

Abstract
Aims
To assess the safety and pharmacokinetics of a new synthetic ozonide antimalarial, OZ439, in a first-in-man, double-blind study in healthy volunteers.
Methods
OZ439 was administered as single oral daily doses of a capsule formulation (50–1200 mg) or an oral dispersion (400–1600 mg, fed and fasted states) and for up to 3 days as an oral dispersion (200–800 mg day−1). Plasma concentrations of OZ439 and its metabolites were measured by LC-MS.
Results
The pharmacokinetic (PK) profile of OZ439 was characterized by a tmax of around 3 h, followed by a multiphasic profile with a terminal half-life of 25–30 h. The PK parameters were approximately dose proportional for each group and profiles of the metabolites followed a similar pattern to that of the parent compound. Following dosing for 3 days, accumulation was less than two-fold but steady-state was not achieved. In the presence of food, no effect was observed on the t1/2 of OZ439 while the exposure was increased by 3 to 4.5-fold. Exposure was higher and inter-subject variability was reduced when OZ439 was administered as an oral dispersion compared with a capsule. The urinary clearance of OZ439 and its metabolites was found to be negligible and OZ439 did not induce CYP3A4. The antimalarial activity profiles of a subset of serum samples suggested that the major antimalarial activity originated from OZ439 rather than from any of the metabolites.
Conclusion
The safety and pharmacokinetic profile of OZ439 merits progression to phase 2a proof of concept studies in the target population of acute uncomplicated malaria.
Keywords: healthy subjects, OZ439, phamacokinetics, safety, synthetic ozonide
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
OZ439 is a synthetic peroxide antimalarial drug candidate that exhibits exceptional antimalarial and pharmacokinetic properties in vitro and in animal models. It is fast-acting against all asexual erythrocytic Plasmodium falciparum stages, with IC50 values comparable with those for the clinically used artemisinin derivatives. Unlike all other synthetic peroxides and semisynthetic artemisinin derivatives, OZ439 completely cures Plasmodium berghei-infected mice with a single oral dose of 20 mg kg−1 and exhibits prophylactic activity superior to a range of currently used antimalarials including that of mefloquine.
WHAT THIS STUDY ADDS
We here report the first administration of OZ439 to human volunteers. The paper presents original data on the tolerability and pharmacokinetics of single doses from 50 to 1600 mg and repeated doses of 200–800 mg for 3 days. These results confirm the predicted prolonged blood concentrations of OZ439 and demonstrate that it is the first endoperoxide drug that has the appropriate profile to become part of a single dose antimalarial treatment.
Introduction
Malaria is a parasitic disease that threatens half of the world's population; 3.3 billion people living in 109 countries are potentially affected [1]. The World Health Organization (WHO) have declared malaria control a global development priority and estimate that in 2009 there were 225 million cases of malaria worldwide, 93% caused by Plasmodium falciparum [2]. In the same year there were 781 000 deaths caused by malaria and 89% of the mortality was in Africa, with children less than 5 years of age attributing 85% of deaths worldwide [2]. These statistics are similar to those reported for the year 2006 [3]. In addition, 70 to 80 million cases of relapsing Plasmodium vivax malaria occur per year adding to the morbidity even if not to the mortality [4].
While global efforts have led to significant progress in the prevention, diagnosis and management of malaria, a number of potential threats to progress still exist, including resistance to insecticides and antimalarial medicines and a lack of alternative treatments [2].
To combat increasing levels of resistance, the WHO currently recommends artemisinin based combination therapy (ACT) as the first line therapy in areas with high prevalence of resistance, whereas chloroquine (CQ) and primaquine (PQ) are the treatments of choice against CQ sensitive P. vivax malaria [2]. The antimalarial peroxide artemisinin and its derivatives are the most potent and rapidly acting antimalarial agents available to date [5] with high parasite kill rates and broad-stage activity producing more rapid clinical and parasitological response than other classes of antimalarial agents. However, due to their short half-life, artemisinins have been associated with high recrudescence rates when used as monotherapy with 3–5 days of treatment [6]. Combination of artemisinin derivatives with other longer acting agents has allowed for shorter courses of ACT therapy, improved treatment outcomes and enhanced patient compliance.
Like artemisinin and its derivatives, ozonides have been proposed to react with Fe2+ in the parasite to produce free radicals leading to alkylation of key parasitic proteins [7]. The first synthetic ozonide, arterolane (RBx11160/OZ277), was identified in a collaborative drug discovery project [8] and found to be safe and well tolerated in humans [9]. Arterolane has been launched in combination with piperaquine phosphate in India by Ranbaxy Laboratories for the treatment of P. falaciparum malaria as a 3 day treatment. However there remains some concern surrounding low exposure of the drug in patients infected with P. falciparum [10]. Because of this finding, a programme to identify and develop new ozonides without this potential liability was initiated. OZ439 (mesylate salt) is a new, orally active, synthetic ozonide that has been shown in both in vitro (P. falciparum) and in vivo (rodent; P. berghei) models to have potential value as a peroxide anti-malarial agent [11]. In preclinical animal models, OZ439 exhibited a prolonged exposure profile resulting in improved efficacy relative to arterolane, artesunate and other comparator antimalarials in murine malaria models (11).
The primary objective of the current study was to investigate the safety and tolerability of single and multiple escalating oral doses of OZ439 in healthy male and female volunteers. The secondary objectives were to characterize the single and multiple dose pharmacokinetic properties of OZ439 and its metabolites and to investigate the effect of food on the PK profile following a single oral dose.
Methods
A phase I, single centre, multipart, double-blind, randomized, placebo-controlled study in healthy male and female subjects was conducted. The study was conducted at Comprehensive Phase One Miramar (3400 Enterprise Way, Miramar, FL 33025, USA) and sponsored by the Medicines for Malaria Venture (MMV), Switzerland. The study protocol was reviewed and approved by an independent ethics committee (Independent IRB, Inc., 6738 West Sunrise Blvd, Suite 102, Plantation, Florida 33313, USA) and carried out under a US Investigational New Drug application (IND). Written informed consent was obtained from each subject prior to the start of any protocol-specific procedures and after full explanation.
Subjects
Healthy, non-smoking, male or female (non-pregnant or lactating) subjects, as determined by pre-study medical history, physical examination (including body temperature) and 12-lead electrocardiogram (ECG), who were between 18 and 55 years of age, inclusive, with body mass index between 18 and 30 kg m−2, inclusive, and body weight >60 kg were eligible for enrolment. Subjects were not allowed to consume alcohol within the 72 h period preceding any dosing period and were to refrain from caffeine, xanthine and grapefruit-containing products for 7 days prior to screening and during admission and follow-up.
The human dose equivalent to the lowest no observed adverse effect level (NOAEL) in dogs (20 mg kg−1 day−1) was calculated to be 10.8 mg kg−1 [12]. Introducing a safety factor of 10-fold, the maximum recommended starting dose was therefore 1.08 mg kg−1, equating to 64.8 mg for a 60 kg human or 75.6 mg in a 70 kg human. Conducting these same calculations based on the NOAEL in the rat (30 mg kg−1 day−1) led to a maximum recommended starting dose of 0.48 mg kg−1 or 29 mg for a 60 kg human (34 mg for a 70 kg human). Based on these considerations, the proposed starting dose was 50 mg. Using the same 10-fold safety factor and the predicted human pharmacokinetic parameters, a similar starting dose was obtained by calculating the anticipated human dose that would give an exposure level (i.e. AUC) comparable with that at the NOAEL in animals.
Study design
Part A – Single rising dose (SRD) escalation
Single oral doses of OZ439 (50–1600 mg OZ439 mesylate) were investigated in a partial crossover, ascending dose study comprising three cohorts (planned eight subjects each). Subjects in cohorts 1 and 2 were enrolled in an alternating panel design. Each subject was randomized to a treatment sequence in which she/he received doses of OZ439 (administered as capsule(s) or as an oral dispersion) on four occasions and placebo on one occasion over a period of at least 9–11 weeks. Progress from one cohort to the next occurred once safety and PK data of the previous cohort were reviewed. A third cohort was added to investigate further the administration of OZ439 as an oral dispersion. All doses were administered at least 1 week apart. Doses were administered after an overnight fast of at least 8 h and standardized meals were commenced from 3 h post-dosing.
Part B – Food effect (FE)
The effect of food was investigated in an additional group of 12 healthy subjects in a two cohort, single dose, open label, two period crossover design, separated by a washout period of at least 7 days. Six subjects were randomized to each sequence group (fasted/fed or fed/fasted). Subjects received a single dose of 800 mg OZ439 as an oral dispersion under either fed or fasted conditions. Both groups were fasted for at least 8 h overnight on the day prior to dosing. In the fed group, dosing occurred 30 min after the start of consumption of a standard high fat meal [13]. Standardized meals were commenced from 3 h post-dosing, in both groups.
Part C – Multiple rising dose (MRD) escalation
Multiple oral doses of OZ439 (200–800 mg) administered as an oral dispersion once daily for 3 days under fasted conditions were investigated in sequential groups of eight healthy subjects (six on active and two on placebo). Subjects were fasted for at least 8 h prior to dosing in the morning, and standardized meals were commenced from 3 h post-dosing, on each dosing day.
Formulations
Two oral formulations of OZ439 were used in the trial; one was a capsule in which OZ439 mesylate powder was filled into gelatine capsules with no additional excipients and the other was an oral dispersion formulation. Capsules were administered with 240 ml water, and the oral dispersion was prepared just prior to dosing by dispersing OZ439 powder in a bottle in either 30 (50–400 mg dose), 60 (800 mg dose), or 120 (1600 mg dose) ml of vehicle comprised of polysorbate 80 (0.8% v/v), sucralose (0.5% w/v), ethyl vanillin (0.05% w/v) and water. The entire volume was administered after which the bottle was rinsed first with 30 ml and then 100 ml water with both volumes also administered to the subjects.
Pharmacokinetic sampling
Blood samples for the assay of OZ439 and its metabolites were collected prior to dosing and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 60, 72 and 96 h post-dose (SRD) and prior to dosing and at 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 24, 36, 48, 72 and 96 h post-dose (FE). For the MRD study, samples were collected on day 1 pre-dose and 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 24 and 36 h post-dose and day 3 pre-dose and 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 24, 36, 48, 72 and 96 h post-dose. Blood samples (5 ml) were collected into tubes containing potassium EDTA as anticoagulant and kept chilled on crushed ice. Plasma was separated within 30 min of sample collection by centrifugation at 4°C. Plasma samples were immediately transferred to polypropylene tubes and stored frozen at −25°C until analysis.
Urine was collected in the SRD study and pooled during the intervals 0–12 h, 12–24 h, 24–36 h and 36–48 h post-dosing. Immediately after collection of each void (total urine volume recorded), a 5 ml urine aliquot from the pooled sample was transferred into a polypropylene tube and 2.5 ml of 100% ethanol (2 : 1, v/v) was added to ensure full, quantitative recovery of OZ439. Samples were stored frozen at or below −25°C prior to assay for OZ439 and its metabolites.
During the MRD study, bioactivity measurements were conducted in a subset of subjects using serum samples collected at 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after administration of a single oral dose (three subjects receiving 200 mg, two subjects receiving 800 mg) of OZ439. Blood samples (2.5 ml) were collected into serum separation tubes (Greiner Vacuette Chemistry Serum Gel Evacuated Tubes no. 454243). The blood was allowed to clot for 30 min on crushed ice (to prevent degradation of the compound) after which the tubes were centrifuged at 4°C and 1500–2000 × g for 15 min. Serum (duplicate aliquots of approximately 300 μl each) was immediately transferred to polypropylene tubes and stored at −70/−80°C prior to analysis.
Bioanalytical methods
Plasma and urine OZ439 and metabolite concentrations were determined using reversed phase liquid chromatography coupled with tandem mass spectrometry operating in the selected reaction monitoring mode.
Chemicals and materials
OZ439 mesylate (Figure 1) was synthesized by Unimark Remedies Ltd, Kerala, India and OZ567, OZ579, OZ580, OZ564 and OZ577 (Figure 1) were obtained from Professor Jonathan Vennerstrom, University of Nebraska Medical Center, Nebraska, USA. HPLC grade solvents of ethanol, methanol, 2-propanol and formic acid were purchased from J. T. Baker, Deventer, the Netherlands. Working solutions of the analytes were prepared from a mixture by serial dilutions in ethanol to concentrations 20 times higher than the corresponding concentrations in the plasma calibration standards. The concentrations were calculated with consideration of purity and the salt factor where applicable. The calibration range was 1.00 to 1000 ng ml−1 for each of the analytes in plasma.
Figure 1.

Structure of OZ439 and its potential metabolites
Sample preparation
To an aliquot of 20 μl human plasma, 100 μl of a mixture of acetonitrile : methanol (4:1, v/v) containing the internal standard (OZ543) was added. After vortex mixing for a few seconds, the samples were centrifuged for about 10 min at approximately 50 000 g at a temperature setting of 8°C. Aliquots of approximately 75 μl of supernatant were transferred to empty autosampler vials and diluted using 75 μl of a mixture of water : acetonitrile (1:1, v/v). An aliquot of 2 μl of the diluted supernatant was injected onto the HPLC column.
Liquid chromatography and mass spectrometry
HPLC was performed using a 1200 binary pump (Agilent Technologies Inc, Santa Clara, CA, USA) which was coupled to a TSQ Quantum Access triple stage quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) operated with Xcalibur and LCquan. The mobile phase consisted of water containing 0.1% formic acid (phase A) and acetonitrile containing 0.1% formic acid (phase B). For chromatographic separation, a Hydrosphere C18, 2.1 × 33 mm, 3 μm column (YMC Co. Ltd, Kyoto, Japan) was used and the content of mobile phase B was increased from 2.0% to 25.0% over 3.3 min, from 25.0% to 30.0% over 2 min, from 30.0% to 65.0% over 2.5 min and further increased from 65.0% to 95.0% over 0.1 min. The composition was held for 0.9 min before reconditioning at the initial conditions. The heated electrospray ion source was used in positive ion mode and data were acquired in the selected ion monitoring mode (SRM) using the following parent and product ions: the SRM transitions were m/z 470.3 to m/z 304.1 for OZ439, m/z 444.3 to m/z 278.1 for OZ564, m/z 486.3 to m/z 320.1 for OZ577, m/z 484.3 to m/z 318.1 for OZ543 (internal standard), m/z 486.3 to m/z 304.1 for OZ567 and OZ579 and m/z 502.3 to m/z 304.1 for OZ580, AA2 and AA3.
The precision, expressed as the coefficient of variation, of the assay was shown to be 5.9–9.3% for plasma OZ439, and ranged between 5.9% and 11.8% for the metabolites. Accuracy was between 98.4 and 101.0% for plasma OZ439 and between 96.3 and 102.5% for the metabolites. Similar precision and accuracy were demonstrated for the analysis of urine samples. The limit of quantification (LOQ) was 1.00 ng ml−1 for OZ439 and its metabolites in both plasma and urine1.
Ex vivo bioactivity assay
For a subset of serum samples, a P. falciparum bioassay was performed with the laboratory strain NF54 to determine the combined antimalarial activity of OZ439 and possible metabolites. The resulting bioactivity data were directly compared with the respective concentrations of OZ439 determined by LC-MS/MS. The bioassay was conducted as described previously [14–16] and was based on the [3H]-hypoxanthine incorporation assay [15] using O+ erythrocytes.
CYP3A4 induction assay
Aliquots (20 ml) of pooled urine samples (0–24 h) from day 1 and day 3 were assayed for the 6β-hydroxycortisol : free cortisol ratio and compared with a day 1 pre-dose sample to look for evidence of CYP3A4 induction.
Safety assessment
Standard safety assessments were carried out at appropriate pre-determined time points during the study and included recording of adverse events, haematology, serum chemistry, coagulation assays, urinalysis, vital signs and 12-lead ECG. In the SRD and MRD parts of the study, continuous lead II ECG monitoring was performed for a minimum of 4 h after dosing and QTc interval was determined from read out of the ECG machine. The ECG readings were reviewed by the Investigator and any significant abnormality referred for review by a single independent cardiologist. Audiometry assessment (vestibular, middle ear and cochlear function, otoscopy and pure tone audiometry) and brainstem auditory evoked potentials were performed during the screening period and at 10 (±2) days after the last dose of study medication. Safety and tolerability results were analyzed descriptively only. Toxicity of laboratory results was assessed based on the WHO Toxicity Grading Scale for Determining the Severity of Adverse Events.
Data and statistical analysis
PK variables, including the maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration–time curves (AUC), the apparent terminal rate constant (λz) and corresponding half-life (t1/2) for OZ439 were derived by non-compartmental analysis using WinNonlin Professional Version 5.2.1 (Pharsight Corporation, Mountain View, CA, USA). For the SRD (day 1 for each period), FE and MRD (day 1 only) studies, AUC(0,t) and AUC(0,∞) were calculated, where AUC(0,t) is the area under the plasma concentration–time curve to the last measurable time point and AUC(0,∞) is the area under the plasma concentration–time curve extrapolated to infinity. For the MRD study, the AUC within the dosing interval (AUC(0,24 h)) on day 1 and day 3 was calculated. The total amount of OZ439 and its metabolites excreted in urine from time zero to 48 h after dosing (Ae(0,48 h)) was calculated. The primary PK variables for assessing dose proportionality following single dose administration were AUC(0,t) (capsule formulation), AUC(0,∞) (oral dispersion) and Cmax, and after multiple dosing were AUC(0,24 h) and Cmax on day 3. A non-linear power model was used to assess dose proportionality. The proportional relationship between each parameter and dose was written as a power function: variable = a × doseb where a is a constant and b is the proportionality constant of the variable of interest. Dose proportionality was also explored graphically by plotting dose-normalized AUC and Cmax against dose.
The primary PK variables for assessing the effect of food (part B) were AUC(0,∞) and Cmax. An equivalence approach was used to investigate whether a statistically relevant difference existed in the PK of OZ439 when administered under fasted or fed conditions. The PK variables AUC and Cmax were log(e) transformed prior to fitting a model. Fixed effect terms for sequence, state (fed/fasted) and treatment period and a random effect for subject were fitted in the model. Least squares geometric means were calculated with the ratio of least squares geometric means (fed/fasted) for AUC and Cmax and 90% confidence limits (CL) for the ratio. In the MRD study, the time to attain steady-state plasma concentrations was assessed visually from plots of mean pre-dose concentrations against sampling day. On multiple dosing, an effect of gender was explored graphically by plotting individual dose-normalized values for Cmax and AUC(0,24h).
Results
Subjects
Single rising dose
A total of 26 subjects were enrolled in the SRD study, 10 in cohort 1, eight in cohort 2 and eight in cohort 3. Four subjects did not complete the study (three due to AEs and one due to a positive drug screen result at period 4 admission). The majority of subjects were male (80.8%) and White (88.5%). All were Hispanic/Latino, with a mean age of 35.6 years (range 21 to 55 years).
Effect of food
A total of 12 subjects received 800 mg OZ439 as an oral dispersion, both fed and fasted, in part B of the study. The majority of subjects were female (69%) and White (92.3%). All were Hispanic/Latino and the mean age was 33.7 years (range 20 to 48 years).
Multiple rising dose
A total of 24 subjects were enrolled in and completed the multiple dose component, six placebo subjects and six subjects each in the 200, 400 and 800 mg OZ439 treatment groups. The majority of subjects were male (62.5%), White (87.5%) and Hispanic/Latino (91.7%), with a mean age of 40.1 years (range 21 to 55 years).
Pharmacokinetics
Visual inspection of the individual plasma concentration–time profiles of OZ439 and of its metabolites indicated a prolonged profile with variability between subjects, particularly for the capsule formulation (individual subject data not shown). The terminal phase was apparent from 48 to 60 h after dosing. Where AUC could be extrapolated to infinity, the extrapolated area generally represented less than 10% of the total AUC.
Single rising dose; capsule formulation
The mean plasma concentration–time profiles of OZ439 administered as an oral capsule to fasted subjects (Figure 2A) were characterized by a median tmax that varied from 3.0 to 5.0 h across the dose groups, with mean Cmax values ranging from 17.4 ng ml−1 (50 mg dose) to 701 ng ml−1 (1200 mg dose) (Table 1). Following attainment of Cmax, plasma concentrations decreased in a multiphasic manner; the mean terminal t1/2 of OZ439 varied from 27.9 to 31.6 h for the 800 and 1200 mg dose levels, respectively (Table 1). Twenty-four hours after dosing, mean OZ439 plasma concentrations were above 20 ng ml−1 at the 800 mg dose and remained above 10 ng ml−1 for 36 h.
Figure 2.

Arithmetic mean plasma concentration vs. time profiles of OZ439 in healthy subjects after administration of (A) a single dose (n = 6–8 per group) of 50, 100, 200, 400, 800 or 1200 mg (capsule) under fasted conditions, (B) a single dose (n = 5–6 per group) of 400, 800 or 1600 mg (oral dispersion) under fasted conditions, (C) a single dose (n = 12 per group) of 800 mg (oral dispersion) under fasted and fed conditions and (D) multiple doses (n = 6 per group) of 200, 400 or 800 mg (oral dispersion) once a day for 3 days under fasted conditions. Error bars represent SDs. (A) •, 50 mg; ▿, 100 mg; ▪, 200 mg; ◊, 400 mg; ▴, 800 mg; ○, 1200 mg; (B) •, 400 mg; ▿, 800 mg; ▪, 1800 mg; (C) •, fasted; ▿, fed; (D) •, 200 mg; ▿, 400 mg; ▪, 800 mg
Table 1.
Plasma PK variables of OZ439 and its metabolites in healthy fasted subjects after administration of a single dose of 50, 100, 200, 400, 800 or 1200 mg of OZ439 (capsule). Values represent the geometric mean (CV%) for all parameters with the exception of tmax which is the median (range)
| Dose of OZ439 | n | Cmax (ng ml−1) | tmax (h) | AUC(0,t) (ng ml−1 h) | AUC(0,∞)* (ng ml−1 h) | t1/2* (h) |
|---|---|---|---|---|---|---|
| OZ439 | ||||||
| 50 mg | 8 | 17.4 (84.7) | 3.00 (2.00–6.00) | 102 (133.7) | NE | NE |
| 100 mg | 8 | 34.2 (173.5) | 5.00 (3.00–12.00) | 249 (145.4) | NE | NE |
| 200 mg | 8 | 102 (82.9) | 3.50 (3.00–6.00) | 890 (89.1) | NE | NE |
| 400 mg | 7 | 135 (164.9) | 3.00 (2.00–6.00) | 1130 (186.6) | NE | NE |
| 800 mg | 8 | 315 (121.8) | 3.00 (2.00–4.00) | 3010 (115.7) | 4690 (43.5) | 27.9 (24.1) |
| 1200 mg | 6 | 701 (154.7) | 3.00 (2.00–6.00) | 6530 (172.4) | 9130 (136.8) | 31.6 (46.9) |
| OZ579 | ||||||
| 50 mg | 8 | 11.3 (75.0) | 4.00 (3.00–4.00) | 75.3 (88.5) | NE | NE |
| 100 mg | 8 | 17.9 (137.2) | 4.00 (3.00–8.00) | 141 (224.8) | NE | NE |
| 200 mg | 8 | 42.2 (69.1) | 4.00 (3.00–6.00) | 472 (95.1) | 743 (10.7) | 27.1 (41.8) |
| 400 mg | 7 | 50.0 (119.5) | 4.00 (3.00–8.00) | 587 (126.2) | 1060 (11.5) | 27.6 (10.1) |
| 800 mg | 8 | 94.5 (81.2) | 4.00 (3.00–4.02) | 1260 (79.4) | 1610 (44.8) | 24.3 (27.4) |
| 1200 mg | 6 | 141 (61.7) | 4.00 (3.00–6.00) | 2090 (83.3) | 2660 (81.9) | 31.6 (29.6) |
| OZ580 | ||||||
| 50 mg | 8 | 26.4 (50.4) | 4.00 (3.00–4.00) | 294 (65.5) | NE | NE |
| 100 mg | 8 | 32.9 (93.2) | 6.00 (4.00–8.00) | 407 (122.9) | NE | NE |
| 200 mg | 8 | 73.6 (64.4) | 6.00 (4.00–8.00) | 1240 (88.7) | NE | NE |
| 400 mg | 7 | 74.4 (86.1) | 4.00 (4.00–8.00) | 1290 (106.9) | NE | NE |
| 800 mg | 8 | 127 (70.1) | 6.00 (4.00–8.00) | 2660 (70.4) | 3700 (39.4) | 13.6 (20.1) |
| 1200 mg | 6 | 166 (66.6) | 7.00 (4.00–8.00) | 3650 (75.1) | 3730 (76.2) | 16.7 (46.5) |
| AA3 | ||||||
| 50 mg | 8 | 8.80 (53.4) | 4.00 (3.00–6.00) | 77.5 (80.3) | NE | NE |
| 100 mg | 8 | 12.3 (94.7) | 6.00 (3.00–8.00) | 135 (130.7) | NE | NE |
| 200 mg | 8 | 26.5 (67.1) | 6.00 (4.00–8.00) | 404 (112.9) | NE | NE |
| 400 mg | 7 | 28.3 (93.2) | 4.00 (3.00–8.00) | 420 (129.1) | NE | NE |
| 800 mg | 8 | 44.5 (67.0) | 6.00 (4.00–6.00) | 903 (69.7) | 1430 (27.3) | 12.7 (31.4) |
| 1200 mg | 6 | 55.4 (65.1) | 6.00 (4.00–8.00) | 1220 (80.2) | 1790 (60.3) | 21.1 (36.9) |
The number of subjects may differ for the variables AUC(0,∞) and t1/2. NE, not estimated.
The shape of the plasma concentration−time profiles of the metabolites generally resembled that of parent compound (profiles not shown). The exposure based on AUC(0,t), was highest for OZ580 followed by, in descending order, OZ579, AA3 (Table 1) and the minor metabolites AA2, OZ567, OZ564 and OZ577 (data not shown). The minor metabolites had AUC(0,t) values which were less than approximately 10% of the value for OZ439. The median tmax for OZ579 was similar compared with OZ439, whereas that of the other metabolites tended to be longer (up to 8 h; Table 1). The mean t1/2 of OZ579 (24.3 to 31.6 h) was in the same range as that for OZ439.
The intersubject variability, as measured by the coefficients of variation (CV%) of the PK variables, was high for OZ439 (Table 1) following administration of the capsule formulation. The calculated intra- and inter-subject variabilities of OZ439 for Cmax, 77.4 and 79.5%, respectively and AUC(0,t), 92.6 and 71.5%, respectively, were also high (OCS Biometric Support; data not shown).
Single rising dose; oral dispersion
Following administration of OZ439 as an oral dispersion, the shape of the mean plasma concentration-time profile (Figure 2B) and the mean terminal t1/2 (25.2 to 31.2 hours; Table 2) of OZ439 were both similar to that seen after administration of the capsule formulation. Values for median tmax were also similar (3 to 5 h, Table 2), but the measured plasma concentrations were higher with the oral dispersion. At a dose of 800 mg, values for Cmax (917 ng ml−1) and AUC(0,t) (9630 ng ml−1 h) were 2.9- and 3.2-fold greater, respectively, following administration of OZ439 as an oral dispersion compared with a capsule. For the oral dispersion, mean plasma concentrations were above 10 ng ml−1 for 36, 60 and 96 h at doses of 400, 800 and 1600 mg, respectively. The shape of the plasma concentration−time profiles of the metabolites again resembled that of parent compound (profiles not shown), with a similar profile for median tmax as seen after capsule administration. The mean t1/2 of the metabolites tended to be in the same range as that of the parent compound (Table 2). As for OZ439, exposure to the metabolites was higher after intake of an oral dispersion compared with a capsule.
Table 2.
Plasma PK variables of OZ439 and its metabolites in healthy fasted subjects after administration of a single dose of 400, 800 or 1600 mg of OZ439 (oral dispersion). Values represent the geometric mean (CV%) for all parameters with the exception of tmax which is the median (range)
| Dose of OZ439 | n | Cmax (ng ml−1) | tmax (h) | AUC(0,t) (ng ml−1 h) | AUC(0,∞)* (ng ml−1 h) | t1/2* (h) |
|---|---|---|---|---|---|---|
| OZ439 | ||||||
| 400 mg | 5 | 566 (53.9) | 3.00 (2.00–4.00) | 5430 (72.0) | 5540 (71.3) | 31.2 (33.5) |
| 800 mg | 6 | 917 (35.6) | 3.00 (3.00–6.00) | 9630 (56.9) | 9790 (56.7) | 25.2 (25.8) |
| 1600 mg | 6 | 1340 (44.5) | 5.00 (2.00–6.00) | 17500 (70.1) | 18400 (68.0) | 30.7 (41.9) |
| OZ579 | ||||||
| 400 mg | 5 | 147 (31.6) | 4.00 (2.00–4.00) | 2120 (32.6) | 2210 (33.4) | 26.5 (21.0) |
| 800 mg | 6 | 165 (48.1) | 3.50 (3.00–6.00) | 2360 (35.5) | 2520 (35.2) | 28.8 (19.9) |
| 1600 mg | 6 | 193 (28.6) | 4.00 (3.00–4.00) | 3570 (15.5) | 4070 (17.4) | 38.0 (34.1) |
| OZ580 | ||||||
| 400 mg | 5 | 171 (38.2) | 6.00 (4.00–8.00) | 3680 (34.1) | 3790 (39.6) | 15.7 (28.5) |
| 800 mg | 6 | 153 (62.6) | 5.00 (4.00–8.00) | 3430 (45.6) | 3490 (45.3) | 15.6 (34.9) |
| 1600 mg | 6 | 145 (56.1) | 5.00 (4.00–8.00) | 4080 (24.8) | 4490 (22.7) | 30.3 (44.3) |
| AA3 | ||||||
| 400 mg | 5 | 63.0 (46.5) | 6.00 (4.00–8.00) | 1360 (37.4) | 1650 (14.8) | 14.2 (12.9) |
| 800 mg | 6 | 58.4 (68.2) | 4.00 (3.00–6.00) | 1290 (53.7) | 1530 (45.8) | 18.8 (25.2) |
| 1600 mg | 6 | 52.4 (57.0) | 5.00 (4.00–8.00) | 1610 (26.8) | 1830 (28.1) | 33.1 (51.6) |
The number of subjects may differ for the variables AUC(0,∞) and t1/2.
The intersubject variability (CV%) of the OZ439 and metabolite PK variables was lower after intake of OZ439 as an oral dispersion when compared with a capsule (Table 2 and Table 1). Similarly, the calculated intra- and inter-subject variabilities of OZ439 for Cmax, 14.0 and 41.8% respectively, and AUC(0,∞), 23.2 and 57.2% respectively, were lower than those for the capsule formulation (OCS Biometric Support; data not shown).
Single rising dose: dose proportionality
A graphical exploration of dose proportionality of OZ439 based on dose-normalized AUC is shown in Figures 3A and 3B for the capsule and oral dispersion formulations. Both appear not to deviate from dose proportionality to a major extent. The dose proportionality constant, β (95% CI), for the capsule formulation was 1.23 (1.01, 1.45) and 1.09 (0.90, 1.28) for AUC(0,t) and Cmax, respectively. Results of the statistical analysis for the oral dispersion gave values for β (95% CI) of 0.89 (0.47, 1.32) and 0.56 (0.29, 0.83) for AUC(0,∞) and Cmax, respectively, indicating dose proportionality for AUC(0,∞) but not for Cmax.
Figure 3.
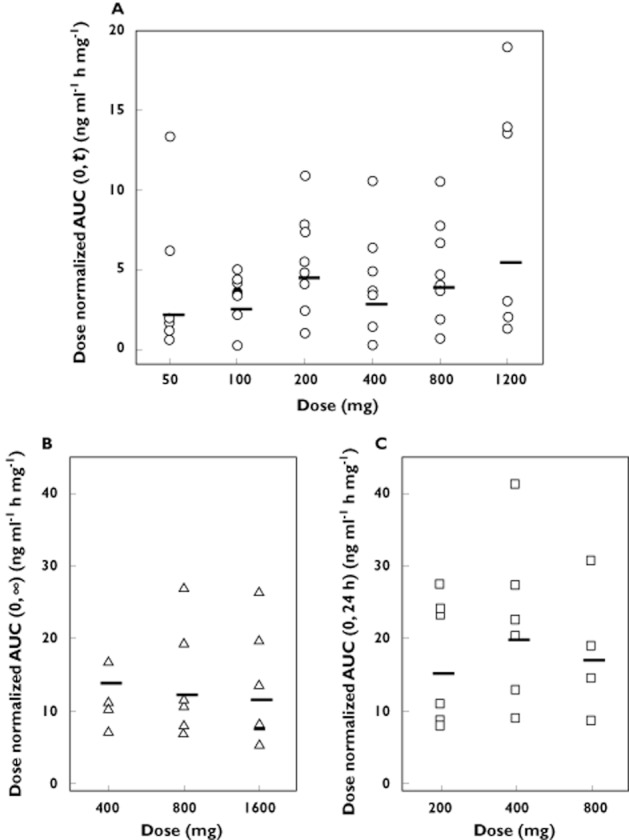
Individual dose normalized values for OZ439 AUC after administration of (A) single doses of 50, 100, 200, 400, 800 or 1200 mg (capsule), (B) single doses of 400, 800 or 1600 mg (oral dispersion) and (C) multiple doses of 200, 400 or 800 mg once daily for 3 days (oral dispersion) to healthy subjects (n = 5–8 per group). Horizontal lines represent the geometric mean for each dose group
Single rising dose: urinary excretion
Except for OZ580, the urinary concentrations of OZ439, OZ564, OZ567, OZ577, OZ579, AA2, and AA3 were low or below the limit of quantitation (BLQ) indicating that renal excretion of these analytes was negligible (data not shown). Concentrations of OZ580 in urine were above the LOQ at the end of the 48 h collection interval in all subjects, regardless of treatment, indicating that the collection interval was too short to capture the total urinary excretion. The mean value for the amount excreted in urine from time 0 h to 48 h after dosing (Ae(0,48 h)) was 2.01 mg of OZ580 at a dose of 1600 mg, which corresponds to 0.12% of the administered dose.
Effect of food
Following administration of a dose of 800 mg OZ439 (oral dispersion) in the fasting condition, the mean plasma concentration−time profile of OZ439 was characterized by a median tmax of 3.0 h followed by a multiphase elimination (Figure 2C and Table 3). In the presence of food, the absorption of OZ439 was marginally delayed, median tmax was 5.0 h and the exposure to OZ439 was markedly increased. Values for Cmax and AUC(0,∞) were approximately 3-fold greater compared with fasting subjects (Table 3). There was no effect of food on the t1/2 of OZ439. In both the fasted and fed states at a dose of 800 mg, mean plasma concentrations remained above 10 ng ml−1 for more than 96 h (Figure 2C). The effect of food on the PK profiles of the metabolites was broadly similar (Table 3). In general, in the presence of food the inter-subject variability (CV%) of the PK variables was lower compared with the fasted state (Table 3). The effect of food on Cmax and AUC(0,∞) was statistically significant, with geometric mean ratios (and 90% CI) of 3.05 (2.23, 4.16) and 2.96 (2.29, 3.83) respectively, falling well outside the bioequivalence limits of 0.80 to 1.25 [13].
Table 3.
Plasma PK variables of OZ439 and its metabolites in healthy subjects after administration of a single dose of 800 mg OZ439 (oral dispersion) under fasted and fed conditions. Values represent the geometric mean (CV%) for all parameters with the exception of tmax which is the median (range)
| Food status | n | Cmax (ng ml−1) | tmax (h) | AUC(0,t) (ng ml−1 h) | AUC(0,∞)* (ng ml−1 h) | t1/2* (h) |
|---|---|---|---|---|---|---|
| OZ439 | ||||||
| Fasted | 12 | 730 (61.1) | 3.00 (2.00–5.00) | 7590 (64.7) | 8080 (68.7) | 38.8 (50.2) |
| Fed | 12 | 2220 (52.6) | 5.00 (2.00–7.00) | 23100 (48.9) | 23900 (49.0) | 31.7 (30.1) |
| OZ579 | ||||||
| Fasted | 12 | 148 (42.7) | 4.00 (3.00–7.00) | 2230 (41.8) | 2480 (44.7) | 34.1 (35.6) |
| Fed | 12 | 232 (30.8) | 5.00 (4.00–6.00) | 4180 (30.4) | 4810 (34.0) | 38.4 (34.3) |
| OZ580 | ||||||
| Fasted | 12 | 166 (45.7) | 5.00 (4.00–10.0) | 3730 (41.2) | 4230 (23.3) | 20.5 (15.9) |
| Fed | 12 | 206 (40.7) | 6.00 (4.00–10.0) | 5870 (38.0) | 6290 (38.5) | 25.7 (23.2) |
| AA3 | ||||||
| Fasted | 12 | 58.9 (43.9) | 5.00 (4.00–7.00) | 1380 (40.1) | 1540 (22.8) | 21.4 (23.4) |
| Fed | 12 | 75.6 (43.4) | 5.50 (4.00–10.0) | 2230 (36.8) | 2410 (37.0) | 23.7 (30.1) |
The number of subjects may differ for the variables AUC(0,∞) and t1/2.
Multiple rising dose
Following multiple dose administration of the oral dispersion formulation (200, 400 or 800 mg OZ439 once daily for 3 days), the mean plasma concentration–time profile of OZ439 was characterized by a median tmax of 3.0 h followed by a multiphase elimination (Table 4 and Figure 2D). The mean t1/2 of OZ439 varied from 37.8 to 41.7 h after the third dose (Table 4). Twenty-four hours after dosing on day 3, mean OZ439 plasma concentrations were above 20 ng ml−1 at all dose levels (Figure 2D) and remained above 30 ng ml−1 for more than 96 h at the 800 mg day−1 dose level. Consistent with the observed trough concentrations was the observation that OZ439 did not accumulate to a major extent, less than 2-fold after 3 days of dosing (Table 4 and Figure 3C). However, steady-state conditions had not been attained on day 3 of dosing, as evidenced by higher trough concentrations on day 4 compared with day 3.
Table 4.
Plasma PK variables of OZ439 in healthy subjects after multiple dose administration of 200, 400 or 800 mg once a day for 3 days of OZ439 (oral dispersion). Values represent the geometric mean (CV%) for all parameters with the exception of tmax which is the median (range)
| Dose of OZ439 | Day | Cmax (ng ml−1) | tmax (h) | AUC(0,24 h) (ng ml−1 h) | t1/2* (h) | Rac |
|---|---|---|---|---|---|---|
| OZ439 | ||||||
| 200 mg | 1 | 314 (55.9) | 3.50 (2.00–5.00) | 2640 (60.6) | NE | |
| 400 mg | 1 | 505 (40.8) | 3.50 (2.00–5.00) | 4560 (48.5) | NE | |
| 800 mg | 1 | 1130 (53.4) | 3.00 (2.00–4.00) | 9570 (51.8) | NE | |
| 200 mg | 3 | 342 (52.5) | 3.00 (2.00–5.00) | 3060 (60.1) | 41.7 (18.2) | 1.16 (18.7) |
| 400 mg | 3 | 764 (46.6) | 3.00 (2.00–5.00) | 7990 (57.5) | 40.8 (31.4) | 1.76 (29.9) |
| 800 mg | 3 | 1390 (51.7) | 3.00 (2.00–5.00) | 13700 (46.0) | 37.8 (28.6) | 1.43 (26.0) |
The number of subjects may differ for the variable t1/2.
NE, not estimated.
The plasma concentration–time profiles of the metabolites resembled that of the parent compound (profiles not shown). Similar to OZ439, the metabolites did not accumulate to a major extent and some (OZ577 and OZ580) did not accumulate at all. The exposure, based on AUC(0,24 h) on day 3, was highest for OZ439 followed by (in descending order) OZ580, OZ579, AA3, OZ567, AA2, OZ564 and OZ577.
The inter-subject variability after multiple dose administration of OZ439 as an oral dispersion, as measured by the CV% of the PK variables, was comparable with that after single dose administration of the oral dispersion formulation (Table 4 and Table 2). The calculated inter-subject variabilities for Cmax and AUC(0,24 h), of 48.9 and 54.1% respectively, were also similar to those after single-dose administration.
Graphically, the dose-normalized AUC of OZ439 up to and including a dose of 800 mg for 3 days appeared dose proportional (Figure 3C). Statistical analysis confirmed dose proportionality, as evidenced by values of β (95% CI) for AUC(0,24 h) of 1.08 (0.63, 1.53) and for Cmax of 1.01 (0.60, 1.42).
Bioactivity measurements
The antimalarial activity profiles of serum samples collected from five healthy subjects after administration of a single oral dose (three subjects at 200 mg, two subjects at 800 mg, all dosed with the oral dispersion) of OZ439 were approximately 15% (with a maximum of 30%) higher compared with the respective concentrations of OZ439 determined by LC-MS/MS analysis (data not shown). IC50 values were in general in the range of 1.3−2.0 ng ml−1, the average value being 1.5 ng ml−1, matching well with the IC50 values that were obtained when pre-dose serum, spiked with a known amount of OZ439, was titrated into the P. falciparum assay (1.7 ng ml−1, data not shown).
CYP3A4 induction assay
No significant changes were observed in the 24-hour urinary 6β-hydroxycortisol : cortisol ratio either by comparing the change in the ratio from baseline with that on day 1 and day 3 or by comparison on those days with the ratios in the placebo group (data not shown). Although no evidence of autoinduction was seen in this study, the dosing duration was relatively short for the impact to be fully assessed.
Safety
Single rising dose
The overall incidence of AEs was highest in the 1600 mg OZ439 oral dispersion group (5/6 subjects) and ranged from 0–2 subjects out of 6–8 in the other groups. In the 1600 mg oral dispersion group, the only AEs experienced by more than two subjects were diarrhoea (3/6 subjects), nausea (3/6 subjects) and gastrointestinal hypermotility (2/6 subjects). All were rated as related to study drug. In all other dose groups, no specific AE was experienced by more than one subject. The only other AEs considered to be related to study drug were abdominal discomfort (1/6 subjects; 1600 mg oral dispersion), dyspepsia (1/17 subjects; placebo), headache (1/8 subjects; 200 mg capsule and 1/6 subjects; 1200 mg capsule) and vasovagal syncope (1/8 subjects; 800 mg capsule).
All AEs were mild or moderate in severity except for one event of first degree atrioventricular block (pre-dose placebo; unrelated) and one of vasovagal syncope in one subject which was considered severe and led to premature discontinuation. One further subject had clinically significant ECG changes, experiencing two episodes of sinus tachycardia (1 h 40 min and 2 h 40 min, respectively) after receiving 800 mg OZ439 capsule and an episode of sinus arrest 3 h 9 min after dosing. Two further subjects were discontinued, one due to an SAE of influenza after dosing with placebo (considered unrelated to study drug) and one following a positive drug screen prior to the fourth period. No clinically meaningful changes were observed in mean clinical laboratory parameters, ECG findings, physical examinations, vital signs or audiometry/BAEP findings. Clinically significant increases in creatine kinase were recorded in a single subject (highest value 1950 U l−1) following OZ439 200 mg capsule and OZ439 800 mg capsule doses which were recorded as mild/moderate AEs. This subject had undertaken recreational physical exercise prior to admission to the corresponding dosing period. The adverse events resolved with hydration and rest and were classified as not related to study drug.
Effect of food
OZ439 was generally well-tolerated in both the fed and fasted regimens. All AEs were mild or moderate in severity. Three of 12 subjects experienced a total of nine AEs in the fed regimen and 4/12 subjects experienced a total of three AEs in the fasted regimen. The only AE reported by more than one subject in either regimen was headache (2/12 subjects in the fed regimen, 1/12 subjects in the fasted regimen, all possibly treatment related). Nausea and oropharyngeal pain (1/12 subjects each) in the fasted regimen were also possibly treatment related. Overall, there were no clinically meaningful changes in the other safety parameters measured, nor clinically significant differences between the fed and fasted regimens. One subject out of 12 had clinically significant increases in creatine kinase results during follow-up on days 18 and 19 which were recorded as an AE of moderate severity not related to study drug and resolved by day 26.
Multiple rising dose
Three of six subjects in the 200 mg group, 1/6 subjects in the 400 mg group and 4/6 subjects in the 800 mg group experienced AEs during the study. The only AEs reported by more than one subject in any group were headache (2/6 subjects in the 200 mg group) and flushing (2/6 in the 800 mg group). All AEs were considered to be mild or moderate in severity. Flushing and throat irritation were the only AEs considered to be related to study drug which occurred in more than one subject (2/6 for each AE in the 800 mg group). Treatment-related AEs experienced by only one subject included one event each of nausea and dizziness in the 200 mg group and one event of nausea in the 800 mg group. No clinically meaningful findings, nor differences between treatment groups, were observed for clinical laboratory parameters, ECG findings, physical examinations vital signs or audiometry/BAEP findings, and there was no significant effect on QTc interval.
Discussion
This first-in-man study was designed to explore the safety, tolerability, and pharmacokinetics of the synthetic ozonide OZ439 after oral administration to healthy subjects.
The SRD and MRD parts of the study employed an alternating panel and sequential group design, respectively. These designs permitted the safety of a lower dose of OZ439 to be evaluated before proceeding to a higher dose. Pharmacokinetic data were also reviewed before proceeding to a higher dose. In the SRD part, six single dose levels were investigated which allowed an estimation of dose proportionality. The starting dose was 50 mg and the maximum single dose to be administered was not to exceed 1600 mg. Doses were reviewed for safety and pharmacokinetics before proceeding to the next higher dose level. Predefined dose escalation criteria were provided in the protocol.
Based on preclinical data, this clinical study of OZ439 focused on the routine monitoring of subjects for clinical signs, haematology (red blood cell count, haematocrit, haemoglobin, reticulocyte count, differential leucocyte count, RBC indices and coagulation parameters) and indicators of haemolysis, liver function tests (ALT, AST, GGT), kidney function tests (BUN and creatinine), ECG (including QTc), electrolyte estimation (sodium, chloride and phosphorus) and urinalysis. Continuous ECG monitoring was conducted at all doses in the SRD study and in at least the two higher doses in the MRD study.
After both single and multiple dose administration, the plasma concentration–time profiles of OZ439 showed that maximum plasma concentrations were reached approximately 3 h after administration and thereafter, OZ439 concentrations decreased in a multiphasic way. Ninety-six hours after a single administration, the mean OZ439 concentration was above 10 ng ml−1 at a dose of 800 mg of the oral dispersion (Figure 2C) (note that preclinical studies indicated that plasma concentrations need to remain above 10–20 ng ml−1 for approximately two parasite cycles to be efficacious). The terminal t1/2 of OZ439 of approximately 30 h is 15-fold greater than that reported previously for the peroxide antimalarial dihydroartemisinin (DHA, t1/2 < 1–2 h) following oral administration of either artesunate [17–19] or DHA [20, 21] to healthy volunteers. Exposure to OZ439 (based on both Cmax and AUC) increased with increasing dose, being approximately dose proportional or deviating only slightly from dose proportionality after both single and multiple dose administration of the oral dispersion formulation.
Following single dose oral administration of OZ439, plasma concentrations were higher and variability was lower following administration of an oral dispersion formulation compared with a capsule formulation containing no excipients. Furthermore, administration of the oral dispersion formulation in the presence of food led to a further increase in exposure, reduction in variability and a delay in tmax from 3 to 5 h, with no effect on the t1/2 of OZ439. Collectively, these results suggest that formulation excipients are necessary to aid in the dispersion and dissolution of OZ439 to ensure reproducible absorption and that administration in the presence of food further increases the solubilization of OZ439 within the gastrointestinal milieu. Studies are currently underway to develop an oral formulation which provides high and reproducible exposure with a reduced food effect.
Following multiple dose administration of the oral dispersion, the mean t1/2 of OZ439 varied from 37.8 to 41.7 h after 3 days dosing. Steady-state conditions were not attained following 3 days of dosing, as evidenced by higher trough concentrations on day 4 compared with day 3. Ninety-six hours after dosing on day 3, OZ439 plasma concentrations were approximately 17 ng ml−1 at a dose of 400 mg, a concentration which exceeds the in vitro IC50 value against P. falciparum by approximately 10-fold [11].
The shapes of the plasma concentration–time curves for the metabolites were generally similar to that of the parent compound, and all but three metabolites had relatively low exposure compared with OZ439. However, as the volumes of distribution of the metabolites is not known, relative plasma exposure may not be reflective of relative total body expposure. As the exposure (i.e. AUC) of OZ439 increased with increasing dose, with the oral dispersion (compared with the capsule) or in the presence food, there was a trend towards a reduction in the metabolite : parent AUC ratio possibly indicating a degree of saturable metabolism that occurs with increasing concentration of the parent compared.
For the principle metabolites (OZ580, OZ579 and AA3), there was minimal antimalarial activity with in vitro IC50 values against P. falciparum being more than 10-fold higher than that of OZ439 (data not shown). The antimalarial activity profiles of serum samples collected from five healthy subjects after intake of a single oral dose (200 or 800 mg oral dispersion) of OZ439 were very similar to the respective pharmacokinetic profiles of OZ439 determined by LC-MS/MS analysis, suggesting that the major antimalarial activity originates from parent OZ439 and not from any of its metabolites.
Following single dose administration, the renal excretion of OZ439 and most metabolites was negligible. The total amount of OZ580 (the highest concentration metabolite in urine) excreted in urine could not be calculated due to the collection interval of 48 h being too short. Only 0.12% of an administered dose of 1600 mg was recovered in urine as OZ580 during this collection interval, indicating that renal excretion is a minor pathway in the elimination of OZ439 and its metabolites.
The safety assessments used in this study were standard and generally recognized as reliable, accurate and relevant for a phase I study. The study included routine monitoring of subjects for clinical signs, haematology parameters and indicators of haemolysis, liver function tests, kidney function tests, ECG (including QTc), electrolyte estimation and urinalysis.
Although there was no preclinical evidence of central nervous system toxicity in the 14 day toxicology or central nervous system safety pharmacology studies conducted with OZ439, artemisinin anti-malarial drugs, which share the same peroxide pharmacophore as OZ439, have been shown to be associated with ototoxicity in some animal models. In addition, there has been a recent report of a retrospective study of adults with uncomplicated malaria treated with lumefantrine–artemether developing a minor degree of change on audiometric testing compared with control subjects when in a noisy environment [21]. Audiometry was therefore carried out before and after dosing in the single dose and repeat dose parts of the study and no clinically significant changes were seen.
In the SRD part of the study, single doses of OZ439 were safe and well tolerated from 50 mg up to 1200 mg given as a capsule and as an oral dispersion up to 1600 mg. Only one subject who received OZ439 was prematurely discontinued due to an AE (vasovagal syncope). This event was preceded by two episodes of sinus tachycardia and was most likely a vagally driven sinus arrest that was associated with a drop in blood pressure (coinciding with standing soon after a blood draw). The sinus arrest occurred 3 h 9 min after the subject received 800 mg OZ439 (the third administration of the sequence). The PK properties of OZ439 suggest that this event occurred at around the tmax and so an association with OZ439 cannot be ruled out, especially as the subject appeared to have no underlying cardiac condition. The duration of the sinus arrest would have been sufficient to cause the syncope. However, this event was an isolated incident and no other subjects who received this or higher doses exhibited anything similar. Follow-up of this subject by a cardiologist confirmed the possibility of underlying sick sinus syndrome which might provoke the reaction after taking the study medication. The final assessment from the cardiologist was that the event was an isolated vasovagal event with no underlying pathology.
In the food effect part of the study, a single dose of 800 mg OZ439 was safe and well tolerated given as an oral dispersion either in the fasted or fed state. In the multiple dose part of the study, OZ439 doses of 200, 400 and 800 mg were safe and well tolerated during once daily dosing for 3 days.
OZ439 did not induce the levels of CYP3A4, as evidenced by no significant changes in the 24 h urinary 6β-hydroxycortisol : cortisol ratio either by comparing the change in the ratio from baseline with that on day 1 and day 3 or by comparison on those days with the ratios in the placebo group. Therefore it is probable that OZ439 will not affect the exposure of any co-administered medications that are CYP3A4 substrates via induction of CYP3A4. Previous in vitro studies in human hepatic microsomes also indicated that OZ439 had a low potential to inhibit the major CYP450 isoforms.
In all three parts of the study, no clinically meaningful results were observed for clinical laboratory parameters, physical examinations, vital signs or ECG findings (with the exception of one subject who received OZ439 [vasovagal syncope] and one who received placebo [atrioventricular block] in the SRD study) and in particular, there appeared to be no effect on the QTc interval. No effect on audiometry/BAEP was observed.
In conclusion, the pharmacokinetic and safety profile of OZ439 in both single and multiple dose studies in healthy subjects provides confidence to progress this agent to patient studies in the target population of acute uncomplicated malaria. This study demonstrated that OZ439 was safe and well tolerated at dose levels that provided prolonged plasma exposure with concentrations exceeding the estimated minimum effective concentration for ∼96 h. Furthermore, the prolonged exposure profile of OZ439 considerably exceeds that of available peroxide antimalarials, thereby having the potential to result in an improved efficacy profile. The variability seen with the capsule formulation and the effect of food on the exposure profile are currently being addressed through reformulation and the authors are confident that the food effect seen in the phase 1 study can be overcome with appropriate formulation management.
Acknowledgments
We thank all volunteers and the staff at Comprehensive Phase One, Miramar, FL., USA. We are grateful to all the funders of MMV who enabled our work to develop this exciting novel compound to change the face of this devastating disease.
Footnotes
The assay was not validated for metabolites AA2 and AA3 as reference samples were not available. Concentrations of these metabolites were estimated using the calibration curve for OZ580 since these three metabolites would be expected to exhibit similar MS response factors.
Competing Interests
JJM and SD are employed by Medicines for Malaria Venture (MMV).
References
- 1.Global Malaria Action Plan. The Roll Back Malaria Partnership. Geneva: World Health Organization; 2008. Available at http://www.rollbackmalaria.org/gmap (last accessed November 2010) [Google Scholar]
- 2.World Malaria Report 2010. Geneva: World Health Organization; 2010. (ISBN 978 92 4 156410 6) [Google Scholar]
- 3.World Malaria Report 2008. Geneva: World Health Organization; 2008. (ISBN 978 92 4 156369 7) [Google Scholar]
- 4.Karunaweera ND, Wijesekera SK, Wanasekera D, Mendis KN, Carter R. The paroxysm of Plasmodium vivax malaria. Trends Parasitol. 2003;19:188–193. doi: 10.1016/s1471-4922(03)00036-9. [DOI] [PubMed] [Google Scholar]
- 5.Ashley EA, White NJ. Artemisinin-based combinations. Curr Opin Infect Dis. 2005;18:531–536. doi: 10.1097/01.qco.0000186848.46417.6c. [DOI] [PubMed] [Google Scholar]
- 6.Bunnag D, Viravan C, Looareesuwan S, Karbwang J, Harinasuta T. Clinical trial of artesunate and artemether on multidrug resistant falciparum malaria in Thailand. A preliminary report. Southeast Asian J Trop Med Public Health. 1991;22:380–385. [PubMed] [Google Scholar]
- 7.Meshnick SR, Taylor TE, Kamchonwongpaisan S. Arteminisin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol Rev. 1996;60:301–315. doi: 10.1128/mr.60.2.301-315.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Tomas JS, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 9.Valecha N, Looareesuwan S, Martensson A, Abdulla SM, Krudsood S, Tangpukdee N, Mohanty S, Mishra SK, Tyagi PK, Sharma SK, Moehrle J, Gautam A, Roy A, Paliwal YK, Kothari M, Saha N, Dash AP. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis. 2010;51:684–691. doi: 10.1086/655831. [DOI] [PubMed] [Google Scholar]
- 10.Ollario P, Wells TNC. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther. 2009;85:584–595. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 11.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FCK, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FDA Guidance for Industry. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, 2005.
- 13.FDA Guidance for Industry. Food-Effect Bioavailability and Fed Bioequivalence Studies, 2002.
- 14.Teja-Isavadharm P, Watt G, Eamsila C, Jongsakul K, Li Q, Keeratithakul D, Sirisopana N, Luesutthiviboon L, Brewer TG, Kyle DE. Comparative pharmacokinetics and effect kinetics of orally administered artesunate in healthy volunteers and patients with uncomplicated falciparum malaria. Am J Trop Med Hyg. 2001;65:717–721. doi: 10.4269/ajtmh.2001.65.717. [DOI] [PubMed] [Google Scholar]
- 15.Snyder C, Chollet J, Santo-Tomas J, Scheurer C, Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp Parasitol. 2007;115:296–300. doi: 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Teja-Isavadharm P, Peggins JO, Brewer TG, White NJ, Webster HK, Kyle DE. Plasmodium falciparum-based bioassay for measurement of artemisinin derivatives in plasma or serum. Antimicrob Agents Chemother. 2004;48:954–960. doi: 10.1128/AAC.48.3.954-960.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis TME, England M, Dunlop A-M, Page-Sharp M, Cambon N, Keller TG, Heidecker JL, Ilett KF. Assessment of the effect of mefloquine on artesunate pharmacokinetics in healthy male volunteers. Antimicrob Agents Chemother. 2007;51:1099–1101. doi: 10.1128/AAC.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orrell C, Little F, Smith P, Folb P, Taylor W, Olliaro P, Barnes KI. Pharmacokinetics and tolerability of artesunate and amodiaquine alone and in combination in healthy volunteers. Eur J Clin Pharmacol. 2008;64:683–690. doi: 10.1007/s00228-007-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binh TQ, Ilett KE, Batty KT, Davis TME, Hung NC, Powell SM, Thu LTA, Thien HV, Phuöng HL, Phuong VDB. Oral bioavailability of dihydroartemisinin in Vietnamese volunteers and in patients with falciparum malaria. Br J Clin Pharmacol. 2001;51:541–546. doi: 10.1046/j.1365-2125.2001.01395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kongpatanakul S, Chatsiricharoenkul S, Sathirakul K, Suputtamongkol Y, Atipas S, Watnasirichaikul S, Pongnarin P, Sangvanich P. Evaluation of the safety and relative bioavailability of a new dihydroartemisinin tablet formulation in healthy Thai volunteers. Trans R Soc Trop Med Hyg. 2007;101:972–979. doi: 10.1016/j.trstmh.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Toovey S, Jamieson A. Audiometric changes associated with the treatment of uncomplicated falciparum malaria with co-artemether. Trans R Soc Trop Med Hyg. 2004;98:261–267. doi: 10.1016/j.trstmh.2003.11.001. discussion 268–9. [DOI] [PubMed] [Google Scholar]