

Abstract
OBJECTIVE
The pro-inflammatory phenotype induced by low laminar shear stress (LSS) is implicated in atherogenesis. The kinin B1 receptor (B1R), known to be induced by inflammatory stimuli, exerts many pro-inflammatory effects including vasodilatation and leukocyte recruitment. We investigated whether low LSS is a stimulus for endothelial B1R expression and function.
METHODS AND RESULTS
Human and mouse atherosclerotic plaques expressed high level of B1R mRNA and protein. In addition, B1R expression was upregulated in the aortic arch (low LSS region) of ApoE-/- mice fed a high fat diet compared to vascular regions of high LSS and animals fed normal chow. Of interest, a greater expression of B1R was noticed in endothelial cells from regions of low LSS in aortic arch of ApoE-/- mice. B1R was also upregulated in human umbilical vein endothelial cells (HUVEC) exposed to low LSS (0-2dyn/cm2) compared to physiological LSS (6-10dyn/cm2): an effect similarly evident in murine vascular tissue perfused ex vivo. Functionally, B1R activation increased prostaglandin and CXCL5 expression in cells exposed to low, but not physiological, LSS. IL-1β and ox-LDL induced B1R expression and function in HUVECs, a response substantially enhanced under low LSS conditions and inhibited by blockade of NFκB activation.
CONCLUSION
Herein, we show that LSS is a major determinant of functional B1R expression in endothelium. Furthermore, whilst physiological high LSS is a powerful repressor of this inflammatory receptor, low LSS at sites of atheroma are associated with substantial upregulation, identifying this receptor as a potential therapeutic target.
CONDENSED ABSTRACT
Low laminar shear stress (LSS) underlies the pro-inflammatory processes in atherogenesis. Herein, we demonstrate that whilst physiological LSS represses inflammatory kinin B1 receptor (B1R) expression/function, low atherogenic LSS is associated with profound upregulation of both in atherosclerosis in both humans and animal models, highlighting B1R as an exciting potential therapeutic target.
Keywords: Atherosclerosis, laminar shear stress, inflammation, kinin B1 receptor
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in developed countries with a major component of these deaths directly related to the consequences of atherogenesis (according to WHO statistics, 17 million people die of CVD each year, http://www.who.int). The past two decades has seen a growing appreciation that inflammatory mechanisms underlie the initiation and progressive development of an atheroma and it is clear that the inner lining of the blood vessel wall, the endothelium, is a pivotal site at which these inflammatory events occur1. In particular, there is recognition that alteration of the phenotype of the endothelium, from protective (and maintaining homeostasis) to damaging, is likely to precipitate the atherogenic process2.
One of the major determinants of endothelial phenotype is laminar shear stress (LSS), defined as the frictional force engendered by blood flow on the endothelium. Indeed, variation in LSS has been identified as determining susceptibility of particular vascular sites to atheroma formation2-4, and, has been proposed to predominate above sex and dietary fat as a risk factor for atherosclerosis5,6. The levels of LSS vary throughout the circulation, however, in large arteries (such as the aorta) the net unidirectional physiological levels of LSS are high (6-20 dyn/cm2 in conduit vessels) and endow the endothelium with an anti-inflammatory phenotype, whilst low LSS (<4dyn/cm2, levels found at sites of atheroma formation i.e. at bifurcations and curvatures such as the aortic arch) is thought to be proinflammatory and pathogenic in atherosclerosis2. However, the exact mechanisms stimulated by LSS that predispose a site to atheroma formation remain unclear.
Of particular relevance to the present study has been the finding that an inducible and pro-inflammatory G protein-coupled receptor (GPCR), the kinin B1 receptor (B1R), is localised to sites of human aortic atheroma7, although the functional significance of this expression has not been explored. The kinins are a family of inflammatory peptides, including bradykinin (BK) or Lys-BK and their metabolites, des-Arg9BK (DBK) and Lys-DBK (LDBK), that interact with two specific GPCRs: B1 receptor (B1R) and B2 receptor8. Whilst the B2R, activated by BK and Lys-BK, is constitutively expressed, the B1R, activated by DBK or Lys-DBK, is weakly expressed normally but is induced under inflammatory conditions8. Functionally, B1R activation induces a number of pro-inflammatory effects, therefore we investigated whether low (atherogenic) LSS might be a stimulus for B1R expression and inflammatory function in the blood vessel wall; these data were complemented by an analysis of the mechanisms involved in endothelial B1R expression and function in atherosclerosis.
MATERIALS AND METHODS
Cell culture and application of shear stress
Steady unidirectional LSS of 10, 6, 2 or 0 dyn/cm2 was applied on human umbilical vein (HUVEC) or aortic (HAEC) endothelial cells using a cone and plate viscometer9,10. Cells were left untreated or treated with B1 agonist: Lys-des-Arg9-BK (10μM), IL-1β (10ng/ml) in the absence or presence of the B1 receptor antagonist SSR24061211 (1μM; 15 min prior to IL-1β application), or with oxidized LDL (oxLDL, 20μg/ml12-14) or with BAY 11-708215 (20μM; 15 min prior to IL-1β application).
Perfused mouse mesentery preparations
The mesentery was mounted in a 37°C water-jacketed organ bath and perfused with warmed physiological salt solution with varying amounts of dextran to achieve high (6 dyn/cm2) or low (2 dyn/cm2) levels of LSS.
Atherosclerosis in ApoE-/- mice
Male atherosclerosis-prone ApoE-/- mice were fed a high fat or chow diet. The whole aorta was removed and in some instances the aortic arch separated from the thoracic aorta for separate analysis of regions subjected to low LSS and high physiological LSS respectively. Blood was collected for lipid analysis.
Immunohistochemical analysis
The aortic arches of ApoE-/- mice were embedded in paraffin and immunohistochemistry analysis performed.
Prostaglandin (PG) and Nitric oxide (NO) measurement in endothelial cell culture supernatant
Concentrations of PGI2 and PGE2 were measured using enzyme immunoassay kits. Nitrite production, as a measure of endothelial NO generation was measured as previously described16.
Quantitative real time PCR (RT-PCR)
Total RNA was extracted from cells or tissue, cDNA synthesised and quantitative RT-PCR conducted.
Western blotting
Western blotting for B1R was determined in human carotid endarterectomy tissue and HUVEC samples using a selective antibody for B1R17. Samples were divided into sections containing lesion (+; Figure SI) and regions devoid of plaque (-).
Radioligand binding assay
In HUVECs, total binding was determined by adding B1R agonist [3H]-LDBK at 0.75nM and non-specific binding was performed by co-treatment with LDBK in excess (10μM, 1h) on ice. Cells were dissolved and the radioactivity determined by liquid β-scintillation count.
RESULTS
B1R is induced in vivo in vascular regions predisposed to atheroma formation
B1R expression was more pronounced in regions of atheromatous plaque from human carotid endarterectomy tissue compared to regions devoid of plaque (Figure 1). B1R mRNA expression was also upregulated in a time-dependent fashion in aorta of ApoE-/- mice fed a high fat diet (p<0.05; Figure SIIA), an effect that was temporally associated with a rise in serum triglyceride level (Figure SIIB). Mice fed a normal chow diet for 12 weeks had normal levels of both serum triglyceride and LDL cholesterol levels (Figure SIIC-D) and no change in B1R mRNA expression (Figure 2A). Further analysis of the different regions of the aorta (i.e. regions of low LSS vs regions of high LSS), demonstrated that in ApoE-/- mice fed a high fat diet a >3-fold increase in expression was evident in the arch region compared to the thoracic aorta (Figure 2B). Immunohistochemical assessment of B1R expression demonstrated expression in both smooth muscle cells and endothelial cells in sections of the inner curvature (i.e. regions of low LSS, Figure 2C), that was less intense in sections of the outer curvature.
Figure 1. B1 receptor is induced in regions of atheroma in humans.
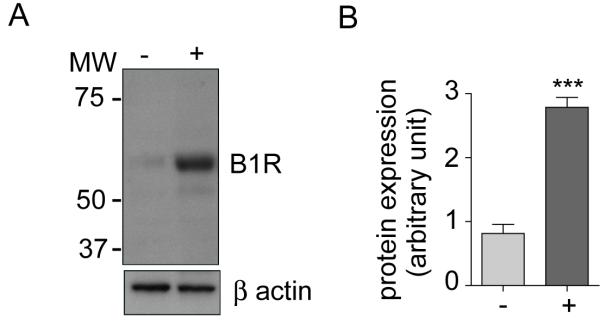
(A) Representative western blot of B1R protein expression in human carotid endarterectomy tissue: sections: with (+; n=4) or without (-, n=4) plaque and (B) quantification of B1R expression normalized to β-actin. Data are shown as mean ± SEM with *** P<0.001.
Figure 2. B1R is induced in regions of vessel curvature predisposed to atheroma formation in ApoE-/- mice.

(A-C) ApoE-/- mice fed a high fat or normal chow diet for 12 weeks. B1R mRNA expression in whole aorta (A) or in aortic arch and thoracic aorta sections (B). (C) B1R imunohistochemical expression in outer and inner curvatures of aortic arch of ApoE-/- mice fed a chow diet. Data are mean ± SEM for n=9, * P<0.05. Arch: aortic arch, Thor: thoracic aorta.
Low LSS induces kinin B1R expression in endothelial cells and in mesenteric tissue ex vivo
Exposure of HUVEC to physiological (high) LSS but not low LSS resulted in a time-dependent alignment of cells (Figure SIIIA). Low LSS was also associated with decreased nitrite production reflecting endothelial dysfunction (Figure SIIIB). Under high LSS a suppression of B1R mRNA expression relative to static HUVECs occurred that was maximal at 8h (by ~50%) and sustained for up to 16h (Figure SIVA). Comparison between cells exposed to high and low LSS for 8h exposed a ~2-fold increase in B1R mRNA expression in cells under low LSS conditions (Figure 3A). In contrast in HAEC, B1R mRNA was undetectable in both conditions (data not shown, n=4). All further cell experiments were conducted after 8h of LSS exposure, and using physiological (high) LSS as a reference control.
Figure 3. Low LSS induces B1R expression.

B1R (A) mRNA expression, (B) protein expression and (C) agonist binding in HUVECs subjected to low LSS or high LSS. Data are mean ± SEM for n=6, ** P<0.01, * P<0.05 high vs low values. (D) B1R mRNA expression in mesenteric tissue from WT mice perfused at high (6 dyn/cm2) or low (2 dyn/cm2) LSS for 4h. Data are mean ± SEM for n=5. *P<0.05.
Western blotting of cell lysates demonstrated upregulated expression following low but not high LSS (Figure 3B and SIVC). Confirmation of antibody selectivity was achieved in preadsorption experiments in HEK-293 (Figure SIVB). In addition [3H]-LDBK binding was increased 20-fold (p<0.01) in cells exposed to low LSS (Figure 3C). HUVEC treated with IL-1β displayed binding with [3H]-LDBK, that was displaced by increasing concentrations of cold LDBK confirming the validity of [3H]-LDBK as tracer for these assays (Figure SIVD).
LSS-induced regulation of B1R expression was also demonstrated in intact blood vessels; B1R mRNA was expressed at a very low level in mouse mesenteries exposed to physiological levels of LSS ex vivo, however exposure to low LSS caused a >5-fold elevation (p<0.05 (Figure 3D).
Low LSS increases B1R functionality
Both PGI2 and PGE2 release and endothelial CXCL5 mRNA expression were significantly enhanced in response to LDBK in HUVECs exposed to low LSS but not high LSS (Figure 4). Neither COX-1 nor COX-2 expression were altered by LSS (Figure SVA-B). CXCL5 mRNA was also upregulated in aortic arch of ApoE-/- mice compared to thoracic aorta (>8-fold increase; data not shown, n=6).
Figure 4. B1R activation stimulates PGE2, PGI2 and CXCL5 production only under low LSS.

HUVECs were subjected to high or low LSS and stimulated or not with B1R agonist Lys-des-Arg9-Bradykinin (LDBK, 10μM) and (A) PGI2 or (B) PGE2 release and endothelial (C) CXCL5 and (D) CXCL6 mRNA expression measured. Data are mean ± SEM for n=6. ns: non-significant, * P<0.05, ** P<0.01.
Additive effects of low LSS and inflammation on B1R expression and function
IL-1β caused a pronounced elevation of B1R mRNA expression in HUVEC under low LSS; an effect that peaked at 1h and returned to basal by 8h (Figure SVIA). In addition, whilst IL-1β (1h) caused ~5-fold increase in B1R mRNA expression under low LSS conditions, this effect was substantially reduced (~50%) under high LSS (Figure 5A). IL-1β treatment also elevated B1R protein expression; an effect enhanced in cells exposed to low LSS vs high LSS, similarly evident in HAEC (Figure 5B), associated with enhanced LDBK-specific binding (Figure SVIB-C) and inhibited by the NFκB inhibitor BAY 11-7082 (Figure 5C).
Figure 5. IL-1β-induced B1R expression is enhanced under low LSS conditions.

B1R mRNA expression in (A) HUVECs (n=6) or (B) HAECs (n=6) subjected to varying LSS (0-10 dyn/cm2, 12h), then stimulated or not with IL-1β (10ng/mL, 4h) prior or not to treatment with (C) the NFκB inhibitor (BAY 11-7082, 20μM, n=6). (D) Following LSS HUVECs were treated with native LDL (nLDL, 20μg/ml) or oxidized LDL (oxLDL, 20μg/ml, 3h, n=3). * P<0.01, ** P<0.05, nd: non-determined, ns: non-significant.
Confirmation that IL-1β-induced B1R expression was associated with enhanced B1R function was demonstrated by the finding that the elevated CXCL5 and CXCL6 mRNA expression evident in response to IL-1β under low LSS was suppressed by ~50% by the B1R antagonist, SR240612 (Figure 6A-B). Finally, oxLDL caused a >3-fold increase in B1R mRNA expression compared to treatment with native LDL under low but not high LSS conditions (Figure 5D).
Figure 6. IL-1β-induced CXCL5 and CXCL6 expression is mediated by B1R activation and enhanced under low LSS conditions.
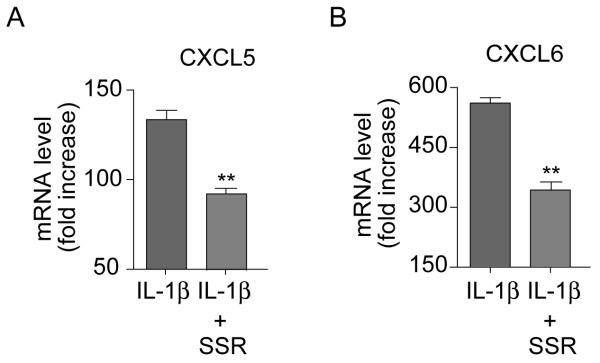
HUVEC were subjected to low LSS for 12h and treated with IL-1β (10ng/mL) for 4h in the absence or presence of the B1R antagonist SSR240612 (1μM, 15 min prior to IL-1β application). (A) CXCL5 or (B) CXCL6 mRNA expression was measured levels normalized to GAPDH. Data are mean ± SEM for n=6, ** for P<0.01.
DISCUSSION
LSS, the uni-directional frictional hemodynamic force, imposed on the endothelial cell surface as a result of blood flow plays a major role in maintaining homeostasis. Herein, we show that low LSS induces expression and functionality of the pro-inflammatory kinin B1R. Moreover, we demonstrate in vitro that inflammatory stimuli and low LSS, combined to mimic the atherogenic environment, synergise to enhance both expression and function of this receptor; a phenomenon also evident at sites of atheroma in both humans and a mouse model of atherosclerosis. Since activation of B1R is a pivotal step in promoting leukocyte recruitment and endothelial permeability in inflammatory responses9,18 and elevated B1R expression is evident at sites of human atheroma7, we propose that the targeting of this receptor represents an exciting prospect for atherosclerotic disease.
Previous evidence has demonstrated that kinin B1R expression is evident in human blood vessels and may be associated with atherosclerosis7. In this study, western blotting of segments of human carotid artery, collected from individuals undergoing endarterectomy, demonstrated a ~3-fold elevation of kinin B1R in those areas associated with substantial atheromatous plaque only. Analysis of the aorta of ApoE-/- mice fed a high fat diet exposed a similar selectivity in localization of B1R expression. Whilst B1R expression was evident basally expressed increased over time, with a near doubling of expression by 12 weeks; an effect not evident in mice fed a chow diet. Comparison of the levels of expression in the aortic arch (a region of substantial atheroma formation) with the longitudinal section of the thoracic aorta (a region of no significant atheroma formation) demonstrated a clear localization of B1R to regions of atheromatous plaque formation (western blotting and uimmunohistochemistry) and, interestingly, at a similar level of intensity evident in human blood vessels (i.e.~3-fold increase). Immunohistochemical analysis also suggested that expression was particularly evident in endothelial cells; the site of LSS sensing in the blood vessel wall (expression was also evident in smooth muscle, as previously reported, and diffuse within the intima likely reflecting inflammatory cell recruitment7).
Indeed, subjecting endothelial cells in culture to low (atherogenic) LSS raised kinin B1R expression above that measured in cells exposed to physiological levels of LSS. That the levels of LSS used accurately reflect atherogenic and physiological levels of LSS was demonstrated by the presence of ‘endothelial dysfunction’ under low LSS, as evidenced by decreased endothelial NO synthesis; a key indicator of this phenomenon in CVD18. Similarly, in the arterial circulation of the mesentery, subjected ex vivo to varying LSS, minimal B1R expression was observed under physiological LSS but a >5-fold increase in expression occurred following exposure to atherogenic LSS. Together, these findings imply a selective upregulation of B1R expression by low atherogenic LSS. An alternative interpretation of these findings, however, is that high LSS represses B1R expression. Indeed, physiological LSS, through specific transcription factor dependent pathways, represses expression of a number of pro-inflammatory proteins in endothelial cells in culture19. However, in the present study B1R expression was suppressed following inhibition of the pro-inflammatory transcription factor NFκB, under low flow conditions, implying an induction by low LSS rather than an inhibition by high LSS.
To investigate whether enhanced expression was associated with enhanced function we measured the expression of downstream inflammatory molecules. Prostaglandins released during inflammation produce local vasodilatation, increasing regional blood flow and microvascular permeability, together facilitating leucocyte infiltration20; and prostaglandins have been implicated in mediating, at least in part, B1R-induced increases in vascular permeability and blood flow in several different vascular beds8,21. PGE2 and PGI2, in particular, are prominent prostaglandins involved in mediating these effects, but are also molecules that have been implicated in atherogenesis22-24. We demonstrated that whilst B1R agonist treatment did not alter PGE2 and PGI2 production by endothelial cells exposed to physiological LSS, both were elevated under low LSS. This effect likely relates to an increase in enzymatic activity since no changes in expression of the principal vascular COX enzymes; COX-1 and -2, was evident. More recently, we have also reported that B1R-induced inflammatory leukocyte recruitment is, at least in part, due to endothelial chemoattractant cytokine ELR-CXCL chemokine, CXCL5/6, synthesis17. In the current study B1R agonist also induced CXCL5 production in cells subjected to low LSS whilst having no effect under high LSS. Indeed, in support of this finding we measured elevated levels of CXCL5 at sites of atheroma formation in ApoE-/- mice. Collectively, these findings intimate that the enhanced expression of B1R under low LSS conditions directly correlates with the enhanced inflammatory phenotype of endothelial cells exposed to B1R agonists.
The kinin B1R promoter possesses several potential shear stress response elements (SSRE) with consensus sequence GAGACC25, Barbie box (CTTT motif) and GAGA (GAGAG motif)26 sites for binding of specific transcription factors, particularly noteworthy being the transcription factor NFκB that binds to GAGACC27. NFκB has been implicated in mediating the enhanced expression of a number of proteins dually regulated by both inflammation and low LSS, including adhesion molecules (E-selectin, VCAM-1)5,6 and chemokines (MCP1, IL8)28-30. In the present study we demonstrate that inhibition of NFκB activation using BAY 11-7082, a selective inhibitor of cytokine-inducible IκBα phosphorylation15,31, inhibited B1R expression in response to IL-1β under both high and low LSS conditions, demonstrating NFκB-dependent for IL-1β-induced B1R expression and low LSS alike.
As mentioned previously inflammation plays a pivotal role in all stages of the atherosclerotic disease process: initiation, progression and plaque rupture1,32. An increasing body of evidence suggests that the prevailing haemodynamic conditions not only alters the expression of inflammatory genes within the endothelium, but also determines the magnitude of the inflammatory response to pathogenic stimuli. Evidence suggests that low LSS is associated with an inflamed phenotype and enhanced responsiveness to diverse inflammatory stimuli including the cytokines IL-1β and TNFα, leading to enhanced expression of adhesion molecules and chemokines (e.g. IL-8 and MCP-1)5,29,33-36 as well as augmented inflammatory cell recruitment33,36. In line with these findings, in the current study, IL-1β produced a greater elevation of B1R expression, reflected by enhanced mRNA expression and agonist binding, and enhanced function in terms of prostaglandin and chemokine CXCL5 synthesis in cells subjected to low LSS compared to high LSS.
This enhanced activity was not limited solely to inflammatory cytokines but also to the molecule currently perceived to be the primary inflammatory stimulus in atherosclerosis: oxLDL37,38. At a concentration in line with those found in patients with CVD, and shown to be pro-inflammatory in endothelial cells12-14, oxLDL substantially elevated kinin B1R expression in endothelial cells exposed to low LSS only. In addition, our studies exposed the existence of a positive loop centered on B1R, whereby the effects produced by the cytokine in combination with low LSS were significantly attenuated by B1R blockade using SR240612. Equally interesting, SR240612 significantly inhibited IL-1β-induced-CXCL6 expression in cells subjected to low LSS, although the B1R agonist did not induce CXCL6 expression in HUVEC. This demonstrates that B1R expression and function was optimal when endothelial cells under low LSS were in a context of inflammation. SR240612 is a selective, non-peptidic antagonist with a Ki of 0.48 nM at B1R and has an estimated pA2 of 9.4 using standard organ bath assays for measurement of antagonist potency11. This antagonist has been tested against >100 other receptors and shows no or negligible activity at concentrations up to 1μM (the concentration we used for our experiments) and is at least 1000 times less potent at the kinin B2R. Thus, these findings clearly demonstrate the pro-inflammatory nature of B1R activation in endothelial cells and are in agreement with our previously published findings in B1R knockout mice17, demonstrating the essential role of the B1/CXCL5 pathway in inflammatory cell recruitment. Together, our data suggest that the contribution of the kinin B1R at sites of inflammation, in terms of both prostaglandins, CXCL5 and CXCL6 expression, is substantially enhanced at sites of low LSS and intimates a potential role for this pathway in the inflammatory events associated with atherogenesis.
A limitation of this work is that most of these data were produced with venular endothelium. However, to mitigate against this criticism, we investigated whether varying LSS could influence B1R expression in HAEC (cells relevant to clinical disease as demonstrated by the high prevalence of aortic lesions in patients i.e. ~60%39,40). Indeed, whilst no expression was evident under basal conditions high LSS was a powerful suppressor of the raised expression under inflammatory conditions, i.e. following IL-1β treatment. These findings intimate that the effect of LSS on endothelial B1R is likely a generalized feature of this cell type irrespective of vessel type.
Recent studies have implicated neutrophil recruitment in atherosclerosis41,42. Depletion of neutrophils, using an anti-PMN antibody, in ApoE-/- mice fed a high fat diet significantly reduced plaque development42. Separate studies have also implicated neutrophil infiltration in promoting erosion and rupture of unstable plaque43,44. Studies investigating the pathways involved in the recruitment of this cell type clearly demonstrate that the interaction between neutrophils and endothelial cells in vivo occurs predominantly at sites of low shear34. Our previous studies have demonstrated an essential role for the kinin B1R in neutrophil recruitment to sites of inflammation, in particular we have demonstrated that Il-1β-induced neutrophil recruitment to the inflamed microvasculature was greatly diminished in B1R knockout mice17,45. Collectively, these data prompt us to speculate that a pathway centred on B1R expression and activation may underlie the neutrophil recruitment recently implicated in the process of atherogenesis in mouse models of disease41,42, although further studies are required to investigate this possibility more fully.
In conclusion, our data suggest that endothelial kinin B1R expression and function is tightly regulated by LSS, with expression being induced by low atherogenic levels of LSS; an effect substantially exacerbated under inflammatory conditions. Furthermore, we have identified a possible role for the kinin B1R in the pathogenesis of inflammatory CVD, particularly atherosclerosis; such findings imply that targeting the B1R pathway may prove beneficial in the therapeutic management of atherosclerotic disease.
Supplementary Material
ACKNOWLEDGEMENTS
Sources of funding: this work was supported by a Wellcome Trust Project Grant. JD was supported by a Basic Science Fellowship of the Barts and the London Charity, AJH by a Wellcome Trust Senior Fellowship, RS by a Wellcome Trust Career Development Award, Sandrine Vessilier by a British Heart Foundation Project Grant, the collection of human tissue was funded by European Community FP6 funding (“Eicosanox”; LSHM-CT-2004-0050333).
We thank Prof Michael Braden for his assistance with measurement of viscosity.
Footnotes
Disclosures: There are no conflicts of interest
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Malek A, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 3.Chien S. Effects of Disturbed Flow on Endothelial Cells. Annals of Biomedical Engineering. 2008;36:554–562. doi: 10.1007/s10439-007-9426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling: Molecular, Cellular, and Vascular Behavior. Journal of the American College of Cardiology. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 5.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr., Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Passerini AG, Shi C, Francesco NM, Chuan P, Manduchi E, Grant GR, Stoeckert CJ, Jr., Karanian JW, Wray-Cahen D, Pritchard WF, Davies PF. Regional determinants of arterial endothelial phenotype dominate the impact of gender or short-term exposure to a high-fat diet. Biochem Biophys Res Commun. 2005;332:142–148. doi: 10.1016/j.bbrc.2005.04.103. [DOI] [PubMed] [Google Scholar]
- 7.Raidoo DM, Ramsaroop R, Naidoo S, Muller-Esterl W, Bhoola KD. Kinin receptors in human vascular tissue: their role in atheromatous disease. Immunopharmacology. 1997;36:153–160. doi: 10.1016/s0162-3109(97)00015-5. [DOI] [PubMed] [Google Scholar]
- 8.Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 9.Bussolari SR, Dewey CF, Jr., Gimbrone MA., Jr. Apparatus for subjecting living cells to fluid shear stress. Rev Sci Instrum. 1982;53:1851–1854. doi: 10.1063/1.1136909. [DOI] [PubMed] [Google Scholar]
- 10.Dewey CF, Jr., Bussolari SR, Gimbrone MA, Jr., Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 11.Gougat J, Ferrari B, Sarran L, Planchenault C, Poncelet M, Maruani J, Alonso R, Cudennec A, Croci T, Guagnini F, Urban-Szabo K, Martinolle JP, Soubrie P, Finance O, Le Fur G. SSR240612 [(2R)-2-[((3R)-3-(1,3-Benzodioxol-5-yl)-3-{[(6-methoxy-2-naphthyl)sulfonyl]amino}propanoyl)amino]-3-(4-{[2R,6S)-2,6-dimethylpiperidinyl]methyl}phenyl)-N-isopropyl-N-methylpropanamide Hydrochloride], a New Nonpeptide Antagonist of the Bradykinin B1 Receptor: Biochemical and Pharmacological Characterization. J Pharmacol Exp Ther. 2004;309:661–669. doi: 10.1124/jpet.103.059527. [DOI] [PubMed] [Google Scholar]
- 12.Itabe H, Ueda M. Measurement of Plasma Oxidized Low-Density Lipoprotein and its Clinical Implications. Journal of Atherosclerosis and Thrombosis. 2007;14:1–11. doi: 10.5551/jat.14.1. [DOI] [PubMed] [Google Scholar]
- 13.Niemann B, Rohrbach S, Catar RA, Muller G, Barton M, Morawietz H. Native and oxidized low-density lipoproteins stimulate endothelin-converting enzyme-1 expression in human endothelial cells. Biochemical and Biophysical Research Communications. 2005;334:747–753. doi: 10.1016/j.bbrc.2005.06.163. [DOI] [PubMed] [Google Scholar]
- 14.Thum T, Borlak J. LOX-1 Receptor Blockade Abrogates oxLDL-induced Oxidative DNA Damage and Prevents Activation of the Transcriptional Repressor Oct-1 in Human Coronary Arterial Endothelium. J Biol Chem. 2008;283:19456–19464. doi: 10.1074/jbc.M708309200. [DOI] [PubMed] [Google Scholar]
- 15.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel Inhibitors of Cytokine-induced Ikappa Balpha Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-inflammatory Effects in Vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 16.Ignarro LJ, Fukuto JM, Griscavage JM, Rogers NE, Byrns RE. Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from L-arginine. Proc Natl Acad Sci U S A. 1993;90:8103–8107. doi: 10.1073/pnas.90.17.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duchene J, Lecomte F, Ahmed S, Cayla C, Pesquero J, Bader M, Perretti M, Ahluwalia A. A novel inflammatory pathway involved in leukocyte recruitment: role for the kinin B1 receptor and the chemokine CXCL5. J Immunol. 2007;179:4849–4856. doi: 10.4049/jimmunol.179.7.4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.GIMBRONE MA, Jr., TOPPER JN, NAGEL TOBI, ANDERSON KR, GARCIA-CARDENA GUIL. Endothelial Dysfunction, Hemodynamic Forces, and Atherogenesis. Ann NY Acad Sci. 2000;902:230–240. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 19.Fledderus JO, van Thienen JV, Boon RA, Dekker RJ, Rohlena J, Volger OL, Bijnens AP, Daemen MJAP, Kuiper J, van Berkel TJC, Pannekoek H, Horrevoets AJG. Prolonged shear stress and KLF2 suppress constitutive proinflammatory transcription through inhibition of ATF2. Blood. 2007;109:4249–4257. doi: 10.1182/blood-2006-07-036020. [DOI] [PubMed] [Google Scholar]
- 20.Williams TJ, Peck MJ. Role of prostaglandin-mediated vasodilatation in inflammation. Nature. 1977;270:530–532. doi: 10.1038/270530a0. [DOI] [PubMed] [Google Scholar]
- 21.McLean PG, Perretti M, Ahluwalia A. Kinin B1 receptors and the cardiovascular system: regulation of expression and function. Cardiovasc Res. 2000;48:194–210. doi: 10.1016/s0008-6363(00)00184-x. [DOI] [PubMed] [Google Scholar]
- 22.Cipollone F, Cicolini G, Bucci M. Cyclooxygenase and prostaglandin synthases in atherosclerosis: Recent insights and future perspectives. Pharmacology & Therapeutics. 2008;118:161–180. doi: 10.1016/j.pharmthera.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 23.McClelland S, Gawaz M, Kennerknecht E, Konrad CS, Sauer S, Schuerzinger K, Massberg S, Fitzgerald DJ, Belton O. Contribution of cyclooxygenase-1 to thromboxane formation, platelet-vessel wall interactions and atherosclerosis in the ApoE null mouse. Atherosclerosis. 2009;202:84–91. doi: 10.1016/j.atherosclerosis.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 24.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane Prostaglandin E Synthase-1: A Novel Therapeutic Target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 25.Resnick N, Collins T, Atkinson W, Bonthron DT, Dewey CF, Gimbrone MA. Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive element. Proc Natl Acad Sci. 1993;90:7908–7900. doi: 10.1073/pnas.90.16.7908-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyakawa AA, Lourdes Junqueira M, Krieger JE. Identification of two novel shear stress responsive elements in rat angiotensin I converting enzyme promoter. Physiol Genomics. 2004;17:107–113. doi: 10.1152/physiolgenomics.00169.2003. [DOI] [PubMed] [Google Scholar]
- 27.Khachigian LM, Resnick N, Gimbrone MA, Collins T. Nuclear factor-kappa B interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J Clin Invest. 1995;96:1169–1175. doi: 10.1172/JCI118106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiu JJ, Chen LJ, Chang SF, Lee PL, Lee CI, Tsai MC, Lee DY, Hsieh HP, Usami S, Chien S. Shear stress inhibits smooth muscle cell-induced inflammatory gene expression in endothelial cells: role of NF-kappaB. Arterioscler Thromb Vasc Biol. 2005;25:963–969. doi: 10.1161/01.ATV.0000159703.43374.19. [DOI] [PubMed] [Google Scholar]
- 29.Partridge J, Carlsen H, Enesa K, Chaudhury H, Zakkar M, Luong L, Kinderlerer A, Johns M, Blomhoff R, Mason JC, Haskard DO, Evans PC. Laminar shear stress acts as a switch to regulate divergent functions of NF-{kappa}B in endothelial cells. Faseb J. 2007;21:3553–3561. doi: 10.1096/fj.06-8059com. [DOI] [PubMed] [Google Scholar]
- 30.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory Shear Stress Stimulates Adhesion Molecule Expression in Cultured Human Endothelium. Circ Res. 1998;82:532–539. doi: 10.1161/01.res.82.5.532. [DOI] [PubMed] [Google Scholar]
- 31.Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappa B induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96:2537–2542. [PubMed] [Google Scholar]
- 32.Ross R. Atherosclerosis -- An Inflammatory Disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 33.Sheikh S, Rahman M, Gale Z, Luu NT, Stone PC, Matharu NM, Rainger GE, Nash GB. Differing mechanisms of leukocyte recruitment and sensitivity to conditioning by shear stress for endothelial cells treated with tumour necrosis factor-alpha or interleukin-1beta. Br J Pharmacol. 2005;145:1052–1061. doi: 10.1038/sj.bjp.0706281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheikh S, Rainger GE, Gale Z, Rahman M, Nash GB. Exposure to fluid shear stress modulates the ability of endothelial cells to recruit neutrophils in response to tumor necrosis factor-alpha: a basis for local variations in vascular sensitivity to inflammation. Blood. 2003;102:2828–2834. doi: 10.1182/blood-2003-01-0080. [DOI] [PubMed] [Google Scholar]
- 35.Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, van Zonneveld AJ, Leenen PJ, van der SA, Serruys PW, de Crom R, Krams R. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest. 2007;117:616–626. doi: 10.1172/JCI28180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shyy YJ, Hsieh HJ, Usami S, Chien S. Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc Natl Acad Sci U S A. 1994;91:4678–4682. doi: 10.1073/pnas.91.11.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braunwald E. Shattuck lecture--cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med. 1997;337:1360–1369. doi: 10.1056/NEJM199711063371906. [DOI] [PubMed] [Google Scholar]
- 38.Ludewig B, Freigang S, Jaggi M, Kurrer MO, Pei YC, Vlk L, Odermatt B, Zinkernagel RM, Hengartner H. Linking immune-mediated arterial inflammation and cholesterol-induced atherosclerosis in a transgenic mouse model. Proc Natl Acad Sci U S A. 2000;97:12752–12757. doi: 10.1073/pnas.220427097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mendel T, Popow J, Hier DB, Czlonkowska A. Advanced atherosclerosis of the aortic arch is uncommon in ischemic stroke: an autopsy study. Neurol Res. 2002;24:491–494. doi: 10.1179/016164102101200230. [DOI] [PubMed] [Google Scholar]
- 40.Meissner I, Khandheria BK, Sheps SG, Schwartz GL, Wiebers DO, Whisnant JP, Covalt JL, Petterson TM, Christianson TJH, Agmon Y. Atherosclerosis of the aorta: Risk factor, risk marker, or innocent bystander?: A prospective population-based transesophageal echocardiography study. Journal of the American College of Cardiology. 2004;44:1018–1024. doi: 10.1016/j.jacc.2004.05.075. [DOI] [PubMed] [Google Scholar]
- 41.van Leeuwen M, Gijbels MJ, Duijvestijn A, Smook M, van de Gaar MJ, Heeringa P, de Winther MP, Tervaert JW. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:84–89. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- 42.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–217. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 43.Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, Maseri A. Widespread coronary inflammation in unstable angina. N Engl J Med. 2002;347:5–12. doi: 10.1056/NEJMoa012295. [DOI] [PubMed] [Google Scholar]
- 44.Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 45.McLean P, Ahluwalia A, Perretti M. Association between kinin B1 receptor expression and leukocyte trafficking across mouse mesenteric post-capillary venules. J Exp Med. 2000;192:367–380. doi: 10.1084/jem.192.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.