

Abstract
N6-methyladenosine (m6A) is a ubiquitous modification in messenger RNA (mRNA) and other RNAs across most eukaryotes. For many years, however, the exact functions of m6A were not clearly understood. The discovery that the fat mass and obesity associated protein (FTO) is an m6A demethylase indicates that this modification is reversible and dynamically regulated, suggesting it has regulatory roles. In addition, it has been shown that m6A affects cell fate decisions in yeast and plant development. Recent affinity-based m6A profiling in mouse and human cells further showed that this modification is a widespread mark in coding and non-coding RNA transcripts and is likely dynamically regulated throughout developmental processes. Therefore, reversible RNA methylation, analogous to reversible DNA and histone modifications, may affect gene expression and cell fate decisions by modulating multiple RNA-related cellular pathways, which potentially provides rapid responses to various cellular and environmental signals, including energy and nutrient availability in mammals.
RNA modifications
RNA plays a central role in multiple cellular processes, including functioning as a carrier of genetic information, catalyzing and regulating biochemical reactions, serving as an adapter molecule in protein synthesis, and providing a structural scaffold in subcellular organelles [1]. To create more diversity to fulfill all the different functions of RNAs, numerous modifications are added to the four canonical bases of the nascent precursor RNA transcripts. More than 100 post-transcriptionally modified ribonucleosides have been identified in almost all types of RNA [2]. In addition, recent evidence has shown that RNA modifications are very dynamic [3,4], which may increase the functional complexity and diversity of RNA and suggests some of these modifications participate in the regulation of gene expression at the post-transcriptional level.
In eukaryotic mRNA, different types of methylation modifications have been documented. Generally demonstrated as m7G(5′)ppp(5′)N1(m)pN2(m)pNpNpN…, mRNA includes N7-methylguanosine (m7G) at the 5′ cap structure [5–7], N6-methyl-2′-O-methyladenosine (m6Am) at the first position of the 5′ terminus (N1) and internal positions [8,9], 2′-O-methylated nucleosides (Nm) on the first two starting positions at the 5′ terminus (N1 and N2) and in internal sequences [8–10], internal 5-methylcytosine (m5C) [11], and internal N6-methyladenosine (m6A) [8,9,12] (Figure 1). Among these, only the modifications on the cap have been well studied: m7G is involved in mRNA processing such as translation initiation, mRNA transport, splicing, and degradation [13,14]; and 2′-O-methylation within the cap structure of mRNA is thought to provide a molecular signature for host cells to distinguish self-mRNAs from non-self RNAs through sensory proteins in viral/mammalian systems [15,16]. By contrast, despite being discovered decades ago, the function of the most prevalent internal modification of eukaryotic mRNA, m6A [17], has remained unclear. The recent discovery of the fat mass and obesity-associated (FTO) protein as an m6A-demethylase demonstrates that m6A is a reversible and dynamic RNA modification that may impact mammalian energy homeostasis [18]. In this review we briefly introduce m6A in mRNA and the enzymes involved in the methylation and demethylation processes, describe recent results obtained from the m6A methylome, and discuss potential biological roles of this dynamic modification.
Figure 1. Modifications in eukaryote mRNA.

The structure and localization of RNA modifications in mRNA are shown. m7G is at the 5′ terminus of the cap structure, m6Am at the first position of the 5′ terminus and internal positions, 2′-O-methylated Nm on the first two starting positions of the 5′ terminus and internal sequence, and m5C and m6A are internal mRNA modifications (green ball indicates the modifications located at the cap, pink ball indicates internal modifications).
m6A methylation
m6A was first found in mammalian mRNA in the mid 1970s (Figure 2). It has been detected in mRNA isolated from eukaryotes including mammals [6,8,9,12,19], plants [20,21], flies [22], and yeast in the meiotic state [23], as well as in viral RNAs that replicate in the nucleus, such as influenza virus and Rous Sarcoma virus (RSV) [24, 25]. The abundance of m6A has been shown to be ~0.1%–0.4% of total adenosine residues in cellular mRNA [6,12], and the average content of m6A has been estimated to be 3–5 residues per mammalian mRNA [9], and 1–15 per RSV viral RNAs [25]. m6A methylation has been shown to preferentially occur at the consensus sequence RRACH (where R represents purine, A is m6A site, and H is a nonguanine base) [8,9]. m6A is also found in introns, which indicates this modification is formed prior to mRNA splicing [26]. However, methylation at a specific site is non-stoichiometric, and only a portion of these consensus sequences are methylated in mRNA; the percentage is ~20–90% in RSV and ~20% in the mRNA encoding bovine prolactin (bPRL) [25, 27, 28].
Figure 2.
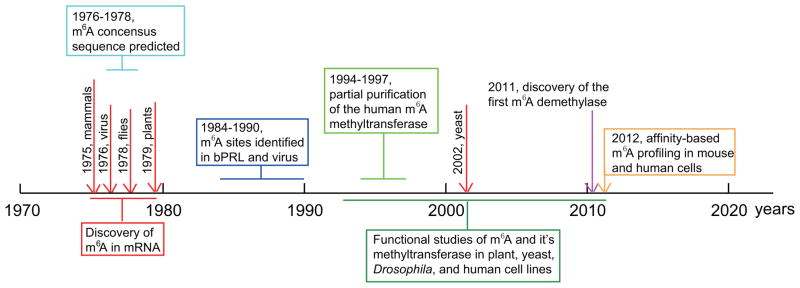
The history of the research on m6A in mRNA.
m6A is formed during processing of the nascent pre-mRNA by N6-adenosine methyltransferase. To date, the only component of the methyltransferase complex identified is the 70kD SAM (S-adenosylmethionine)-binding subunit MT-A70 (also known as METTL3) [29,30]. Although the rest of the complex has not been characterized, the mechanism of the methyl transfer is most likely the conserved SN2 type reaction from SAM to the amino group of adenosine, based on the high similarity of the catalytic domain of MT-A70 to those of the other methyltransferase enzymes (Figure 3) [31]. MT-A70 co-localizes with nuclear speckles in HeLa cells, which indicates that the methylation may happen co-transcriptionally. Loss of MT-A70 does not lead to cell cycle arrest, but appears to cause cell death, highlighting its critical role in mammalian cells [17].
Figure 3. The reversible adenosine methylation reaction in mRNA and non-coding RNA.

(a) The reversible m6A methylation catalyzed by N6-adenosine methyltransferase and demethylases (FTO and ALKBH5). (b) The proposed SN2 methylation reaction of N6-adenosine and the oxidative demethylation through a proposed N6-hydroxymethyladenosine intermediate. SAM: S-adenosylmethionine, SAH: S-adenosyl-L-homocysteine.
Because the presence of a methyl group at the N6 position of adenosine does not alter its Watson-Crick base-pairing property, m6A cannot be mapped by reverse transcription-based methods, which hindered the functional study of m6A in mRNA [32–35]. So far, only thirteen m6A base-resolution sites from RSV viral RNA and one m6A site from the cellular mRNA bPRL have been mapped despite the fact that this modification has been known for more than 30 years [25–28]. Two approaches were applied to probe the biological function of m6A [36–38]. The first was to construct an expression system with RSV or bPRL sequences and then mutate the m6A sites within them to study its effect on mRNA biogenesis. Although little new insight was gained, one interesting observation was that the mutation of the only m6A site in the bPRL sequence led to a new methylation site in an adjacent consensus sequence that was not normally methylated. The second method used general methylation inhibitors (S-tubericidinylhomocysteine or cycloleucine) in human HeLa cells [37] or mouse sarcoma cells [38], and the results suggest that these chemicals affect the efficiency of pre-mRNA splicing or transport of mRNA from the nucleus to the cytoplasm; however, they inhibit both m6A and cap-associated 2′-O-methyl formation as well as many other biological methylation events, making it impossible to distinguish the effects of just loss of m6A. It was reported that methylated dihydrofolate reductase transcript was translated 20% more efficiently than the non-methylated transcript in an in vitro translation assay, indicating that the presence of m6A in mRNA may affect translation efficiency [38].
Additional clues into the function of this modification have come from the recent identification of several putative m6A-binding proteins [32], suggesting that methylation of RNA may modulate the affinity of certain RNA-binding proteins to mRNA and thus likely impact aspects of mRNA metabolism. Because m6A is enriched within 3′-UTRs that contain miRNA-binding sites (discussed below), miRNAs may affect the methylation levels of their target transcripts [33]. However, further experimental investigations are required to validate these hypotheses.
m6A RNA methylomes in mammals
New insight into the role of RNA methylation has come from the recent development of affinity-based m6A profiling, which has revealed the transcriptome-wide maps of m6A distributions in human [32] and mouse cells [33]. This technology was enabled by advances in high-throughput sequencing combined with the m6A antibody that was successfully developed and used to investigate m6A modifications in snRNAs in HeLa cells [39]. In this new method, mRNA isolated from mouse liver or brain or human cell lines was chemically fragmented into approximately 100 nt long stretches, followed by m6A-antibody-based immunoprecipitation. The captured methylated RNA fragments were then subjected to high-throughput sequencing, providing the first initial view of the human and mouse m6A modification landscape in a transcriptome-wide manner.
These studies identified m6A in more than 7,000 mammalian mRNA transcripts and 300 non-coding RNAs (ncRNAs), indicating that m6A is a wide-spread modification. The data revealed a preference for m6A deposition around stop codons, in 3′UTRs, and within long internal exons, and that the m6A sites are highly conserved between human and mouse [29,30]. Many of these modifications appear to be quite stable, as 70–95% of m6A peak positions remained constant under various treatments such as ultra-violet radiation, heat shock, hepatocyte growth factor, and interferon-γ, but some of them were stimulus-dependent and dynamically modulated by interferon-γ treatment [32]. The m6A modification also exhibits tissue-specific regulation and is markedly increased throughout brain development [33]. Although m6A is not enriched at splice junctions [33], knockdown of MT-A70 (the SAM binding subunit of the putative mRNA N6-adenosine methyltransferase complex) affects gene expression and alternative splicing patterns as well as modulates p53 signaling and induction of apoptosis [17,32]. In general, the RRACH consensus sequence was confirmed, but the previous suggestion of the preferential occurrence of m6A within the loop of a stem-loop structure was not supported, as no strong secondary structures were found in regions harboring newly identified m6A peaks [29]. However, due to the lack of single-base resolution, we still cannot rule out the possibility that other m6A-containing motifs may exist in mRNA and ncRNA and are installed by other N6-adenosine methyltransferases. It should be noted that the m6A sites in human rRNA and U6 small nuclear RNA do not match the RRACH consensus sequences found in mRNA [40].
m6A in other organisms
In the yeast Saccharomyces cerevisiae, m6A is not detectable in mRNA from mitotic log phase cells; however, during meiosis, m6A can be easily detected in the mRNAs. The ratio of m6A to A is 1.0% in the poly(A) RNA isolated from cells growing for 3 h in sporulation medium [41]. Ime4, the homolog of MT-A70 in S. cerevisiae, is required for m6A modification and has a role in the initiation and nutritional control of meiosis [23,41,42]. The antisense ncRNA RME2 (Regulator of Meiosis 2) prevents IME4 gene expression by blocking the elongation of the full-length IME4 transcript, whereas in diploid cells, the a1-α2 complex represses the transcription of RME2, thus allowing IME4 induction during meiosis [43,44]. During meiosis, IME4 mRNA expression increases to a significant level by 6 h and decreases substantially to near baseline levels by 24 h after yeast cells are transferred to sporulation medium [23]. Targets for methylation include the mRNA of Ime4 itself, together with the transcripts encoding Ime1 and Ime2, two key regulators of meiosis [41]. In a recent study [45], two meiotic proteins, Mum2 and Slz1, were identified as Ime4 interacting partners, and it was found that these proteins are required for m6A formation. In addition, the study revealed that the m6A level increases when yeast cells enter meiosis in nutrient-poor liquid medium, and that the m6A level drops rapidly when nutrients are returned and cells begin foraging and undergo pseudohyphal growth. These observations indicate that this RNA methylation controls cell fate and the initiation of meiosis in yeast.
m6A was found in the mRNA of the monocot plants maize [20], wheat [46], and oat [47] more than thirty years ago. The potential function of m6A has been studied relatively more thoroughly in the model organism Arabidopsis thaliana [21,48]. Arabidopsis mRNA contains a similar amount of m6A to that harbored in mammalian cells, with a ratio of m6A to A of 1.5% from 2-week-old seedlings. Similar to the enrichment of m6A in the 3′-UTR in mammals, m6A in plants is shown to be located at the 3′ end of transcripts in a region 100–150 bp before the poly(A) tail [48]. m6A content varies in different tissues, and a higher ratio of m6A to A was found in the flower buds relative to the roots and leaves, which correlates with MTA (a homolog of human MT-A70 and yeast Ime4, encoded by At4g10760) expression levels [21]. MTA expression is also strongly associated with dividing tissues, suggesting that RNA methylation is involved in cell division. In agreement with this, inactivation of MTA in Arabidopsis leads to failure of the developing embryo to progress past the globular stage, indicative of cell division defects, and the arrested seeds from the MTA knockout line lack m6A-containing mRNAs [21]. Further insight into the role of m6A in plants has come from studying a mutant with decreased levels of MTA expression. These plants exhibit decreased m6A levels and abnormal growth with reduced apical dominance, abnormal organ definition, and an increased number of trichome branches [48]. Microarray data from plants with reduced m6A levels showed that a significant portion of the down-regulated mRNAs are involved in transport or targeted transport, and most of the up-regulated genes are associated with stress and stimuli responses [48]. These data indicate that this modification may be a dynamic response to environmental cues, which is particularly important for plants. Interestingly, MTA was also found to interact and colocalize with the splicing factor FIP37 in splicing speckle-like nuclear compartments in plants [21]. Arabidopsis FIP37 is a homolog of yeast Mum2, Drosophila female-lethal-2-D (FL(2)D), and human wilms’ tumor 1-associating protein (WTAP). Both FL(2)D and WTAP are suggested to be associated with the spliceosome. Drosophila FL(2)D is required for the accumulation of correctly spliced forms of two critical sexual determination gene (sex lethal and transformer) [49,50], and human WTAP interacts with wilms’ tumour1 (WT1) protein, one isoform of which (+KTS WT1) binds to RNA and is incorporated into spliceosomes [51,52]. The interaction of MTA with splicing factors in plants suggests a role for m6A in regulating RNA splicing, and moreover, that this may be conserved in flies and humans.
The internal m6A modification in Drosophila mRNA was first discovered in 1978 [22]. A study in 2011 focused on Dm ime4, the Drosophila homolog of MT-A70, but did not evaluate the levels of m6A in wild-type and Dm ime4 knockout flies [53]. Dm ime4 is expressed in ovaries and testes and localizes to soma and germ-line cells in ovarioles. The levels of Notch signaling are reduced in follicle cells of Dm ime4 mutants, which phenotypically lead to fused egg chambers with follicle-cell defects. A complete deletion of Dm ime4 is lethal, suggesting that m6A is essential for viability and participates in Notch signaling during egg chamber development. The studies of m6A in these other organisms further point to significant involvement of this prevalent modification in various aspects of mRNA metabolism.
The m6A demethylases—FTO and ALKBH5 proteins
Several genome-wide association studies (GWAS) showed a strong association of single nucleotide polymorphisms (SNPs) in the fat mass and obesity-associated (FTO) gene with body mass index (BMI) and risk of obesity and type 2 diabetes in multiple populations [54–56]. The trend from these studies has continued, and FTO has recently been linked with the phenotypic variability of BMI [57]. Mice overexpressing Fto display increased food intake and increased adiposity in a dose-dependent manner [58], in agreement with the dose-dependent effects of FTO in adiposity observed in humans [54]. Mice lacking Fto show increased postnatal lethality, postnatal growth retardation, reduced fat mass, lower body weight, increased energy expenditure, and a relative increase in food intake [59–61]. Deletion of Fto in the brain recapitulates the reduced body weight of Fto nulls, indicating that Fto functions in the central nervous system to regulate postnatal energy homeostasis [61]. GWAS have also shown that common variants in the FTO gene have been linked with glucose metabolism [62–64], neurological diseases including depression [65] and Alzheimer’s disease [66], cardiovascular disease [64], cancer [67,68], and end-stage renal disease [69]. In addition, a loss-of-function homozygous mutation (R316Q) in the FTO gene typically leads to postnatal growth retardation, microcephaly, psychomotor delay, facial dysmorphism, certain brain malformations, cardiac abnormalities, and cleft palate. In all cases, lethality usually occurred before the age of 3 years [70].
Despite the strong genetic association between FTO and body weight observed by GWAS, little was known about the molecular mechanism leading to the phenotypes. FTO is a homolog of the non-heme Fe(II)/α-ketoglutarate (α-KG)-dependent AlkB family dioxygenases, which typically catalyze demethlyation/hydroxylation, and has been shown to oxidatively demethylate m3T in single-stranded DNA (ssDNA) and m3U in single-stranded RNA (ssRNA) in vitro [71,72], although the observed activity is exceedingly low compared to those of the other AlkB family proteins. The arginine at position 316 is one of the α-KG ligands, and the R316Q mutant causes reduced FTO catalytic activity in vitro. The phenotypes associated with this mutation described above are direct evidence that the enzymatic activity of FTO is critical. However, because m3T is very rare in mammalian genomic DNA, and m3U only exists in a deeply buried position in folded rRNA, they are unlikely to be the physiological substrates of FTO. The preference of FTO for ssDNA/ssRNA can be explained by the presence of an extra loop covering one side of the conserved jelly-roll motif, which selectively competes with the complementary strand of the DNA/RNA duplex [73]. In a surprising discovery, FTO was found to be an m6A RNA demethylase [18].
FTO can efficiently convert m6A to adenosine in synthetic ssRNA and ssDNA with much higher activity compared to m3U at neutral pH in vitro (Figure 3a). m6A is the most prevalent modification in mRNA of eukaryotes, but is undetectable in genomic DNA in higher eukaryotes [74], suggesting that the physiological substrate of FTO is RNA, not DNA. To test this, FTO was either knocked down or overexpressed in two human cell lines, HeLa and 293FT. mRNA was then isolated by using poly(dT) beads followed by an rRNA depletion step, and m6A was quantified by three different methods: LC-MS/MS, 2D-TLC, and m6A-antibody-based dot-blot, respectively. All results showed that m6A in mRNA is subject to FTO-mediated demethylation inside these cells, confirming it as a physiological substrate of FTO. It was further found that FTO is partially co-localized with nuclear splicing speckle factors (SART1 and SC35) and RNA polymerase II (phosphorylated at Ser2), but not with markers for other nuclear subregions such as telomeres, replication sites, Cajal bodies, cleavage bodies, or P-bodies, which indicated that FTO is involved in the processing of nascent transcribed mRNA [18].
The discovery that FTO is an mRNA m6A demethylase added RNA modification to the list of reversible and dynamic modifications, similar to DNA methylation and histone modifications, but exerting its functions post-transcriptionally. It further suggests physiological roles of m6A in human biological processes, in particular energy homeostasis regulation. Additional m6A demethylases may exist and play functional roles in biological regulation. Indeed, another human m6A demethylase was recently identified in mammals that has significant effects on spermatogenesis [75]. ALKBH5 (alkB, alkylation repair homolog 5) is a homologue of FTO [76]. Its m6A demethylation activity impacts total RNA synthesis and mRNA (polyA-tailed RNA) export inside mammalian cells. Alkbh5-knockout male mice exhibited increased m6A in mRNA and impaired fertility resulting from apoptosis that affects meiotic metaphase-stage spermatocytes. The discovery of this second m6A demethylase for mRNA and ncRNA reinforces the notion that this modification plays dynamic regulatory roles in fundamental process.
To date, FTO homologs have been found in vertebrates (from fish to mammals) and marine algae (from unicellular photosynthetic picoplankton to multicellular seaweed), but not in invertebrate animals, fungi, plants, bacteria, or archaea [77]. Although ALKBH5 is also only conserved in vertebrates [76], it is not unlikely that m6A demethylases other than FTO and ALKBH5 exist in other organisms.
Perspectives on the role of reversible RNA methylation
There is growing evidence that adenosine methylation in RNA (mRNA and ncRNAs) plays important regulatory roles, and that mis-regulation ultimately affects physiologic processes in mammals. These data support the hypothesis that dynamic/reversible RNA modifications may function analogously to reversible DNA and protein modifications, heralding the arrival of “RNA epigenetics” [3,4].
The dynamics of m6A can be temporally and spatially modulated in different manners depending on the localization and concentration of methyltransferase/demethylase enzymes and RNA substrates. The majority of FTO localizes in nuclear speckles, where methyltransferases reside. Because adenosine methylation is non-stoichiometric, the methylated/unmethylated ratio at specific adenosine sites on a given mRNA can be tuned by differentially expressing methyltransferase/demethylase enzymes as well as by regulating the subcellular localization of both the transcript and the enzymes. Apart from the equilibrium model, a potential “irreversible” mechanism may exist through the generation of short-lived intermediates during demethylation (Ye Fu, Guifang Jia, Chuan He, et al. unpublished), which prevents direct re-methylation shortly after oxidation of m6A by FTO; the formation of fully demethylated adenosine from the oxidation intermediate could be delayed until mRNA is transported out of speckles (Figure 4a). Additionally, certain RNAs may be transported to concentrated FTO loci, possibly via a specific sequence recognized by an RNA-binding protein that also interacts with FTO or FTO partner proteins. This would result in greater demethylation of those targeted RNAs.
Figure 4. Proposed functions of reversible m6A methylation.

(a) The intermediate modification created by FTO could be recognized by a specific RNA-binding protein to impact RNA metabolism. If this interaction occurred in the nuclear speckle, where FTO is localized, it might protect the modification until it was exported from the nuclear speckle (b) The m6A methylation may be recognized by specific m6A-binding proteins, or preclude the binding by certain RNA-binding proteins that preferably bind to unmethylated RNA. The presence of m6A can therefore impact various aspects of RNA metabolism such as pre-mRNA splicing, mRNA transport from nuclear to cytoplasm, translation, mRNA turnover, mRNA stability or sub-cellular localization. In addition, the presence of m6A in the 3′UTR of mRNA might weaken or enhance mRNA association with miRNA (yellow box) via modulation of protein-RNA recognitions.
The comparison between RNA methylation and other epigenetic marks also extends to the enzymes and proteins involved: the methyltransferases and demethylases are “writers” and “erasers”, and the m6A modification on RNA can be recognized by “readers”, m6A-specific binding proteins [32] that recognize the m6A modification and affect pre-mRNA splicing, mRNA transport from nuclear to cytoplasm, translation, mRNA turnover, mRNA stability or sub-cellular localization [78] (Figure 4b). m6A might also be involved in siRNA or miRNA pathways as a reversible chemical handle. As mentioned above, a significant portion of m6A occurs in the 3′UTR of the transcripts, which correlates with miRNA-binding sites. About two thirds of 3′UTRs that contain m6A peaks also contain at least one predicted miRNA binding site, and the most highly expressed miRNAs have a significantly greater percentage of target transcripts that contain m6A [33]. Because m6A is not exactly on the miRNA binding sites but near them, the presence of m6A in 3′UTRs of target transcripts might recruit specific binding proteins to weaken or enhance the subsequent interactions of the transcripts with the miRNA (Figure 4b).
Over 90% of the human genome is likely to be transcribed, of which only 2% (around 20,000 genes) is translated into proteins and the remaining 98% are ncRNAs, including tens of thousands of long noncoding RNAs (lncRNAs). Although the majority of lncRNAs have yet to be characterized, some have been found to serve key regulatory roles [79, 80]. An emerging theme from multiple model systems is that lncRNAs form extensive networks of ribonucleoprotein (RNP) complexes with numerous chromatin regulators and subsequently target these enzymatic activities to appropriate locations in the genome [80]. The finding that m6A is also widespread in lncRNAs [33] establishes a possible connection between RNA modification and chromatin regulations. The potential link between m6A to the function of lncRNA is an intriguing area to explore. Regulation at the RNA level could allow for rapid response to signaling and stimuli and might be heritable, in particular if the methylation status of certain lncRNA or mRNA affects histone and/or DNA modifications.
Interestingly, the human methyltransferase BCDIN3D has been shown to methylate the 5′ phosphate of a miRNA precursor, pre-miR-145, and prevent its processing by Dicer to produce the mature miRNA product [81]. It would be interesting to see whether this methylation can be reversed to re-activate the methyl-trapped pre-miRNA as another example of reversible methylation on RNA. Thus, pre-miRNAs, and possibly even pre-tRNAs and pre-rRNAs, could be subjected to similar reversible methylation, which may affect maturation and transport of these RNAs and add additional layers of complexity and potential regulation mechanisms.
Concluding remarks
Although the exact mechanism and function of m6A in mRNA and ncRNA remain obscure, it is certain that this reversible adenosine methylation has important regulatory roles across eukaryotes. The discovery of at least two m6A mRNA demethylases strongly indicates that this reversible modification has dynamic roles in fundamental biological regulation. It is highly likely that other methyltransferases and demethylases also exist. Investigations of these writers and erasers will help to establish related cellular regulatory pathways. For example, characterization of m6A distribution within the wild-type and FTO mutant cells will help reveal the mechanisms of FTO-based regulation. In addition, exploration of m6A-binding proteins is essential to the understanding of the functional roles of the m6A modification. The ability to perform transcriptome-wide profiling of m6A is a significant step forward and has provided new insight into the biology of RNA methylation [32,33]. The next challenge is to develop single-base resolution sequencing, which could quantify the relative abundance of m6A at each modification site and greatly expand our understanding of RNA epigenetics.
Acknowledgments
The authors thank the US National Institutes of Health (NIH) for support (GM071440, GM088599). We thank Dr. Tao Pan for insightful discussions and Sarah F. Reichard, MA for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Cantara WA, et al. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He C. Grand challenge commentary: RNA epigenetics? Nat Chem Biol. 2010;6:863–865. doi: 10.1038/nchembio.482. [DOI] [PubMed] [Google Scholar]
- 4.Yi CQ, Pan T. Cellular dynamics of RNA modification. Acc Chem Res. 2011;44:1380–1388. doi: 10.1021/ar200057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rottman FM, et al. Sequences containing methylated nucleotides at the 5′-termini of messenger RNAs: possible implications for processing. Cell. 1974;3:197–199. doi: 10.1016/0092-8674(74)90131-7. [DOI] [PubMed] [Google Scholar]
- 6.Wei CM, et al. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4:379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- 7.Shatkin AJ. Capping of eukaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- 8.Wei CM, et al. 5′-terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochem. 1976;15:397–401. doi: 10.1021/bi00647a024. [DOI] [PubMed] [Google Scholar]
- 9.Schibler U, et al. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol. 1977;115:695–714. doi: 10.1016/0022-2836(77)90110-3. [DOI] [PubMed] [Google Scholar]
- 10.Dong HP, et al. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. Plos Pathog. 2012;8:e1002642. doi: 10.1371/journal.ppat.1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Squires JE, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry RP, et al. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′-terminus. Cell. 1975;4:387–394. doi: 10.1016/0092-8674(75)90159-2. [DOI] [PubMed] [Google Scholar]
- 13.Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daffis S, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Züst R, et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bokar JA. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA. In: Grosjean H, editor. Fine-tuning of RNA functions by modification and editing. Springer-Verlag; Berlin, Heidelberg, NY: 2005. pp. 141–177. [Google Scholar]
- 18.Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desrosiers R, et al. Characterization of Novikoff mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry. 1975;14:4367–4374. doi: 10.1021/bi00691a004. [DOI] [PubMed] [Google Scholar]
- 20.Nichols JL. “Cap” structures in maize poly(A)-containing RNA. Biochim Biophys Acta. 1979;563:490–495. doi: 10.1016/0005-2787(79)90067-4. [DOI] [PubMed] [Google Scholar]
- 21.Zhong S, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levis R, Penman S. 5′-terminal structures of poly(A)+ cytoplasmic messenger RNA and of poly(A)+ and poly(A)− heterogeneous nuclear RNA of cells of the dipteran Drosophila melanogaster. J Mol Biol. 1978;120:487–515. doi: 10.1016/0022-2836(78)90350-9. [DOI] [PubMed] [Google Scholar]
- 23.Clancy MJ, et al. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30:4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krug RM, et al. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kane SE, Beemon K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol. 1985;5:2298–2306. doi: 10.1128/mcb.5.9.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carroll SM, Narayan P, Rottman FM. N6-methyladenosine residues in an intron-specific region of prolactin pre-mRNA. Mol Cell Biol. 1990;10:4456–4465. doi: 10.1128/mcb.10.9.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horowitz S, et al. Mapping of N6-methyladenosine residues in bovine prolactin mRNA. Proc Natl Acad Sci USA. 1984;81:5667–5671. doi: 10.1073/pnas.81.18.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayan P, et al. Context effects of N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res. 1994;22:419–426. doi: 10.1093/nar/22.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bokar JA, et al. Characterization and partial purification of mRNA N6-adenosine methylatransferase from HeLa cell nuclei. J Biol Chem. 1994;269:17697–17704. [PubMed] [Google Scholar]
- 30.Bokar JA, et al. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 31.Scavetta RD, et al. Structure of Rsrl methyltransferase, a member of the N6-adenine β class of DNA methyltransferases. Nucleic Acids Res. 2000;28:3950–3961. doi: 10.1093/nar/28.20.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 33.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′UTRs and near stop codons. Cell. 2012;150:1–12. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X, Yu YT. Detection and quantitation of RNA base modifications. RNA. 2004;10:996–1002. doi: 10.1261/rna.7110804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai Q, et al. Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. 35:6322–6329. doi: 10.1093/nar/gkm657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Csepany, et al. Sequence specificity of mRNA N6-adenosine methyltransferse. J Biol Chem. 1990;265:20117–20122. [PubMed] [Google Scholar]
- 37.Camper SA, et al. Effect of undermethylation on mRNA cytoplasmic appearance and half-life. Mol Cell Biol. 1984;4:538–543. doi: 10.1128/mcb.4.3.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuck MT, et al. Inhibition of 6-methyladenine formation decreases the translation efficiency of dihydrofolate reductase transcripts. Int J Biochem Cell Biol. 1999;31:837–851. doi: 10.1016/s1357-2725(99)00041-2. [DOI] [PubMed] [Google Scholar]
- 39.Bringmann P, Lührmann R. Antibodies specific for N6-methyladenosine react with intact snRNPs U2 and U4/U6. FEBS Lett. 1987;213:309–315. doi: 10.1016/0014-5793(87)81512-0. [DOI] [PubMed] [Google Scholar]
- 40.Shimba S, et al. Accurate and efficient N6-adenosine methylation in splicesomal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res. 1995;23:2421–2426. doi: 10.1093/nar/23.13.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bodi Z, et al. Yeast targets for mRNA methylation. Nucleic Acids Res. 2010;38:5327–5335. doi: 10.1093/nar/gkq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah JC, Clancy MJ. IME4, a gene that mediates MAT and nutritional control of meiosis in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:1078–1086. doi: 10.1128/mcb.12.3.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hongay CF, et al. Antisense transcription controls cell fate in Saccharomyces cerecisiae. Cell. 2006;127:735–745. doi: 10.1016/j.cell.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 44.Gelfand B, et al. Regulated antisense transcription controls expression of cell-type-specific genes in yeast. Mol Cell Biol. 2011;31:1701–1709. doi: 10.1128/MCB.01071-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agarwala SD, et al. RNA methylation by the MIS complex regulates a cell fate decision in yeast. Plos Genet. 2012;8:e1002732. doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy TD, Lane BG. Wheat embryo ribonucleates. XIII Methyl-substitutes nucleosites constituents and 5′-terminal dinucleotide sequences in bulk poly(A)-rich RNA from imbibing wheat embryos. Can J Biochem. 1979;57:927–931. doi: 10.1139/o79-112. [DOI] [PubMed] [Google Scholar]
- 47.Haugland RA, Cline MG. Post-transcriptional modifications of oat coleoptile ribonucleic acids. 5′-terminal capping and methylation of internal nucleosides in poly(A)-rich RNA. Eur J Biochem. 1980;104:271–277. doi: 10.1111/j.1432-1033.1980.tb04425.x. [DOI] [PubMed] [Google Scholar]
- 48.Bodi Z, et al. Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front Plant Sci. 2012;48:1–9. doi: 10.3389/fpls.2012.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Granadino B, et al. The Drosophila melanogaster fl(2)d gene is needed for the female-specific splicing of Sex-lethal RNA. EMBO J. 1990;9:2597–2602. doi: 10.1002/j.1460-2075.1990.tb07441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granadino B, et al. The gene fl(2)d is needed for the sex-specific splicing of transformer pre-mRNA but not for double-sex pre-mRNA in Drosophila melanogaster. Mol Gen Genet. 1996;253:26–31. doi: 10.1007/s004380050292. [DOI] [PubMed] [Google Scholar]
- 51.Larsson SH, et al. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell. 1995;81:391–401. doi: 10.1016/0092-8674(95)90392-5. [DOI] [PubMed] [Google Scholar]
- 52.Davies RC, et al. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998;12:3217–3225. doi: 10.1101/gad.12.20.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hongay CF, Orr-Weaver TL. Drosophila Inducer of Meiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc Natl Acad Sci USA. 2011;108:14855–14860. doi: 10.1073/pnas.1111577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frayling TM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dina C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 56.Hinney A, et al. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. Plos ONE. 2007;2:e1367. doi: 10.1371/journal.pone.0001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490:267–272. doi: 10.1038/nature11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Church C, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42:1086–1092. doi: 10.1038/ng.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fischer J, et al. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- 60.Church C, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 2009;5:e1000599. doi: 10.1371/journal.pgen.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao X, et al. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE. 2010;5:e14005. doi: 10.1371/journal.pone.0014005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobsson JA, et al. Novel genetic variant in FTO influences insulin levels and insulin resistance in severely obese children and adolescents. Int J Obes. 2008;32:1730–1735. doi: 10.1038/ijo.2008.168. [DOI] [PubMed] [Google Scholar]
- 63.Do R, et al. Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec Family Study. Diabetes. 2009;57:1147–1150. doi: 10.2337/db07-1267. [DOI] [PubMed] [Google Scholar]
- 64.Lappalainen T, et al. Association of the FTO gene variant (rs9939609) with cardiovascular disease in men with abnormal glucose metabolism-the Finnish Diabetes Prevention Study. Nutr Metab Cardiovasc Dis. 2011;21:691–698. doi: 10.1016/j.numecd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Rivera M, et al. Depressive disorder moderates the effect of the FTO gene on body mass index. Mol Psychiatr. 2012;17:604–611. doi: 10.1038/mp.2011.45. [DOI] [PubMed] [Google Scholar]
- 66.Keller L, et al. The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzhermers Dis. 2011;23:461–469. doi: 10.3233/JAD-2010-101068. [DOI] [PubMed] [Google Scholar]
- 67.Kaklamani V, et al. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med Genet. 2011;12:52–61. doi: 10.1186/1471-2350-12-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tarabra E, et al. The obesity gene and colorectal cancer risk: a population study in Northern Italy. Eur J Intern Med. 2012;23:65–69. doi: 10.1016/j.ejim.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 69.Hubacek JA, et al. The FTO gene polymorphism is associated with end-stage renal disease: two large independent case-control studies in a general population. Nephrol Dial Transplant. 2012;27:1030–1035. doi: 10.1093/ndt/gfr418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boissel S, et al. Loss-of-function mutant in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–111. doi: 10.1016/j.ajhg.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerken T, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jia G, et al. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008;582:3313–3319. doi: 10.1016/j.febslet.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han Z, et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature. 2010;464:1205–1209. doi: 10.1038/nature08921. [DOI] [PubMed] [Google Scholar]
- 74.Ratel D, et al. Undetectable levels of N6-methyl adenine in mouse DNA: Cloning and analysis of PRED28, a gene coding for a putative mammalian DNA adenine methyltransferase. FEBS Lett. 2006;580:3179–3184. doi: 10.1016/j.febslet.2006.04.074. [DOI] [PubMed] [Google Scholar]
- 75.Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. doi: 10.1016/j.molcel.2012.10.015. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurowski MA, et al. Phylogenomic identification of five new human homologs of the DNA repair enzyme AlkB. BMC Genomics. 2003;4:48. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robbens S, et al. The FTO gene, implicated in human obesity, is found only in vertebrates and marine algae. J Mol Evol. 2008;66:80–84. doi: 10.1007/s00239-007-9059-z. [DOI] [PubMed] [Google Scholar]
- 78.Pawlicki JM, Steitz JA. Nuclear networking fashions pre-messenger RNA and primary microRNA transcripts for function. Trends Cell Biol. 2010;20:52–61. doi: 10.1016/j.tcb.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilusz JE, et al. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xhemalce B, et al. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell. 2012;151:278–288. doi: 10.1016/j.cell.2012.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]