many possibilities could explain why HvJAMM1 mediates linear cleavage of Ubl domain fusions. While H. volcanii does not encode translational fusions of SAMP1/2, many archaea do encode SAMP homologs with short and/or long C-terminal extensions. For example, the SAMP homolog Nmag_1914 of Natrialba magadii has two amino acids C-terminal to its diglycine motif (-GGSR) (Siddaramappa et al., 2012), while most Sulfolobus spp. encode homologs of linear linked SAMP-MoaE. Thus, the linear cleavage activity observed for HvJAMM1 may be common to archaeal JAMM domain proteins of group I (cd08070). In eukaryotic cells, linear-linked Ub/Ub-like domains are synthesized not only by translation of Ub/Ub-like gene fusions but also by attachment of the C-terminal glycine of Ub to the α-amino group of a protein after translation (Hay, 2007; Iwai and Tokunaga, 2009; Emmerich et al., 2011; Walczak et al., 2012). Thus, H. volcanii may generate linear linked SAMPs at the post-translational level that necessitate regulation by HvJAMM1 (although these types of modifications have yet to be detected in archaea). Another reason for the broad substrate specificity of HvJAMM1 may be that hydrolysis of peptide bonds carboxyl to Ubl domains (e.g., SAMPs, CysO, QbsE) is an ancient activity that predates the bacterial/archaeal delineation for group I (cd08070). Examples in bacteria that are consistent with this possibility include Mec+ that cleaves CysO-cysteine adducts (Burns et al., 2005), QbsD that may remove cysteine and phenylalanine from the C-terminus of QbsE (Godert et al., 2007), and MoaX (a MoaD-MoaE domain fusion) that can restore MoCo biosynthesis in moaD moaE deletion strains suggesting cleavage of this Ubl-domain fusion (Williams et al., 2011).
EXPERIMENTAL PROCEDURES
Materials
Biochemicals were purchased from Sigma-Aldrich (St. Louis, MO). Other organic and inorganic analytical grade chemicals were purchased from Fisher Scientific (Atlanta, GA). Desalted oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Molecular biology grade enzymes were purchased from New England Biolabs (Ipswich, MA) unless otherwise indicated.
Strains and growth conditions
The strains used in this study are summarized in Suppl. Table S1. E. coli TOP10and XL-1 Blue were used for routine recombinant DNA experiments, and E. coli Rosetta (DE3) was used for heterologous expression of H. volcanii genes. E. coli GM2163 was used for replication of plasmid DNA prior to transformation into H. volcanii strains according to standard methods (Dyall-Smith, 2009). E. coli strains were grown in Luria-Bertani (LB) medium supplemented with ampicillin (Amp, 0.1 mg·ml−1), ZnCl2 (100 μM) and isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.4 mM) as needed. H. volcanii strains were routinely grown in ATCC974 complex medium and supplemented with novobiocin (Nv, 0.2 μg·ml−1) when carrying pJAM plasmids. For synthesis of SAMP-protein conjugates, H. volcanii strains were grown in GMM-Ala with Nv as previously described (Humbard et al., 2010). For growth with DMSO as a terminal electron acceptor, H. volcanii strains were grown as previously described (Miranda et al., 2011). Pop-in/pop-out method and culture conditions used for generation of H. volcanii deletion strains were as previously described (Allers et al., 2004). E. coli and H. volcanii were grown at 37 and 42 °C, respectively, in liquid cultures with rotary shaking at 150–200 rpm and on solid medium (15 % [w/v] agar plates).
DNA cloning and site-directed mutagenesis
Plasmids and primers used in this study are summarized in Suppl. Table S1–2. In general, PCR was according to standard methods with H. volcanii DS70 genomic DNA or appropriate plasmid DNA as a template and primer pairs as indicated in Table S2. Phusion DNA polymerase was used for high-fidelity PCR-based cloning, and Taq DNA polymerase was used for colony screening. PCR generated-DNA fragments of appropriate size were isolated from 0.8 % (w/v) SeaKem GTG agarose (FMC Bioproducts, Rockland, ME) gels in TAE [40 mM Tris, 20 mM acetic acid, and 1 mM ethylenediaminetetraacetic acid (EDTA)] buffer at pH 8.0 using the QIAquick gel extraction kit (Qiagen, Valencia, CA) as needed. The fidelity of all DNA plasmid constructs was verified by Sanger DNA Sequencing (Eton Bioscience, Inc. and UF ICBR DNA sequencing core).
Amino acids residues of HvJAMM1 were selected for site-directed mutagenesis by structural modeling (see later section) and by use of the ConFunc Prediction Server (Wass and Sternberg, 2008). Plasmid pJAM991 (encoding HvJAMM1) was purified from E. coli TOP10 and used as template for PCR-based mutagenesis with primer pairs indicated in Suppl. Table S2. Pfu DNA polymerase was used to generate amplicons with the desired single-point mutations as described in the QuikChange Lightning mutagenesis protocol (Stratagene, La Jolla, CA). PCR products were treated with DpnI and incubated at 37°C for 2 h, purified using QIAquick purification kit (Qiagen), and transformed into E. coli XL1-Blue or E. coli XL10-Gold. For site-directed mutagenesis of SAMP1, the desired single-point mutation (encoding S85K) was incorporated into the primer annealing to the 5′ end of the gene that was used for PCR with pJAM947 as template. Further details on plasmid construction are provided in Suppl. Table S2.
Dendgrogram construction
Sequences of JAMM/MPN+ domain proteins (Suppl. Table S3) including HVO_2505 (HvJAMM1) and HVO_1016 (HVJAMM2) were retrieved from NCBI, InterPro and Member Databases by Blast and InterProScan signature sequences. JAMM/MPN+ domains were aligned using ClustalW (Larkin et al., 2007) after N-and C-terminal trimming of protein sequences. Pairwise comparisons were performed between sequences and mean genetic distance was evaluated using p-distance with gaps analyzed using pairwise deletion. The neighborhood-joining tree that best fit distance data was constructed using MEGA 4.0 (Tamura et al., 2007). Gene neighborhood and co-occurrence patterns of archaeal JAMM domain proteins were determined using STRING 9.0 (Szklarczyk et al., 2011).
SDS-PAGE and immunoblotting
For reducing SDS-PAGE, proteins were boiled (10–15 min) in loading buffer [at final concentration of 50 mM Tris-HCl pH 6.8, 2 % (w/v) SDS, 10 % (v/v) glycerol, 1 % (v/v) β-mercaptoethanol, 12.5 mM EDTA and 0.02 % (w/v) bromophenol blue] and separated by SDS-PAGE (10 to 12 % gels) according to Laemmli (Laemmli, 1970). Proteins were stained in gel by Commassie Blue R-250 or SYPRO Ruby (BioRad). Epitope-tagged proteins were detected by immunoblot using alkaline phosphatase-linked anti-Flag (α-Flag) M2 monoclonal antibody (Sigma) and rabbit anti-StrepII (α-StrepII) polyclonal antibody (GenScript) followed by goat anti-rabbit IgG (H+L)-alkaline phosphatase-linked antibody (SouthernBiotech). Alkaline phosphatase activity was detected colorimetrically using nitroblue tetrazolium chloride (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and by chemiluminescence using CDP-Star (Applied Biosystems) with X-ray film (Hyperfilm; Amersham Biosciences).
Protein synthesis and purification
All protein purification steps were performed at 4°C or on ice unless otherwise indicated, and cells were harvested by centrifugation (10 to 15 min at 9,200 × g and 4°C). With exception of circular dichroism (see below for details), protein concentration was determined by bicinchoninic acid (BCA) assay using bovine serum albumin as the standard (Pierce, Thermo Scientific).
HvJAMM protein purification
HvJAMM1/2 wild-type and variant proteins were purified from recombinant E. coli as follows. E. coli Rosetta (DE3) strains were freshly transformed with the appropriate pET15b-based expression plasmids (listed in Suppl. Table S1) and grown in0.5 to 1 liter LB Amp in 2.8 L Fernbach flasks at 37 °C. At log phase (OD600 of 0.4–0.6 units), IPTG (0.4 mM) and ZnCl2 (100 μM) were added and cultures were shifted to 25 °C for 4.5–12 h. Cells were harvested, washed in ice-chilled Tris-low salt buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) and stored at −80 °C until lysed. Cell pellets were resuspended in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 40 mM imidazole, 1 mM PMSF) (4 ml per 1 g wet wt cells) and lysed thrice by French Press (2,000 psi). An equal volume of dilution buffer (20 mM Tris-HCl, pH 7.5, 4 M NaCl) was added to cell lysate. Protein sample was clarified by centrifugation (twice for 10–20 min at 9,200 × g and 4°C) and filtration (0.2 μm). His6-tagged HvJAMM proteins were applied to a HisTrap HP column (71-5027-68AH, GE Healthcare) pre-equilibrated in Tris-salt buffer (20 mM Tris-HCl, pH 7.5, 2 M NaCl) with 40 mM imidazole and washed with the same buffer. HvJAMM1 proteins were eluted using Tris-salt buffer with 500 mM imidazole. Purified protein fractions (of single protein band based on Coomassie Blue stained SDS-PAGE) were pooled and buffer-exchanged twice by dialysis into HEPES-salt buffer (20 mM HEPES buffer, pH 7.5, 2 M NaCl). The HvJAMM proteins were further purified by size exclusion chromatography to estimate their native molecular mass and to prepare the samples for circular dichroism as well as activity assay in the presence and absence of metal supplementation. Dialyzed samples were applied to a Superdex 75 10/300 GL column (GE Healthcare) equilibrated in Tris-salt buffer and eluted in the same buffer. Molecular mass standards were applied to the same column equilibrated in Tris-low salt buffer to avoid denaturation/aggregation of the mesohalic standards [bovine serum albumin (66 kDa), ovalbumin (43 kDa), thrombin (36 kDa), α-casein (23.6 kDa), cytochrome c (12.38 kDa) and aprotinin (6.5 kDa)] (Sigma).
SAMP conjugate enrichment
H. volcanii strains H26-pJAM947 (Flag-SAMP1), H26-pJAM1777 (Flag-SAMP1 S85K) and H26-pJAM949 (Flag-SAMP2) were grown to stationary phase in 100 ml of GMM-Ala with Nv (0.2 μg·ml−1). HEPES-salt buffer was ice-chilled for use in all subsequent steps. Cells were harvested, washed twice with 20 ml of HEPES-salt buffer and resuspended in the same buffer (1 ml per 1 g wet wt cells). Cell suspension was sonicated (3 × 30 to 60 sec at140 W on ice), and the lysate was clarified by centrifugation (9,200 × g for 15 min at 4 °C). The supernatant was boiled for 20 min and further clarified by centrifugation (9,200 × g for 15 min at 4 °C). Total protein concentration was adjusted to 5 μg·μl−1, and samples were stored at −80 °C until further use.
Purification of isopeptide linked and linear forms of SAMP1-MoaE
For purification, H. volcanii strains were grown to stationary phase in ATCC medium with Nv (0.2 μg·ml−)(1 L in 2.8 L Fernbach flask). Strains included HM1109-pJAM1777 (that separately encodes Flag-SAMP1 S85K and MoaE-StrepII), HM1109-pJAM1314 (that separately encodes Flag-SAMP1 and MoaE-StrepII) and HM1052-pJAM1796 (that encodes a linear fusion of Flag-SAMP1-MoaE-StrepII). Cells were harvested, washed once with ice-chilled Tris-salt buffer, and stored at −80 °C until used. Cell pellets were resuspended in lysis buffer (4 ml per 1 g wet wt cells) and lysed thrice by French Press (2,000 psi). Cell-free extract was obtained by centrifugation (twice for 10 min at 9,200 × g and 4°C) followed by filtration (0.2 μm). Sample was applied to a Strep-Tactin column (Qiagen) pre-equilibrated in Tris-salt buffer. The column was washed with Tris-salt buffer, and proteins (including unconjugated MoaE-StrepII as well as Flag-SAMP1-MoaE-StrepII conjugates) were eluted with Tris-salt buffer supplemented with 5 mM d-desthiobiotin. Purified protein fractions were pooled and buffer-exchanged by dialysis into TBS buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl). Strep-Tactin-purified proteins were applied to a 0.7 × 1.25 cm column of α-FLAG M2-affinity beads (0.5 ml, Sigma) pre-equilibrated with TBS. Bound proteins were washed with 20 column volumes of TBS. Flag-SAMP1-conjugated to MoaE-StrepII was eluted with FLAG peptide (Sigma)(at 0.1 mg FLAG peptide per 1 ml of TBS). Samples were dialyzed into HEPES-salt buffer and stored at 4 °C.
Enzyme activity assays
Temperature, buffer, pH condition, chemical effectors and enzyme and substrate concentrations were varied to optimize and develop a standard condition for assay of desampylation. Standard assay for desampylation of ‘total’ SAMP conjugates was monitored in 10 μl reaction volume with 10 μM enzyme and 5 μg of Flag-SAMP conjugate enrichment in 20 mM HEPES buffer at pH 7.5 with 2 M NaCl and 50–500 μM ZnCl2. To monitor desampylation of SAMP1-MoaE conjugates, standard assays (10 μl) included 5 μM HvJAMM1, 12 μM SAMP1-MoaE and 50 μM ZnCl2 in HEPES-salt buffer. Reactions were incubated for 2–3 h at 50 °C. Negative controls included incubation of Flag-SAMP conjugates alone, use of heat-killed enzyme (boiled 15 min) and addition of 50 μM EDTA. Assays to examine the hydrolysis of unmodified proteins were in 10 μl final volume and were incubated for 2 h at 37 °C. Reactions included 3.5–5 μM enzyme and 2 μg unmodified protein (hemoglobin, cytochrome c, carbonic anhydrase, creatine phosphokinase, β-amylase and bovine serum albumin)(Sigma) in 20 mM HEPES buffer, pH 7.5 supplemented with 2 M NaCl and 50 μM ZnCl2 for HvJAMM1 and with 150 mM NaCl without ZnCl2 for proteinase K (from Engyodontium album, formerly Tritirachium album)(Sigma P6556). For desampylase and other protease assays, reaction products were separated by reducing SDS-PAGE (12 % gels). Total protein was detected by staining with Sypro Ruby and Coomassie Blue R-250. Free and conjugated forms of Flag-and StrepII-tagged proteins were detected by immunoblot (see above for details). Release of 7-amino-4-methylcoumarin from Ub and diglycine linked to 4-methyl-coumaryl-7-amide (AMC) (Ub-AMC from Enzo Life Sciences, Farmingdale, NY)(diglycine-AMC from Bachem, Torrance, CA) was monitored fluorimetrically as previously described (Barrett, 1980). The assay mixture (0.25 ml) contained 2.5 μM enzyme and 2.5 μM fluorigenic substrate in HEPES-salt buffer supplemented with 10 mM DTT and 0.4% (v/v) DMSO and was incubated for 2 h at 25 °C. As a positive control, Ub-AMC cleavage by isopeptidase T (USP5) (Calbiochem, San Diego, CA) was similarly monitored at55 nM enzyme (minus 2 M NaCl in buffer).
Circular dichroism
Circular dichroism (CD) data were acquired on an Aviv 202 spectrometer from Aviv Biomedical (Lakewood, NJ) similar to previously described (Greenfield, 2006). Protein was at 5 μM based on UV-Vis spectroscopy absorbance at 280 nm (using an extinction coefficient of 17,085 M−1cm−1 for HvJAMM1 and 11,460 M−1cm−1 for HvJAMM2). The CD signal from the buffer was subtracted from the protein signal and converted to mean residue ellipticity (deg cm2 dmol−1) using equation 1.
| (1) |
Here, θ is ellipticity (mdeg) [defined as θ = tan−1 (b/a), where a and b are the major and minor axes of the resulting elliptically polarized light]; N is the number of amino acid residues in the protein; C is the protein molar concentration; and l is the path length in cm with l = 0.1 cm, based on the path length of Hellma CD cuvettes (Müllheim, Germany) used in this experiment. Spectra were recorded in the far UV (200 to 250 nm) at 25 °C, with sampling points every 1 nm and averaging time of 4 s. The reported CD data were averages of four scans and corrected by subtracting buffer baseline.
Determining protein mass by MALDI-TOF and ESI-MS
For Matrix Assisted Laser Ionization (MALDI)-Time-of-Flight (TOF) analysis, an AB Sciex 4700 proteomics analyzer was used that was equipped with TOF/TOF ion optics and a diode pumped Nd:YAG laser with 200 Hz repetition rate. Sinapinic acid [10 mg/mL in 50% (v/v) acetonitrile and 0.1% (v/v) trifluoric acid] was used as matrix, which was mixed at a 1:1 volume ratio with each protein and spotted on a MALDI target plate. The operating conditions were as follows: linear mode, detection of positive ions, laser intensity between 5000 and 6350, and mass range between 8000 and 20000 m/z with focus mass of 15000. The acceleration voltage and grid voltage were 20000 and 18450 volts, respectively. The spectra were generated by accumulating 400 laser shots into a single spectrum. The instrument was calibrated using a ProteoMass™ Peptide and Protein MALDI-MS Calibration Kit (Sigma Aldrich Inc.). For electrospray ionization (ESI)-TOF analysis, an AB Sciex QSTAR XL quadrupole (Q)TOF MS equipped with a nanoelectrospray source (Protana XYZ manipulator) was used. Protein (5 μl) was applied to a processed borosilicate nanoelectrospray needle, and a nanoelectrospray was generated from the needle at 1500 volts. The spray needle was connected to a 5 ml syringe through a 50 cm long Teflon tube (0.5 mm id). The syringe was driven forward at a rate of 50 μl·min−1 by the integrated QSTAR Harvard 22 syringe pump. The m/z response of the mass spectrometer was calibrated with renin (10 pmol·μl−1)(AB SCIEX), and BSA [20 pmol·μl−1 in 50% (v/v) acetonitrile, 0.1% (v/v) formic acid] (Sigma) was used as a standard. Signals were analyzed using Analyst QS software (AB Sciex Inc., CA).
Mapping sampylation sites by MS/MS
SAMP1-MoaE conjugate purified from H. volcanii HM1109-pJAM1777 was digested with trypsin overnight, followed by injection onto a capillary trap (LC Packings PepMap) and desalting by equilibration for 5 min with a flow rate 3 μl·min−1 of 0.1% (v/v) acetic acid. The samples were loaded onto an LC Packing® C18 Pep Map nanoflow HPLC column. The elution gradient of the HPLC column started at 3% solvent A, 97% solvent B and finished at 60% solvent A, 40% solvent B for 30 min for protein identification. Solvent A consisted of 0.1% (v/v) acetic acid, 3% (v/v) acetonitrile, and 96.9% (v/v) H2O. Solvent B consisted of 0.1% (v/v) acetic acid, 96.9% (v/v) acetonitrile, and 3% (v/v) H2O. LC-MS/MS analysis was carried out on a LTQ Orbitrap XL mass spectrometer (Thermo Scientific, Bremen, Germany). The instrument under Xcalibur 2.07 with LTQ Orbitrap Tune Plus 2.55 software was operated in the data dependent mode to automatically switch between MS and MS/MS acquisition. Survey scan MS spectra (from m/z 300 – 2000) were acquired in the LTQ Orbitrap with a resolution of 60,000 at m/z 400. The five most intense ions were sequentially isolated and fragmented in the linear ion trap by collisionally induced dissociation (CID) at a target value of 5,000 or maximum ion time of 150 ms. Dynamic exclusion was set to 60 seconds. Typical mass spectrometric conditions include a spray voltage of 2.2 kilovolts, no sheath and auxiliary gas flow, a heated capillary temperature of 200°C, a capillary voltage of 44 volts, a tube lens voltage of 165 volts, an ion isolation width of 1.0 m/z, a normalized collision energy of 35% for MS2 in LTQ. The ion selection threshold was 500 counts for MS2. An activation q = 0.25 and activation time of 30 ms were set. The MS/MS spectra were analyzed using ProteinPilot software v. 4.1 (AB Sciex Inc., CA) and manually inspected for confirmation.
Metal analysis
The zinc and nickel content of purified protein samples was measured according to Method 200.7 (Martin et al., 1994). The samples were diluted and analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Garratt-Callahan Company, CA).
Protein structure modeling
The structure and function of HvJAMM1 were predicted using the Protein Homology/Analogy Recognition Engine (Phyre2) through modeling and docking approaches (Kelley and Sternberg, 2009). Generated 3-D images, superimposed with AfJAMM (PDB: 1R5X) and human AMSH (PDB: 3RZU) as templates, were visualized using PyMOL 1.2r1 (Schrödinger)(DeLano, 2002).
Supplementary Material
Acknowledgments
The authors would like to thank Carolyn Diaz and Ran Zheng at the UF ICBR Proteomics Core and Maria Cristina Dancel at the Mass Spectrometry Facility of UF Department of Chemistry for assistants with MS. The authors would also like to thank Savita Shanker at the UF ICBR Genomics Core for Sanger DNA sequencing. This work was funded in part by NIH R01 GM057498 to JMF for analysis of HvJAMM1/2 and DOE DE-FG02-05ER15650 to JMF for analysis of SAMP-MoaE conjugates. Thanks to the Tamil Nadu Agricultural University for extending study leave to S.U. The authors do not have a conflict of interest to declare.
Footnotes
The corresponding author and co-authors do not have a conflict of interest to declare.
References
- Allers T, Ngo HP, Mevarech M, Lloyd RG. Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl Environ Microbiol. 2004;70:943–953. doi: 10.1128/AEM.70.2.943-953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggio XI, Rees DC, Deshaies RJ. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004;2:E2. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Barrett AJ. Fluorimetric assays for cathepsin B and cathepsin H with methylcoumarylamide substrates. Biochem J. 1980;187:909–912. doi: 10.1042/bj1870909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KE, Baumgart S, Dorrestein PC, Zhai H, McLafferty FW, Begley TP. Reconstitution of a new cysteine biosynthetic pathway in Mycobacterium tuberculosis. J Am Chem Soc. 2005;127:11602–11603. doi: 10.1021/ja053476x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Balaji S, Iyer LM, Aravind L. Small but versatile: the extraordinary functional and structural diversity of the β-grasp fold. Biol Direct. 2007;2:18. doi: 10.1186/1745-6150-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connaris H, Chaudhuri JB, Danson MJ, Hough DW. Expression, reactivation, and purification of enzymes from Haloferax volcanii in Escherichia coli. Biotechnol Bioeng. 1999;64:38–45. [PubMed] [Google Scholar]
- Cooper EM, Boeke JD, Cohen RE. Specificity of the BRISC deubiquitinating enzyme is not due to selective binding to Lys63-linked polyubiquitin. J Biol Chem. 2010;285:10344–10352. doi: 10.1074/jbc.M109.059667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28:621–631. doi: 10.1038/emboj.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, et al. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- Davies CW, Paul LN, Kim MI, Das C. Structural and thermodynamic comparison of the catalytic domain of AMSH and AMSH-LP: nearly identical fold but different stability. J Mol Biol. 2011;413:416–429. doi: 10.1016/j.jmb.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. PyMOL molecular graphics system. 2002 Available at: http://www.pymol.org.
- Dyall-Smith M. The Halohandbook: Protocols for Halobacterial Genetics. 2009 http://www.haloarchaea.com/resources/handbook/Halohandbook_2009_v7.1.pdf.
- Emmerich CH, Schmukle AC, Walczak H. The emerging role of linear ubiquitination in cell signaling. Sci Signal. 2011;4:re5. doi: 10.1126/scisignal.2002187. [DOI] [PubMed] [Google Scholar]
- Feng L, Wang J, Chen J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem. 2010;285:30982–30988. doi: 10.1074/jbc.M110.135392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godert AM, Jin M, McLafferty FW, Begley TP. Biosynthesis of the thioquinolobactin siderophore: an interesting variation on sulfur transfer. J Bacteriol. 2007;189:2941–2944. doi: 10.1128/JB.01200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Ortiz M, Gomis-Ruth FX, Huber R, Aviles FX. Inhibition of carboxypeptidase A by excess zinc: analysis of the structural determinants by X-ray crystallography. FEBS Lett. 1997;400:336–340. doi: 10.1016/s0014-5793(96)01412-3. [DOI] [PubMed] [Google Scholar]
- Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guterman A, Glickman MH. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem. 2004;279:1729–1738. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- Hay RT. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 2007;17:370–376. doi: 10.1016/j.tcb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland DR, Hausrath AC, Juers D, Matthews BW. Structural analysis of zinc substitutions in the active site of thermolysin. Protein Sci. 1995;4:1955–1965. doi: 10.1002/pro.5560041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbard MA, Miranda HV, Lim JM, Krause DJ, Pritz JR, Zhou G, et al. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature. 2010;463:54–60. doi: 10.1038/nature08659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Tokunaga F. Linear polyubiquitination: a new regulator of NF-κB activation. EMBO Rep. 2009;10:706–713. doi: 10.1038/embor.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz EJ, Isasa M, Crosas B. A new map to understand deubiquitination. Biochem Soc Trans. 2010;38:21–28. doi: 10.1042/BST0380021. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Komander D. Mechanism, specificity and structure of the deubiquitinases. Subcell Biochem. 2010;54:69–87. doi: 10.1007/978-1-4419-6676-6_6. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol. 2000;20:2367–2377. doi: 10.1128/mcb.20.7.2367-2377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TD, Brockhoff CA, Creed JT. Method. Vol. 200.7. Environmental Monitoring Systems Laboratory, Office of Research and Development, U.S. Environmental Protection Agency; Cincinnati, OH: 1994. Determination of metals and trace elements in water and wastes by inductively coupled plasma-atomic emission spectrometry. Rev. 4.4. [Google Scholar]
- Maupin-Furlow J. Proteasomes and protein conjugation across domains of life. Nat Rev Microbiol. 2012;10:100–111. doi: 10.1038/nrmicro2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Clague MJ, Urbe S. AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol. 2004;166:487–492. doi: 10.1083/jcb.200401141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevarech M, Frolow F, Gloss LM. Halophilic enzymes: proteins with a grain of salt. Biophys Chem. 2000;86:155–164. doi: 10.1016/s0301-4622(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Miranda HV, Nembhard N, Su D, Hepowit N, Krause DJ, Pritz JR, et al. E1-and ubiquitin-like proteins provide a direct link between protein conjugation and sulfur transfer in archaea. Proc Natl Acad Sci U S A. 2011;108:4417–4422. doi: 10.1073/pnas.1018151108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullakhanbhai MF, Larsen H. Halobacterium volcanii spec. nov., a Dead Sea halobacterium with a moderate salt requirement. Arch Microbiol. 1975;104:207–214. doi: 10.1007/BF00447326. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Nunoura T, Takaki Y, Kakuta J, Nishi S, Sugahara J, Kazama H, et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 2011;39:3204–3223. doi: 10.1093/nar/gkq1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Fortin J, Shao G, Bretscher H, Messick TE, Greenberg RA. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J Biol Chem. 2010;285:30971–30981. doi: 10.1074/jbc.M110.135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls KS, Yacovone SK, Maupin-Furlow JA. GlpR represses fructose and glucose metabolic enzymes at the level of transcription in the haloarchaeon Haloferax volcanii. J Bacteriol. 2010;192:6251–6260. doi: 10.1128/JB.00827-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Pyzyna B, Atrasz RG, Henderson CA, Morrill KL, Burd AM, et al. Growth kinetics of extremely halophilic archaea (family Halobacteriaceae) as revealed by Arrhenius plots. J Bacteriol. 2005;187:923–929. doi: 10.1128/JB.187.3.923-929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MJ, Wuebbens MM, Turque O, Rajagopalan KV, Schindelin H. Structural studies of molybdopterin synthase provide insights into its catalytic mechanism. J Biol Chem. 2003;278:14514–14522. doi: 10.1074/jbc.M300449200. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Nagase H. Proteolytic enzymes: a practical approach. 2. 2001. Inhibition of proteolytic enzymes; pp. 105–130. [Google Scholar]
- Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, et al. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Wuebbens MM, Rajagopalan KV, Leimkühler S. Role of the C-terminal Gly-Gly motif of Escherichia coli MoaD, a molybdenum cofactor biosynthesis protein with a ubiquitin fold. Biochemistry. 2007;46:909–916. doi: 10.1021/bi062011w. [DOI] [PubMed] [Google Scholar]
- Sherwood KE, Cano DJ, Maupin-Furlow JA. Glycerol-mediated repression of glucose metabolism and glycerol kinase as the sole route of glycerol catabolism in the haloarchaeon Haloferax volcanii. J Bacteriol. 2009;191:4307–4315. doi: 10.1128/JB.00131-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N. Post-translational modification of cellular proteins by a ubiquitin-like protein in bacteria. J Biol Chem. 2012;287:17568–17577. doi: 10.1074/jbc.M112.359844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddaramappa S, Challacombe JF, De Castro RE, Pfeiffer F, Sastre DE, Gimenez MI, et al. A comparative genomics perspective on the genetic content of the alkaliphilic haloarchaeon Natrialba magadii ATCC 43099T. BMC Genomics. 2012;13:165. doi: 10.1186/1471-2164-13-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tran HJ, Allen MD, Löwe J, Bycroft M. Structure of the Jab1/MPN domain and its implications for proteasome function. Biochemistry. 2003;42:11460–11465. doi: 10.1021/bi035033g. [DOI] [PubMed] [Google Scholar]
- Van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annu Rev Biochem. 2012 doi: 10.1146/annurev-biochem-093010-153308. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, Koonin EV, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- Walczak H, Iwai K, Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. doi: 10.1186/1741-7007-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wass MN, Sternberg MJ. ConFunc--functional annotation in the twilight zone. Bioinformatics. 2008;24:798–806. doi: 10.1093/bioinformatics/btn037. [DOI] [PubMed] [Google Scholar]
- Williams MJ, Kana BD, Mizrahi V. Functional analysis of molybdopterin biosynthesis in mycobacteria identifies a fused molybdopterin synthase in Mycobacterium tuberculosis. J Bacteriol. 2011;193:98–106. doi: 10.1128/JB.00774-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HL, Aldrich HC, Maupin-Furlow J. Halophilic 20S proteasomes of the archaeon Haloferax volcanii: purification, characterization, and gene sequence analysis. J Bacteriol. 1999;181:5814–5824. doi: 10.1128/jb.181.18.5814-5824.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- Zhou G, Kowalczyk D, Humbard MA, Rohatgi S, Maupin-Furlow JA. Proteasomal components required for cell growth and stress responses in the haloarchaeon Haloferax volcanii. J Bacteriol. 2008;190:8096–8105. doi: 10.1128/JB.01180-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Zhou W, Wang J, Puc J, Ohgi KA, Erdjument-Bromage H, et al. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell. 2007;27:609–621. doi: 10.1016/j.molcel.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.