

Abstract
Background
Hypereosinophilic syndromes (HES) are chronic disorders that require long-term therapy to suppress eosinophilia and clinical manifestations. Corticosteroids are usually effective, yet many patients become corticosteroid-refractory or develop corticosteroid toxicity. Mepolizumab, a humanised monoclonal anti-interleukin-5 antibody, demonstrated corticosteroid-sparing effects in a double-blind, placebo-controlled study of FIP1L1/PDGFRA-negative, corticosteroid-responsive subjects with HES.
Objective
To evaluate long-term safety and efficacy of mepolizumab (750 mg) in HES.
Methods
MHE100901 is an open-label extension study. The primary endpoint was the frequency of adverse events (AEs). Optimal dosing frequency, corticosteroid-sparing effect of mepolizumab, and development of anti-mepolizumab antibodies were also explored.
Results
Seventy-eight subjects received 1–66 mepolizumab infusions each (including mepolizumab infusions received in the placebo-controlled trial). Mean exposure was 251 weeks (range 4–302). The most common dosing interval was 9–12 weeks. The incidence of AEs was 932 events per 100 subject-years in the first year, declining to 461 events per 100 subject-years after 48 months. Serious AEs, including one death, were reported by the investigator as possibly due to mepolizumab in three subjects. The median daily prednisone dose decreased from 20.0 to 0 mg in the first 24 weeks. The median average daily dose for all subjects over the course of the study was 1.8 mg. Sixty-two percent of subjects were prednisone-free without other HES medications for ≥12 consecutive weeks. No neutralizing antibodies were detected. Twenty-four subjects withdrew prior to study completion for death (n=4), lack of efficacy (n=6), or other reasons.
Conclusion
Mepolizumab was well tolerated and effective as a long-term corticosteroid-sparing agent in PDGFRA-negative HES.
Keywords: eosinophil, monoclonal antibody, interleukin-5, corticosteroid
Introduction
Hypereosinophilic syndromes (HES) are a rare group of disorders characterized by persistent blood eosinophilia >1500/mm3 and evidence of eosinophil-related end-organ pathology.1 Corticosteroids are the mainstay of treatment and are effective, at least initially, in most patients with FIP1L1/PDGFRA-negative HES.2 Long-term corticosteroid therapy is problematic, however, since many patients become refractory to treatment and/or develop drug-related side effects. Conventional second-line agents, including hydroxyurea and interferon-α, are effective in only a subset of patients and are associated with significant toxicity.
Mepolizumab is a fully humanized monoclonal IgG1 antibody specific for human interleukin-5 (IL-5). In a randomized, double-blind, placebo-controlled study (MHE100185), mepolizumab (750 mg given intravenously every 4 weeks for 36 weeks) was safe and effective as a corticosteroid-sparing agent in adults with FIP1L1/PDGFRA-negative HES.3 However, subjects received only a maximum of nine monthly doses of mepolizumab. Since HES is a chronic disease, subjects are likely to require long-term, maintenance therapy. Consequently, long-term safety and efficacy, including the effects of prolonged eosinophil depletion, the development of resistance, and optimal dosing frequency, need to be assessed. The current study was designed as an open-label extension to MHE100185 to address these issues.
Methods
Study population
Study subjects were 18–75 years of age with HES, as defined by >1500 eosinophils/mm3 for ≥6 months, eosinophil-related organ involvement, and no identifiable cause of eosinophilia. Information pertaining to HES history, clinical manifestations, and treatment prior to enrolment in MHE100185 has been reported previously.3
Study design
This open-label, multicenter study (ClinicalTrials.gov, number NCT00097370, Supplemental Table 1) was designed as an extension to a double-blind, placebo-controlled study (MHE100185) conducted from March 2004 to March 2006.3 Subjects who completed 9 months of treatment or withdrew from MHE100185 after receiving at least two doses of study drug were eligible. Exclusion criteria included life-threatening disease or other serious illness, serum creatinine ≥3 times institutional upper limit of normal, aspartate aminotransferase or alanine aminotransferase ≥5 times institutional upper limit of normal, platelet count <50 000/μL, development of abnormal cardiac function within the past 3 months, allergic reaction to study MHE100185 investigational product, or history or suspicion of drug or alcohol abuse.
Subjects receiving >10 mg prednisone daily were enrolled into Stage 1. After two consecutive monthly infusions of mepolizumab 750 mg, subjects progressed to Stage 2 if they met the following criteria: 1) stable dose of concomitant HES medication; 2) absolute eosinophil count (AEC) <600/mm3; and 3) no new or worsening symptoms of HES. Subjects receiving ≤10 mg prednisone at entry were enrolled directly into Stage 2. In Stage 2, mepolizumab dosing was guided by the individual subject’s HES signs and symptoms and AEC, with each 750 mg dose given at least 4 weeks apart and only if signs and symptoms were present or the AEC had risen to >600/mm3. Subjects progressed to Stage 3 when an optimal dosing frequency was selected, as defined by two consecutive intervals between three infusion time points that were equal in length (±7 days).
All subjects underwent clinical and laboratory evaluation, recording of daily prednisone dose and HES medication use, and serum or urine pregnancy testing (in women of childbearing potential) at study baseline, each infusion visit, and the end of study/early withdrawal visit. AEC was assessed weekly during Stages 1 and 2 and prior to each infusion in Stage 3. Complete blood count was performed at baseline and at every infusion visit until Stage 3, when the frequency was decreased to every 3 months. Routine serum chemistries were assessed at baseline, every 2 months in Stage 1, and every 3 months thereafter. Electrocardiograms were performed every 2 months during Stage 1 and every 6 months thereafter. Other measurements of end-organ involvement were performed as clinically indicated.
Safety and efficacy endpoints
The primary endpoint of the study was the overall frequency of adverse events (AEs), which were coded using the Medical Dictionary for Regulatory Activities by System Organ Class and within System Organ Class by preferred term. AEs and serious AEs (SAEs) were summarised by incidence, investigator’s assessment of possible relationship to the study medication, severity, and whether the events led to discontinuation of mepolizumab. Age-gender-adjusted incidence rates from the Surveillance Epidemiology and End Results (SEER) Registry in the United States4 were applied to the person-time (287 total person-years; 136.7 in males) accrued by mepolizumab-exposed subjects to obtain the expected numbers of cancers in this population. SEER is a program of the National Cancer Institute (NCI) and is an authoritative source of information on cancer incidence and survival in the United States. SEER currently collects and publishes cancer incidence and survival data from population-based cancer registries covering approximately 28 percent of the US population.
Secondary endpoints included the proportion of subjects maintaining a daily dose of ≤10 mg prednisone for ≥12 weeks, the proportion of subjects corticosteroid-free for ≥12 weeks, AEC, and the proportion of subjects in each dosing frequency group at the end of Stage 2.
Anti-mepolizumab antibody assay
Blood samples for immunogenicity testing were obtained at the end of study/early withdrawal visit from subjects who received at least three mepolizumab infusions. Samples were screened for anti-mepolizumab antibody using an assay that detects antibodies blocking binding of mepolizumab to human IL-5. The assay was developed and validated using the Meso-Scale Discovery plate-reader at Tandem Laboratories (Weston Trenton, NJ, USA) and has a relative sensitivity of 2 ng/mL. All confirmed positive samples were tested for neutralizing antibody using a ligand competitive binding assay (relative sensitivity 0.4 μg/mL) developed and validated at GlaxoSmithKline (Philadelphia, PA, USA).
Statistical analysis
The intent-to-treat population was defined as all subjects who received at least one infusion of mepolizumab in the current study. Dosing intervals and the effects of anti-mepolizumab antibodies on efficacy were analyzed post-hoc using Wilcoxon matched- pairs signed-rank test and a χ2 test, respectively.
Results
Baseline characteristics of the study population
Seventy-eight of the 80 eligible subjects enrolled in the open-label extension study (table 1). Of these, 37 entered in Stage 1 (prednisone dose >10 mg at entry) and 41 in Stage 2 (prednisone dose ≤10 mg at entry). Eight subjects with a baseline daily prednisone dose ≤10 mg were mistakenly enrolled into Stage 1. Baseline characteristics were similar between the subjects entering in Stage 1 or Stage 2, with the exception of prednisone dose at entry (by definition) and prior exposure to mepolizumab (table 1).
Table 1.
Baseline characteristics of the study population
| Entry at Stage 1 (n=37) | Entry at Stage 2 (n=41) | All subjects (n=78) | |
|---|---|---|---|
| Sex | |||
| Men | 19 (51%) | 23 (56%) | 42 (54%) |
| Women | 18 (49%) | 18 (44%) | 36 (46%) |
|
| |||
| Age, years | 49 (19–69) | 54 (18–75) | 50 (18–75) |
|
| |||
| Race | |||
| White | 29 (78%) | 38 (93%) | 67 (86%) |
| Black | 5 (14%) | 1 (2%) | 6 (8%) |
| Other | 3 (8%)* | 2 (5%) | 5 (6%) |
|
| |||
| Received prior mepolizumab therapy | 7 (19%) | 33 (80%) | 40 (51%) |
|
| |||
| Prednisone dose at entry, mg | 20 (0–50) | 0 (0–10) | 7·5 (0–50) |
|
| |||
| Patients prednisone-free at entry | 2 (5%) | 22 (54%) | 24 (31%) |
|
| |||
| Eosinophil count at entry,/mm3 | 380 (20–5420) | 100 (10–910) | 150 (10–5420) |
Data are number (%) of subjects or median (range).
Other includes Arabic/North African, Asian.
Exposure to mepolizumab
The mean duration of mepolizumab exposure in subjects who received more than one infusion (including prior exposure during MHE100185) was 251 weeks (range 4–302 weeks). Two subjects received only one infusion during MHE100901. One subject developed an urticarial reaction during the first exposure to mepolizumab. The second subject did not require a second infusion due to persistent suppression of HES symptoms and eosinophil count following the first infusion. The mean number of mepolizumab infusions per subject (including those administered during MHE100185) was 25 (range 1–66), and 26 (33%) subjects received more than 30 infusions. Sixty-nine percent of subjects (54/78) were receiving mepolizumab at the study end (figure 1).
Figure 1.
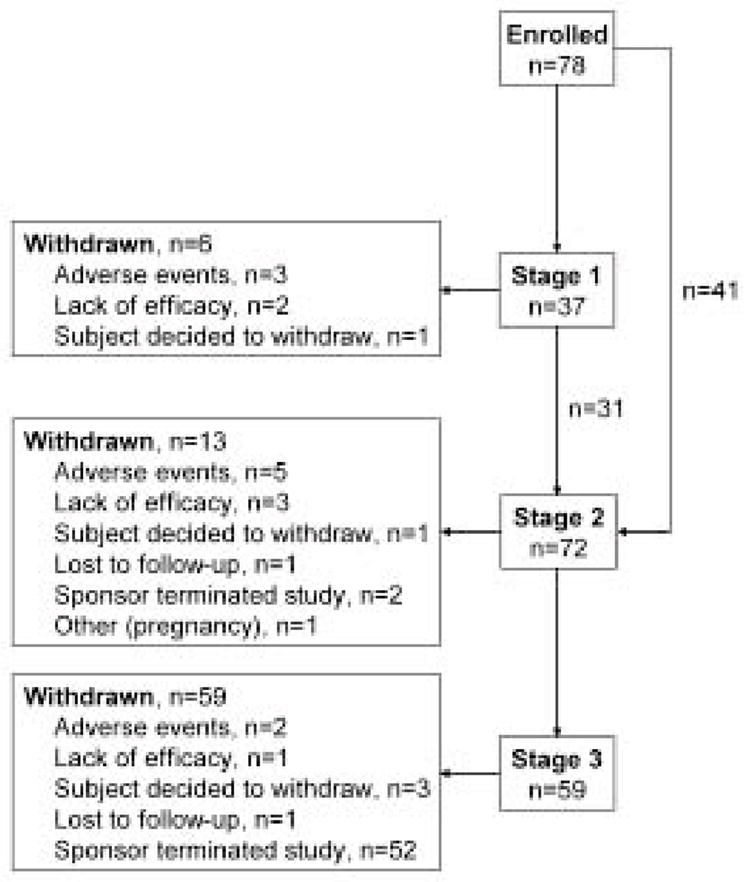
Flow diagram of subject follow-up with transition to different stages. Note: all subjects who were withdrawn due to termination of the study by the sponsor were eligible to continue receiving mepolizumab on a compassionate use protocol.
Safety
Ninety-seven percent of the subjects (76/78) reported at least one AE during the trial, of which cough (33%), fatigue (31%), headache (29%), upper respiratory tract infection (29%), and sinusitis (28%) were most common (Supplemental Table 2). Forty-one subjects experienced at least one severe AE: severe arthralgia and pneumonia were reported for four subjects each, and severe nausea, abdominal pain, dyspnoea, pruritus, and pyrexia were reported for three subjects each. Twenty (26%) subjects experienced AEs reported by the local investigator as possibly related to mepolizumab, of which fatigue (n=6, 8%) and nausea (n=3, 4%) were most common. The incidence of AEs adjusted for exposure was 932 events per 100 subject-years in the first 12 months of the study, declining to 461 events per 100 subject-years after 48 months (table 2 and supplemental figure 1). The incidence of AEs adjusted for exposure was 2127 events per 100 subject-years in the placebo arm of the prior mepolizumab trial (MHE100185).
Table 2.
Adverse event rates per 12 month period adjusted for exposure.
| Period (months) | Control* | 0–12 | 12–24 | 24–36 | 36–48 | >48 |
|---|---|---|---|---|---|---|
|
| ||||||
| Subject-years exposure | 15.0 | 69.0 | 61.1 | 59.2 | 57.8 | 39.9 |
|
| ||||||
| Adverse Events | ||||||
| Number of events | 319 | 643 | 374 | 300 | 349 | 184 |
| Event rate** | 2127 | 931.9 | 612.1 | 506.8 | 603.8 | 461.1 |
|
| ||||||
| Serious AEs | ||||||
| Number of events | 7 | 31 | 28 | 15 | 26 | 7 |
| Event rate** | 46.7 | 44.9 | 45.8 | 25.3 | 45.0 | 17.5 |
Subjects who received placebo in the placebo-controlled, double-blind mepolizumab trial (MHE100185)
Events per 100 subject-years exposure within period.
Fifty-one percent of subjects (n=40) experienced a total of 108 non-fatal SAEs (table 3). The most common of these were pneumonia (n=4) and pyrexia (n=3). Five non-fatal SAEs in three subjects were reported as possibly related to mepolizumab (table 3). Four deaths occurred during the study (table 3), of which one, angioimmunoblastic T cell lymphoma (AITL) with cardiopulmonary failure, was reported as possibly drug-related. A second subject experienced multiple episodes of hypotension ascribed to adrenal insufficiency, and during the final episode, severe vomiting resulted in aspiration pneumonia and respiratory failure. A third subject with a history of atrial fibrillation died suddenly of an unknown cause (no autopsy was performed). A fourth subject died of cardiac failure, sepsis, and multi-organ failure. The incidence of serious AEs adjusted for exposure was 45 events per 100 subject-years in the first 12 months of the study and remained relatively stable over the course of the study (table 2).
Table 3.
Summary of serious adverse events
| Subjects (n=78) | |
|---|---|
|
| |
| Serious adverse events occurring in ≥2 subjects | |
| Pneumonia | 4 (5%) |
| Pyrexia | 3 (4%) |
| Cardiac failure | 2 (3%) |
| Acute cholecystitis | 2 (3%) |
| Diarrhoea | 2 (3%) |
| Dyspnoea | 2 (3%) |
| Eosinophilia | 2 (3%) |
| Prostate cancer | 2 (3%) |
|
| |
| Fatal serious adverse events | |
| Total | 4 (5%) |
| Cardiac failure, sepsis, multi-organ failure | 1 (1%) |
| Aspiration pneumonia, respiratory failure | 1 (1%) |
| Sudden death | 1 (1%) |
| Angioimmunoblastic T cell lymphoma*, cardiopulmonary failure | 1 (1%) |
|
| |
| Non-fatal serious adverse events leading to withdrawal | |
| Total | 3 (4%) |
| Hypotension | 1 (1%) |
| Transverse myelitis* | 1 (1%) |
| Disease progression | 1 (1%) |
|
| |
| Non-fatal serious adverse events possibly related to mepolizumab | |
| Total | 3 (4%) |
| Motor neuron disease* | 1 (1%) |
| Transverse myelitis*, optic neuritis*, spinal disorder* | 1 (1%) |
| Increased T-lymphocyte count* | 1 (1%) |
Data are number (%) of subjects.
Possibly treatment related according to investigator.
Seven subjects withdrew from the study because of a non-fatal AE or SAE (Supplemental Table 2 and Table 3), two of which, transverse myelitis and urticaria, were reported as possibly related to treatment. No other potential drug-related hypersensitivity or infusion reactions associated with mepolizumab treatment were reported. In two cases, the AE leading to withdrawal was related to persistence of HES despite therapy.
During the more than 5 years of the study, a single opportunistic infection was reported (Pneumocystis jiroveci infection in a patient who was infected with HIV during the study). Although systematic screening for viral, bacterial, or parasitic infections was not performed as part of the study, investigators were encouraged to pursue infectious diagnoses where medically appropriate. The incidence of infectious AEs adjusted for exposure decreased over the course of the study from 150 to 100 events per 100 subject-years (Supplemental Table 3).
Seven subjects developed neoplasms: prostate cancer and basal cell carcinoma in two subjects each; mycosis fungoides, multiple myeloma, and fatal AITL in one subject each. Based on the SEER rates from the general population from 2005–2009 (154.8/100,000), the expected number of prostate cancers was 0.21 versus 2 observed. The SEER rate from the general population (2001 to 2009) for multiple myeloma, 5.8/100,000, when applied to the person-time accrued by the trial subjects, gave 0.02 expected cases versus 1 observed. The combined rates for mycosis fungoides and angioimmunoblastic lymphoma, 0.5/100,000, gave 0.001 expected number of cases versus 2 observed.
Five subjects developed non-allergic immune-mediated disorders: rheumatoid arthritis (n=1), polymyalgia rheumatica (n=1), temporal arteritis (n=1), lichen planus (n=1), and autoimmune thrombocytopenia (n=1). Corticosteroid tapering resulted in the development of severe adrenal insufficiency in two subjects, as well as steroid withdrawal syndrome and low serum cortisol, reported as moderate for one subject each. One subject became pregnant twice during the study. The first pregnancy was electively terminated, and the second resulted in the birth of a healthy neonate.
Given the long duration of the study and frequency of laboratory testing, minor laboratory abnormalities were common. However, with the exception of eosinophilia, which occurred by design during the adjustment of infusion intervals in stage 2, laboratory abnormalities of clinical concern were relatively infrequent (Supplemental Table 4). The most common laboratory abnormality flagged for clinical concern was an elevated gamma glutamyl transferase (GGT) level (n=11). It should be noted, however, that the GGT level was elevated at baseline in 6 subjects, was elevated on more than one occasion in only 5 subjects and resolved despite continuation of mepolizumab in 8 subjects. Similarly, the second most commonly flagged laboratory abnormality, elevated bilirubin, was present in all 6 subjects at baseline and resolved in 4 by the end of study.
Efficacy
Prednisone use
The median baseline daily prednisone dose was 20.0 mg (range 0–50 mg) and 0 mg (range 0–10 mg) for subjects entering the study in Stage 1 and Stage 2, respectively. Twenty-four subjects were corticosteroid-free at baseline. Almost all subjects who entered the study with a daily prednisone dose ≤10 mg (44/49; 90%) and most subjects who entered with a daily prednisone dose >10 mg (21/29; 72%) maintained a daily prednisone dose of ≤10 mg for ≥12 weeks as sole therapy for HES (other than mepolizumab). In subjects who previously received placebo in MHE100185, the median daily prednisone dose decreased from 20.0 to 0 mg in the first 24 weeks of the study and remained stable thereafter (figure 2). The median average daily prednisone dose for all 78 subjects over the course of the study was 1.8 mg.
Figure 2.

Initial decline in corticosteroid use in the first 6 months of the study
The median daily prednisone dose is represented at regular intervals (weekly from week 1 to week 12, then monthly thereafter) in patients who had received mepolizumab (closed circles) or placebo (open circles) in the double-blind, placebo-controlled trial prior to their enrollment in the open-label extension.
Forty-eight of 78 subjects (62%) received mepolizumab monotherapy for HES for ≥12 consecutive weeks. Among subjects who participated for over 1 year, 41/67 (61%) received mepolizumab monotherapy for at least 365 consecutive days; among subjects who participated for more than 3 years, 24/60 (40%) received mepolizumab monotherapy for at least 3 years. After the expected increase in the number of subjects who were prednisone-free due to corticosteroid tapering in subjects who entered in Stage 1, the number of subjects who were prednisone-free remained constant at >50% throughout the study (figure 3).
Figure 3.

Proportion of subjects off corticosteroid therapy over the course of the study
The proportion of subjects taking no corticosteroids is shown at 12 week intervals beginning with study entry. The black and gray bars indicate subjects who entered the study in stage 1 (daily prednisone dose greater than 10 mg) and stage 2 (daily prednisone dose less than or equal to 10 mg), respectively.
Of the 19 subjects on prednisone at the time of study termination (median dose 5 mg, range 2.5–13.75 mg), three were receiving corticosteroid therapy for conditions other than HES: rheumatoid arthritis, polymyalgia rheumatica, and temporal arteritis.
Other immunosuppressive medications
Immunosuppressive medications other than corticosteroids were used for treatment of HES in three subjects during the study (azathioprine [n=1], methotrexate [n=2], cyclosporine [n=1], and mycophenolate mofetil [n=1]). Six additional subjects were treated for other disorders as follows: azathioprine for active polyneuropathy (n=1); methotrexate for asthma exacerbation (n=1) and rheumatoid arthritis (n=2); mycophenolate mofetil, cyclophosphamide, and cyclosporine for nephrotic syndrome (n=1); and cyclophosphamide and vincristine for AITL (n=1).
Peripheral blood eosinophilia
AEC measured at 2 or 4 weeks post-mepolizumab infusion in Stage 2 was used as a proxy for long-term efficacy of mepolizumab in suppressing peripheral blood eosinophilia. Stage 2 data were selected for analysis because the AEC was measured weekly, and subjects had achieved a stable dose of concomitant HES therapy. At 2 weeks following mepolizumab infusion, only 7/68 subjects had one or more AEC >500/mm3, and no subject had an AEC >1000/mm3. At 4 weeks post-infusion, four subjects had one or more AEC >500/mm3, all of whom had at least one elevated AEC at 2 weeks post-infusion. Two of these subjects had a single AEC >1000/mm3 (1230/mm3 and 1020/mm3). Overall, mean AEC remained below 500/mm3 in all but one subject during Stage 2 (figure 4).
Figure 4.

Mean absolute eosinophil count (AEC) at 2 and 4 weeks post-mepolizumab infusion. Each symbol represents the mean of all of the AEC measurements for an individual subject at 2 or 4 weeks post-infusion during Stage 2
Dosing interval
Of the 59 subjects who reached Stage 3, 30 (51%) received mepolizumab with a final Stage 2 dosing interval of >12 weeks and six (10%) with an interval of ≥24 weeks (figure 5). The median final Stage 2 dosing interval for all subjects was 12.8 weeks (range 21 days to 37 weeks).
Figure 5.

Histogram of final dosing intervals in Stage 2
The optimal interval between mepolizumab infusions (i.e. optimal dosing frequency) was determined for each patient at the end of stage 2. The bars show the proportion of subjects who received their next mepolizumab infusion at a given interval, ranging from ≤ 4 weeks to > 24 weeks.
Response to mepolizumab therapy
Only six subjects withdrew from the study due to lack of efficacy. The initial dose of mepolizumab was ineffective in lowering the eosinophil count in one subject who had previously received placebo. Five additional subjects withdrew due to persistent HES-related symptoms involving the skin (n=2), respiratory system (n=2), and digestive tract (n=1).
To explore whether the requirement for mepolizumab infusions changed as the duration of treatment increased, the interval between the last two infusions in Stage 2 was compared with the mean interval between the last three infusions in Stage 3 for subjects who received ≥3 infusions in Stage 3. Although the duration between infusions decreased slightly from a median of 90·5 to 86 days (p=0.046), the most common dosing frequency (9–12 weeks) did not change between Stages 2 and 3.
Anti-mepolizumab antibodies were assessed in 136 samples from 69 subjects. Thirty-nine (57%) subjects had at least one positive result with titres ranging from 5 to 1600. A post hoc analysis was performed comparing pre-defined efficacy endpoints in antibody-positive and -negative subjects. The percent of subjects receiving ≤10 mg prednisone (as sole background therapy) at the end of study was comparable between the two groups (32/39 [82%] vs 25/30 [83%]; p=0.889).
Discussion
The primary objective of this open-label extension study was to evaluate the long-term safety of mepolizumab 750 mg at a maximum frequency of once per month in subjects with HES. As in the prior placebo-controlled trial,3 nearly all subjects reported AEs, of which less than one-third were considered possibly related to mepolizumab. The AE profile was also similar. Only sinusitis and cough were reported more frequently in this open-label study. The high overall frequency of AEs likely reflects both a cumulative increase in common intercurrent conditions during the long period of observation and AEs due to clinical manifestations of HES and corticosteroid toxicity. The fact that the incidence rates for AEs adjusted for exposure were similar to those in the placebo group from the earlier trial and decreased over the course of the study supports the latter hypothesis, although a lack of efficacy of mepolizumab in treating selected tissue manifestations of HES cannot be excluded. Indeed, mepolizumab has been shown to be effective in nasal polyposis and severe asthma only when there is evidence of tissue or blood eosinophilia and/or IL-5 expression.5–7
Despite an increased overall frequency of SAEs observed in the open-label extension compared with the earlier placebo-controlled trial (51% vs 16%), the incidence rate of SAEs adjusted for exposure was similar to that of the placebo group from the earlier trial and remained relatively stable over the course of the study. Furthermore, most SAEs (106/112; 95%) were reported as unrelated to mepolizumab by local investigators. Among the four subjects who experienced SAEs possibly related to mepolizumab, two had expansion of previously documented clonal T cell populations. An additional subject developed mycosis fungoides that was reported as unrelated to mepolizumab. Development of T cell lymphoma can occur in up to 5% of patients with HES,8 especially in the setting of an abnormal T cell clone (lymphocytic variant HES (L-HES)).9 In other cases, occult lymphoma is misdiagnosed as HES.10 In the present study, neither of the two subjects who developed T cell lymphoma was classified as L-HES, although both had extremely elevated serum thymus-and-activation-regulated chemokine levels.11 Furthermore, none of the 13 study subjects with L-HES developed lymphoma, although one experienced a marked increase in the number of clonal CD3−CD4+ T cells with prednisone tapering.
Since there was no control arm in the present study, the SEER database4 was used to provide estimates of cancer incidence for comparative purposes. Although the SEER data suggest an increased incidence of prostate cancer and multiple myeloma in the study population, this comparison assumes that the risk of cancer in patients with HES is the same as that of the general population and that this risk is constant over time. These assumptions are likely to be incorrect, since HES is a systemic condition that affects multiple organs, and consequently, many of the study subjects were taking immunosuppressive therapies that could put them at a higher risk for cancer. In addition, the detailed medical surveillance required by the protocol likely increased the efficiency of detection of disease, and, as indicated above, patients with HES are at increased risk of developing lymphoma. Nevertheless, vigilance for the development of malignancies is warranted.
Another major limitation of the current study was that most subjects were receiving or had recently discontinued long-term corticosteroid treatment at the time of enrolment. The effects of corticosteroid tapering on AE interpretation are straightforward in some instances (ie, the development of severe adrenal insufficiency). However, the relative contribution of mepolizumab treatment and/or eosinophil depletion versus corticosteroid tapering to the development of autoimmune and/or lymphoproliferative disorders is less clear. This debate is reminiscent of the controversy over the role of leukotriene antagonists in the emergence of Churg-Strauss syndrome in patients with pre-existing asthma.13
Efficacy endpoints were also affected by prolonged corticosteroid use, since several investigators chose to maintain subjects on low-dose corticosteroid throughout the study to prevent complications of corticosteroid withdrawal, despite complete clinical and haematological response to mepolizumab. Nevertheless, mepolizumab was an effective long-term corticosteroid-sparing agent in a high proportion of subjects. In fact, at the time of study termination, 34/54 (63%) subjects were on mepolizumab monotherapy for HES. Furthermore, there was no evidence of attenuation of response and, although anti-mepolizumab antibodies were detected in 57% of subjects, these antibodies did not have neutralising activity in vitro and there was no evidence that their presence had an effect on prednisone dose at the end of study. Predictors of response could not be assessed due to the small number of non-responding subjects; however, in a subgroup analysis of MHE100185, mepolizumab was equally effective as a corticosteroid-sparing agent in subjects with L-HES as those with idiopathic HES.11
The present study confirms the data from the prior placebo-controlled trial demonstrating the safety and efficacy of mepolizumab as a corticosteroid-sparing agent in HES and extends the findings over a period of time that is clinically relevant in the management of a chronic disease. Nevertheless, difficulties in conducting clinical trials in small patient populations, and persisting challenges, including the validation of biomarkers and/or endpoints for disease characterisation, have hampered the approval of drugs for rare diseases, such as HES.13 In fact, in 2004, only 7.1% of the European Union-designated potential orphan drugs were approved for marketing.14 Whether the findings in the current study move mepolizumab one step closer to this goal remains to be seen.
Clinical Implications.
This study demonstrates that targeted anti-eosinophil therapy with mepolizumab can provide a long-term, well-tolerated alternative to corticosteroid treatment in patients with PDGFRA-negative, corticosteroid-responsive HES.
Acknowledgments
Funding This study (protocol MHE100901) was supported by GlaxoSmithKline, the Division of Intramural Research, NIAID, NIH (ADK), and the Belgian National Fund for Scientific Research (FNRS, grant number 3.4545.11) (FER).
In addition to the authors, the members of the Mepolizumab HES study Group are as follows: Australia — L Coyle (Royal North Shore Hospital, Sydney); Belgium — D Blockmans, G Verhoef (University Hospital Gasthuisberg, Leuven); Canada — K Peters, V Taraska (Winnipeg Clinic, Winnipeg, MB); S Couban, V Dorcas (Dalhousie University and Capital District Health Authority, Halifax, NS); N Shear (University of Toronto, Toronto, ON); France — P Y Hatron (Claude Huriez Hospital, CHRU, Lille); F Ackermann, O Blétry (Service de Médecine Interne, Hôpital Foch, Suresnes); Germany — W Gross, F Moosig (University of Lübeck, Department of Rheumatology and Klinikum Bad Bramstedt, Bad Bramstedt); A Ganser, M Port (Hannover Medical School, Hannover); U Darsow, J Huss-Marp, B Hüttig, C Merkel (Technische Universität München, Munich); Italy — M Baccarani, G Martinelli (Department of Hematology-Oncology “L. and A. Seràgnoli”, University of Bologna, Bologna); United States — T Greene, J Ramsdell (University of California San Diego School of Medicine, San Diego, CA); R Weber (National Jewish Health, Denver, CO); K E Spates, C Talar-Williams (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD); D Codispoti, P F Weller (Beth Israel Deaconess Medical Center, Boston, MA); K A Bachman, J H Butterfield (Mayo Clinic, Rochester, MN); B K Buckmeier Butz (Division of Allergy and Immunology, Department of Pediatrics, Cincinnati Children’s Hospital and Medical Center, Cincinnati, OH); D D Hagaman (Vanderbilt University Medical Center, Nashville, TN); K M Leiferman (University of Utah School of Medicine, Salt Lake City, UT) S L Burton, S L Page (Virginia Commonwealth University, Richmond, VA); W W Busse, S Mathur (University of Wisconsin School of Medicine and Public Health, Madison, WI).
The authors also thank Elaine F Griffin, DPhil, at Evidence Scientific Solutions for editorial support.
Abbreviations
- AEC
absolute eosinophil count
- AE
adverse event
- AITL
angioimmunoblastic T cell lymphoma
- HES
hypereosinophilic syndromes
- Ig
immunoglobulin
- IL-5
interleukin-5
- FIP1L1
Fip-1-like 1
- L-HES
lymphocytic variant HES
- NCI
National Cancer Institute
- PDGFRA
platelet-derived growth factor receptor alpha
- SAE
serious adverse event
- SEER
Surveillance Epidemiology and End Results
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, et al. Contemporary consensus proposal on criteria and classification of eosinophil disorders and related syndromes. J Allergy Clin Immunol. 2012 doi: 10.1016/j.jaci.2012.02.019. [EPub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogbogu PU, Bochner BS, Butterfield JH, Gleich GJ, Huss-Marp J, Kahn JE, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009;124:1319–25.e3. doi: 10.1016/j.jaci.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, Simon HU, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358:1215–28. doi: 10.1056/NEJMoa070812. [DOI] [PubMed] [Google Scholar]
- 4.Howlander N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, et al., editors. Vintage 2009 Populations. National Cancer Institute; Bethesda, MD: SEER Cancer Statistics Review, 1975–2009. http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, April 2012. [Google Scholar]
- 5.Gevaert P, Van Bruaene N, Cattaert T, Van Steen K, Van Zele T, Acke F, et al. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J Allergy Clin Immunol. 2011;128:989–95.e8. doi: 10.1016/j.jaci.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 6.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–84. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–93. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 8.Simon HU, Plotz SG, Dummer R, Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. 1999;341:1112–20. doi: 10.1056/NEJM199910073411503. [DOI] [PubMed] [Google Scholar]
- 9.Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am. 2007;27:389–413. doi: 10.1016/j.iac.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Romero Fernández E, Pardo JR, Doyle A, Albendea MC, de la Rúa AR. Hemophagocytic syndrome associated NK/T nasal type lymphoma presenting as hypereosinophilic syndrome: a case report and literature review. Leuk Res. 2011;35:e97–9. doi: 10.1016/j.leukres.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Roufosse F, de Lavareille A, Schandené L, Cogan E, Georgelas A, Wagner L, et al. Mepolizumab as a corticosteroid-sparing agent in lymphocytic variant hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126:828–35.e3. doi: 10.1016/j.jaci.2010.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lilly CM, Churg A, Lazarovich M, Pauwels R, Hendeles L, Rosenwasser LJ, et al. Asthma therapies and Churg-Strauss syndrome. J Allergy Clin Immunol. 2002;109:S1–19. doi: 10.1067/mai.2002.120854. [DOI] [PubMed] [Google Scholar]
- 13.Stolk P, Willemen MJC, Leufkens HGM. Rare essentials: drugs for rare diseases as essential medicines. Bull World Health Organ. 2006;84:745–51. doi: 10.2471/blt.06.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joppi R, Bertele V, Garattini S. Orphan drug development is progressing too slowly. Br J Clin Pharmacol. 2006;61:355–60. doi: 10.1111/j.1365-2125.2006.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]