

Abstract
Seizures are the manifestation of highly synchronized burst firing of a large population of cortical neurons. Epileptiform bursts with an underlying plateau potential in neurons are a cellular correlate of seizures. Emerging evidence suggests that the plateau potential is mediated by neuronal canonical transient receptor potential (TRPC) channels composed of members of the TRPC1/4/5 subgroup. We previously showed that TRPC1/4 double-knockout (DKO) mice lack epileptiform bursting in lateral septal neurons and exhibit reduced seizure-induced neuronal cell death, but surprisingly have unaltered pilocarpine-induced seizures. Here, we report that TRPC5 knockout (KO) mice exhibit both significantly reduced seizures and minimal seizure-induced neuronal cell death in the hippocampus. Interestingly, epileptiform bursting induced by agonists for metabotropic glutamate receptors in the hippocampal CA1 area is unaltered in TRPC5 KO mice, but is abolished in TRPC1 KO and TRPC1/4 DKO mice. In contrast, long-term potentiation is greatly reduced in TRPC5 KO mice, but is normal in TRPC1 KO and TRPC1/4 DKO mice. The distinct changes from these knockouts suggest that TRPC5 and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Furthermore, the reduced seizure and excitotoxicity and normal spatial learning exhibited in TRPC5 KO mice suggest that TRPC5 is a promising novel molecular target for new therapy.
Introduction
Seizures are the manifestation of highly synchronized burst firing of a large population of cortical neurons (Dichter and Ayala, 1987). The epileptiform burst firing with an underlying plateau potential is a cellular correlate of the hypersynchronous burst. How the plateau potential is generated is central to the pathophysiology of seizure. However, the molecular and cellular mechanisms underlying the plateau potential remain uncertain.
Group I metabotropic glutamate receptors (mGluRs) have been implicated in seizure and excitotoxicity (Sacaan and Schoepp, 1992; Barton and Shannon, 2005). Activation of group I mGluRs elicits epileptiform bursts with a large underlying plateau potential in lateral septal neurons (Zheng and Gallagher, 1991; Zheng et al., 1996) and hippocampal pyramidal neurons (Caeser et al., 1993). The ion channels mediating this plateau potential were thought to be the Ca2+-activated nonselective cation (CAN) channels (Zheng and Gallagher 1992; Luthi et al., 1997; Gee et al., 2003), but there has been growing evidence that the CAN channels activated by group I mGluRs are actually canonical transient receptor channels (TRPC) channels.
TRPC channels are “polymodal”: they can be activated by G protein-coupled receptors, such as mGluR1 (Kim et al., 2003), and they can also be activated by a rise in intracellular free calcium (Gee et al., 2003; Stroh et al., 2012). The mammalian TRPC family can be divided into TRPC1/4/5 and TRPC3/6/7 subgroups based on sequence homology and functional properties (Birnbaumer et al., 2003). TRPC channels may be either homomeric or heteromeric channels formed by members in the same subgroup (Strübing et al., 2001; Hofmann et al., 2002).
We previously had shown that heteromeric TRPC1/4 channels are major contributors to the plateau potential in lateral septal neurons (Phelan et al., 2012), but in vivo data from that study presented surprising results. In the pilocarpine-induced status epilepticus model, TRPC1/4 double knockout (DKO) mice showed reduced neuronal cell death in the lateral septum and the hippocampal CA1 region after severe seizures; despite this, the severity of the seizures was not significantly altered (Phelan et al., 2012). One explanation for this conundrum is that the plateau potential underlying the epileptiform bursts in other limbic areas is not mediated by TRPC1/4 channels. This explanation is supported by the differences in TRPC expression: TRPC5 expression is more widespread, but high-level expression of TRPC4 is largely limited to the lateral septum (Mori et al., 1998; Lein et al., 2007; Fowler et al., 2007).
This motivated us to investigate the role of TRPC5 in seizure and excitotoxicity with an independently generated TRPC5 knockout (KO) line. We show that, surprisingly, TRPC5 KO mice exhibit normal epileptiform burst firing but a significant reduction in pilocarpine-induced seizures. Conversely, TRPC1 KO mice, like TRPC1/4 DKO mice, show greatly reduced epileptiform bursts yet exhibit normal pilocarpine-induced seizures. In addition, long-term potentiation is greatly reduced in TRPC5 KO mice, but it is normal in TRPC1 KO and TRPC1/4 DKO mice. In all, our data suggest that TRPC5 and TRPC1/4 contribute to epileptogenesis and excitotoxicity through distinct mechanisms, despite the high sequence homology and similar gating properties between TRPC4 and TRPC5.
Materials and Methods
Generation of TRPC5 Knockout Mice.
TRPC5 KO mice were generated by first introducing, by homologous recombination in embryonic stem cells, loxP sites into regions flanking exon 5, deriving conditional KO mice in an 129EvSv genetic background using techniques previously described elsewhere (Jiang et al., 2002) and then removing the segment between the two loxP sites by crossing with Sox2-cre mice on a C57BL/6J background (Jackson Laboratories, Bar Harbor, MA).
Immunofluorescence Staining of TRPC5.
Adult male mice under anesthesia were intracardially perfused with 4% paraformaldehyde fixative in 0.1 M phosphate buffer. The brain was then removed and sectioned with a Vibratome (Pelco 1500; Ted Pella Inc., Redding, CA) at 50 μm thickness. Coronal sections containing the hippocampus were blocked first with Mouse on Mouse reagent (Vector Laboratories, Burlingame, CA), and then were incubated with a monoclonal antibody N67/15 (NIH/NeuroMab) targeting the cytosolic carboxyl tail (827-845 amino acids) of TRPC5 and a commercial secondary antibody and fluorescein-based visualization kit (Vector Laboratories). Confocal images were acquired on a Zeiss Axiovert 200M microscope equipped with a spinning disk CARV II (BD Biosciences, San Jose, CA), a Neo Fluor 63× oil-immersion objective, and a Hamamatsu Orca ER camera (Hamamatsu Photonics, Hamamatsu City, Japan).
Pilocarpine-Induced Seizures.
Age-matched wild-type (WT), TRPC1, or TRPC5 KO mice in a 129Sv/C57Bl6 mixed genetic background (3–6 months old) were injected with a single dose of pilocarpine (i.p.) at one of the following dosages: 175, 222, 280 mg/kg 30 minutes after an injection of methylscopolamine nitrate (1 mg/kg; i.p.) as described previously elsewhere (Phelan et al., 2012). Seizures induced by pilocarpine were recorded by a digital camcorder and were scored based on the modified Racine scale independently by two investigators (Phelan et al., 2012).
Screening for Neurodegeneration with Fluoro-Jade C Staining.
Two days after pilocarpine-induced seizures, the mice were anesthetized with ketamine (60 mg/kg, i.m.) and then were intracardially perfused with 4% paraformaldehyde as described previously elsewhere (Phelan et al., 2012). The brains were removed, postfixed for at least 48 hours, and then cut into 50 μm serial coronal sections using a Vibratome (Pelco 1500; Ted Pella Inc.). Free-floating sections were stained with Fluoro-Jade C (FJC), as previously reported elsewhere (Schmued et al., 2005). Four forebrain sections at least 300 μm apart that contain typical vulnerable regions (i.e., the cingulate cortex, septum, striatum, hippocampus, amygdala, piriform cortex, and thalamus) were processed to determine whether there were FJC-positive neurons. If any FJC-positive neurons were found, a complete series of 150-μm spaced sections were further stained with FJC. Images of FJC-positive sections were captured using a Coolsnap fx camera (Photometrics, Tucson, AZ) mounted on an Olympus fluorescent microscope using a green fluorescent protein (GFP) filter (Olympus America, Center Valley, PA).
Nissl Counterstaining and Analysis of Cell Death.
Coronal sections (50-μm thick) were stained with 0.1% cresyl violet solution. Unbiased cell counting of the hippocampal region was obtained using Stereologer (Stereology Resource Center, Chester, MD) in serial Nissl-stained sections spaced 150 μm apart extending from stereotaxic coordinates of bregma −1.3 to −2.3 mm, as described previously elsewhere (Phelan et al., 2012). The coefficients of error for all three regions in all groups were acceptable (range: 0.06–0.14 for CA1, 0.06–0.11 for CA3, and 0.11–0.18 for the hilar region).
Electrophysiologic Recordings.
Transverse slices of adult mouse brain containing the hippocampus were obtained from 2- to 3-month-old WT, TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice in a 129Sv/C57Bl6 mixed genetic background. The mice were anesthetized with ketamine (60 mg/kg, i.m.) followed by decapitation. Serial 500 μm thick sections were cut with a Vibraslice (WPI, Sarasota, FL) as described previously, and were allowed to recover in oxygenated artificial cerebrospinal fluid (ACSF) for at least 1 hour at room temperature before recording. A single hippocampal slice was held submerged in the recording chamber between two nylon meshes and was superfused with oxygenated ACSF warmed to 32° ± 1°C at a rate of 1–2 ml/min. For intracellular recordings, glass microelectrodes filled with 3 M sodium acetate were used as described previously elsewhere (Phelan et al., 2012).
For field potential recordings, glass pipettes were pulled from filamented borosilicate glass on a Flaming/Brown Micropipette Puller (Model P-97; Sutter Instrument, Novato, CA) to a final tip resistance of 6 to 9 MΩ when filled with ACSF. A concentric stimulus electrode was placed in the striatum radiatum. The field excitatory postsynaptic potential (fEPSP) was recorded in the current-clamp mode with an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA), was digitized using a model 1322A Digidata interface and pClamp 10 (Molecular Devices), and was stored on a computer hard drive. For long-term potential (LTP) experiments, the stimulus intensity was adjusted to produce a fEPSP that is approximately half of the maximal amplitude, and the average stimulus intensity used was 10.8 ± 0.7 V (mean ± S.E.M.) for WT (n = 14) and 10.1 ± 0.6 V for TRPC5 KO (n = 9).
Radial Arm Water Maze Test.
WT and TRPC5 KO mice in a 129Sv/C57Bl6 mixed genetic background were tested for cognitive function with a radial arm water maze using a protocol described previously elsewhere (Alamed et al., 2006) with slight modifications. This 2-day test consisted of nine trials on day 1 (alternating between a visible and hidden platform for the first few trials) and 12 trials on day 2 (using only a hidden platform); for each mouse, the platform was placed in the same lane for all trials. Each mouse was given up to 60 seconds to find the platform based on visual cues around the pool; each entry into a lane without the platform, 15 seconds in the incorrect lane, or 15 seconds without entering a lane was considered an error. The number of errors each mouse made before escaping to the platform was averaged over each block of three trials. The water temperature in the pool was kept constant at 21°C.
The experimenters performing behavioral analysis were blinded to the genotype of the mice, and all animal experimental protocols were approved by the institutional animal care and use committee.
Statistical Analysis.
One-way or two-way analysis of variance (ANOVA) with post hoc analysis were used unless stated otherwise.
Results
Confirmation of the Genetic Ablation of TRPC5.
The genetic ablation of TRPC5 was achieved by deleting exon 5 (Fig. 1). The truncated TRPC5 mRNA is predicted to encode the cytosolic N-terminus plus all of transmembrane domain 1 (TM1) and about 75% of TM2, terminating after VLG before the FIWG motif. This protein, if made, lacks the TM5-pore-TM6 ion channel domain and is predicted to be nonfunctional. The deletion of exon 5 was confirmed by reverse-transcription polymerase chain reaction (RT-PCR) analysis and sequencing of the amplicon that spanned the deletion (Fig. 1C). Furthermore, the lack of TRPC5 protein was confirmed by immunofluorescent staining using a monoclonal anti-TRPC5 antibody (Fig. 1, D and E).
Fig. 1.

Confirmation of TRPC5 knockout. (A) Intron-exon organization of the Mus musculus TRPC5 gene. Open boxes, untranslated exonic sequence. Closed boxes, translated open reading frame. *Stop codon. Closed triangles between exons 4 and 5, and between exons 5 and 6, loxP sites (ATAAC T TCGT ATAGC ATACA TTATA CGAAG TTAT). 3F, 4R, 6F, and 7R, PCR primers (sequences available upon request). The lengths of the amplicons, including primers, are depicted. (B) Diagram of the expected disruption after excision of exon 5 by the action of the cre recombinase expressed as a transgene under control of the Sox2 promoter (Sox2-cre; Jackson Laboratories). (C) RT-PCR analysis of brain mRNA from a TRPC5 control mouse (WT) and a TRPC5 knockout (KO) mouse. The depicted image is of the electrophoretic migration in a 1.5% agarose gel of the amplicons obtained using the primers depicted in A and B. The results confirmed that in TRPC5 KO mice a TRPC5 mRNA is made that is predicted to lack the coding sequence corresponding to exon 5. This was confirmed by sequencing the amplicon. The mRNA is predicted to encode a 414-amino-acid protein that corresponds to TRPC5[1-413] plus a non-natural valine. (D–E) Confocal images showing immunohistochemical staining of TRPC5 in the hippocampal CA1 and CA3 region. Note the cytoplasmic TRPC5 immunostaining (green) in the soma surrounding the nucleus (blue, stained with DAPI) and the lack of TRPC5 immunostaining in the TRPC5 KO mice. Scale bar: 10 μm.
TRPC5 KO Mice Exhibit Significantly Reduced Pilocarpine-Induced Seizure.
Pilocarpine induces severe seizures by acting on the m1 muscarinic acetylcholine receptor (AChR) in the hippocampus (Sheffler et al., 2009). At the cellular level, activation of m1 AChR results in a plateau potential similar to the one induced by activation of mGluRs. Although several studies have suggested that this plateau potential may be mediated by neuronal TRPC channels (Yan et al., 2009; Zhang et al., 2011; Tai et al., 2011), knocking out both TRPC1 and TRPC4, two highly expressed TRPCs, did not diminish the severity of pilocarpine-induced seizures (Phelan et al., 2012). This result suggests that heteromeric TRPC1/4 channels are not required for pilocarpine-induced seizures; however, TRPC5-containing channels could still be required. To investigate this, we examined pilocarpine-induced seizures in TRPC5 KO and TRPC1 KO mice. As shown in Fig. 2A, the severity of pilocarpine-induced seizures was significantly reduced during the late phase (50 minutes and more after the initial pilocarpine injection) in TRPC5 KO mice whereas the severity of seizures was comparable during the early phase. This reduction in seizure severity during the late phase results in a decrease in average seizure scores from 4.43 to 3.26 (Fig. 2B). Seizure-induced mortality was also reduced (Fig. 2C), with only the occasional death at higher pilocarpine doses (1 of 10 at 222 mg/kg, and 1 of 13 at 280 mg/kg) in TRPC5 KO mice. These data clearly suggest that TRPC5 plays a critical role in pilocarpine-induced seizures. In contrast, the severity of pilocarpine-induced seizures in TRPC1 KO mice was identical to that observed in WT mice (Fig. 2, A and B), implying that the ubiquitously expressed TRPC1 does not play a critical role in the generation of pilocarpine-induced seizures. Taken together, our data suggest that TRPC5 plays a critical role in pilocarpine-induced seizures; furthermore, this role of TRPC5 is not shared with either TRPC1 or TRPC4.
Fig. 2.

Pilocarpine-induced seizures were significantly reduced in TRPC5 KO mice. (A) The time course of pilocarpine-induced seizures in WT, TRPC5KO, and TRPC1KO mice after a single injection of pilocarpine (280 mg/kg, i.p.). Pooled data (mean ± S.E.M.) was plotted (n = 18, 14, and 19 for WT, TRPC5KO, and TRPC1 KO, respectively). See Phelan et al. (2012) for description of seizure scale. Note statistically significantly reduced seizure scores in TRPC5 KO mice at the late phase after pilocarpine injection [P < 0.001, two-way analysis of variance (ANOVA)]. *P < 0.05; **P < 0.01, Bonferroni post hoc tests against WT. (B) Average seizure scores were lower in TRPC5 KO compared with WT mice (P < 0.01, two-way ANOVA). *P < 0.05, Bonferroni post hoc tests against WT. Pooled data (mean ± S.E.M.) was plotted (n = 24, 10, and 18, for WT at 175, 222, and 280 mg/kg pilocarpine, respectively; n = 8, 10, and 14 for TRPC5 KO at 175, 222, and 280 mg/kg pilocarpine, respectively; n = 9, 15, and 19 for TRPC1 KO at 175, 222, and 280 mg/kg pilocarpine, respectively). (C) Mortality in the first 24 hours after pilocarpine injections was reduced in TRPC5 KO mice (n = 8, 10, and 14) compared with WT mice (n = 24, 10, and 18) and TRPC1KO mice (n = 9, 15, and 19).
TRPC5 KO Mice Exhibit Greatly Reduced Seizure-Induced Neuronal Cell Death in the Hippocampus.
Do TRPC5 channels also contribute to seizure-induced cell death? To answer this question, we compared the seizure-induced neuronal cell death in WT and TRPC5 KO mice. Previous studies showed that only severe seizures (i.e., with an average seizure score above 3) lead to neuronal cell death in WT mice (Borges et al., 2003; Phelan et al., 2012). Eight of 14 TRPC5 KO mice treated with 280 mg/kg pilocarpine did not exhibit severe seizures, and we did not observe any FJC-positive neurons in these mice. Six TRPC5 KO mice treated with 280 mg/kg pilocarpine exhibited severe seizures, and five showed FJC-positive neurons in typically vulnerable brain areas such as the lateral septum, piriform cortex, cingulate cortex, and thalamus; the remaining one had no FJC staining in a series of sections spanning from the septum to the hippocampus. Although there was widespread neuronal cell death in many areas, a highly significant reduction in pilocarpine-induced neuronal cell death was observed in the hippocampus. Therefore, we focused on the hippocampus in the subsequent analysis.
In contrast to the widespread FJC-positive neurons in WT mice with severe seizures (Fig. 3, A and C–E), the FJC-positive neurons in TRPC5 KO mice with severe seizures were largely in the hilar region (Fig. 3, B and H) and scattered in the CA3 region (Fig. 3, B and F) and the subiculum (Fig. 3B). FJC-positive neurons were absent in the CA1 region in four out of five TRPC5 KO mice (Fig. 3, B and G) whereas a few scattered FJC-positive neurons were observed in the CA1 region in the remaining mouse. Nissl staining of adjacent sections confirmed the lack of seizure-induced neuronal cell death in the CA1 region (Fig. 4, B–D).
Fig. 3.

TRPC5 KO mice exhibited greatly reduced FJC-positive neurons in the hippocampus after pilocarpine-induced severe seizures. Neuronal cell death induced by pilocarpine-induced seizures was assessed in selected groups of mice with comparable average seizure scores above 3 for WT (3.79 ± 0.14, n = 6) and TRPC5 KO mice (3.48 ± 0.09, n = 6). Mice with average seizure scores ≤3 did not show any FJC-positive neurons. Representative images of FJC-stained neurons in the hippocampus of WT (A, C–E) and TRPC5 KO mice (B, F–H) (2-day survival: WT, 175 mg/kg; TRPC5 KO, 280 mg/kg). Scale bars: 0.25 mm (A, B); 0.05 mm (C, D, F, G); 0.04 mm (E, H).
Fig. 4.
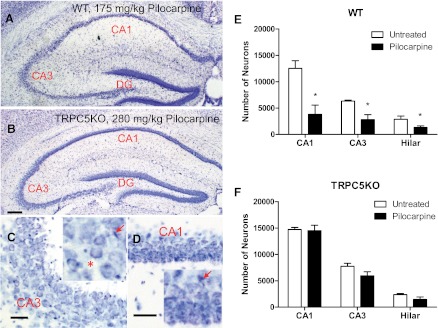
Reduced neuronal cell death after pilocarpine-induced seizures in the hippocampus of TRPC5 KO mice. (A–B) Nissl staining in a representative transverse section through the hippocampus in WT and TRPC5 KO mice after severe seizures induced by pilocarpine. Note the normal appearance of the pyramidal cell layer in (B) TRPC5 KO in contrast to the severe gliosis and a loss of normal pyramidal neurons in (A) WT. (C–D) High-power photomicrographs illustrating the lack of gliosis and sparing of CA1 and CA3 neurons in TRPC5 KO mice. Scale bars: 0.5 mm (A–B), 25 μm (C–D). (E–F) Serial coronal sections (50 μm) from mice with similar pilocarpine-induced seizures were stained with Nissl and surviving neurons (with stained cytoplasm and round nuclei) were counted using Stereologer with a 100× oil-immersion objective. The manifestation of seizure-induced degeneration typically is a loss of normal pyramidal cells (*) and occasionally the condensed pyramidal cell nucleus indicated by arrows in the insets of C and D. Pooled data (mean ± S.E.M.) were plotted (n = 4, 5 for untreated and pilocarpine-treated WT mice; n = 3, 5 for untreated and pilocarpine-treated TRPC5 KO mice). In WT mice, the number of neurons was statistically significantly reduced in the CA1, CA3, and hilar regions after severe seizures induced by pilocarpine (P < 0.05, unpaired t tests). On the other hand, in TRPC5 KO mice, the number of neurons was not statistically significantly reduced after pilocarpine-induced severe seizures in any of the three areas (unpaired t tests).
To quantify the degree of neuronal loss induced by pilocarpine-induced seizures, we counted the surviving neurons in a series of Nissl-stained sections using stereologic methods described previously elsewhere (Phelan et al., 2012). In WT mice, the cell count in all three hippocampal regions analyzed showed statistically significant differences between untreated and treated mice, indicating neuronal cell death caused by pilocarpine-induced seizures (Fig. 4E). On the other hand, the cell count in all three hippocampal regions analyzed showed no statistically significant differences between the treated and untreated TRPC5 KO mice (Fig. 4F). Consistent with a lack of FJC staining, the cell count in the CA1 region in pilocarpine-treated TRPC5 KO mice was indistinguishable from the cell count in the CA1 region in untreated TRPC5 KO mice, suggesting a general absence of seizure-induced neuronal cell death in the CA1 region in TRPC5 KO mice.
Epileptiform Burst Firing Induced by mGluR Agonists Is Reduced in TRPC1 KO and TRPC1/4 DKO Mice, but Is Unaltered in TRPC5 KO Mice.
What are the underlying cellular mechanisms for the reduced pilocarpine-induced seizures and neuronal cell death observed in TRPC5 KO mice? Kandel and Spencer (1961) observed three types of firing patterns in hippocampal pyramidal neurons: single spikes, short bursts, and long-lasting bursts with a prominent plateau. These firing patterns are not mutually exclusive, as single spikes can be intermingled with bursts in the same pyramidal neuron. The contribution of excitatory synaptic input and intrinsic membrane properties to the plateau potential has been long debated. Three candidates remain in contention as the ion channels that mediate the plateau potential underlying the epileptiform bursts: persistent Na+ channels with uncertain molecular identity (Chen et al., 2011), T-type voltage-gated Ca2+ channels (Cain & Snutch, 2010), and TRPC channels (Gee et al., 2003; Wang et al., 2007; Yan et al., 2009; Phelan et al., 2012).
Several studies have suggested that the plateau underlying burst firing induced by activation of mAChRs or mGluRs could be mediated by TRPC5-containing channels (i.e., TRPC1/5, TRPC4/5, or TRPC1/4/5 channels) in cortical pyramidal neurons (Yan et al., 2009; Tai et al., 2011; Zhang et al., 2011). To test whether this is the case in the hippocampus, we recorded the spontaneous burst firing induced by 1S,3R-ACPD, an mGluR agonist, in pyramidal neurons of the CA1 region in adult mice (2 to 3 months old). Under control conditions, CA1 pyramidal neurons (n = 9) showed significant spike adaptation (Fig. 5A), and the spontaneous firing patterns of these pyramidal neurons were random single spikes with variable spike intervals (Fig. 5B). In the presence of 30-μM 1S,3R-ACPD, five of the nine CA1 pyramidal neurons exhibited evoked burst firing (Fig. 5A) or spontaneous burst firing (Fig. 5B) characterized by a burst of spikes with an underlying plateau. The spontaneous firing in remaining neurons consisted of single spikes. Spontaneous burst firing with a underlying plateau potential (Fig. 6A) was observed in the presence of 30-μM 1S,3R-ACPD in half of the CA1 pyramidal neurons in TRPC5 KO mice (n = 8). As in WT, the spontaneous firing in the remaining neurons in TRPC5 KO mice consisted of single spikes. Furthermore, a quantitative analysis showed that the number of spikes per burst and the amplitude of the plateau underlying the burst were not altered in TRPC5 KO mice (Fig. 6, B and C). The number of spikes per bursts was 9.9 ± 1.2 for WT (n = 5) and 9.7 ± 1.7 for TRPC5 KO mice (n = 4), and the amplitude of plateau was 16.2 ± 2.4 mV for WT and 15.8 ± 1.6 mV for TRPC5 KO mice.
Fig. 5.

The effect of 1S,3R-ACPD on the firing pattern of CA1 pyramidal neurons. (A) Representative traces showing the firing patterns evoked by a depolarizing current step (500 ms) in a CA1 pyramidal neuron recorded from adult WT mice (Vh= −70 mV). Note the spike adaptation under control conditions and the burst in the presence of 30 μM 1S,3R-ACPD, a mGluR agonist. (B) Spontaneous firing in the same CA1 pyramidal neuron under the control conditions and in the presence of 30 μM 1S,3R-ACPD.
Fig. 6.

Spontaneous burst firing induced by mGluR activity is normal in TRPC5 KO mice, but reduced in TRPC1 KO and TRPC1/4 DKO mice. (A) Representative current-clamp recordings showing the spontaneous burst firing induced by 30 μM 1S,3R-ACPD in CA1 pyramidal neurons in adult TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice. (B) The amplitude of the plateau underlying the burst is comparable in the WT and TRPC5 KO mice, but significantly reduced in TRPC1 KO and TRPC1/4 DKO mice. Amplitudes were measured for three randomly selected bursts in each neuron and then averaged. Pooled data (mean ± S.E.M.) were plotted (n = 4, 4, 5, 5 for WT, TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice, respectively). (C) The duration of each burst was quantified by the number of action potentials within each burst, and three random bursts from each CA1 pyramidal neuron were analyzed to obtain the average number of spikes per burst. Pooled data (mean ± S.E.M.) were plotted (n = 4, 4, 5, 5 for WT, TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice, respectively). **P < 0.01, ANOVA and Tukey’s post hoc test.
Carbachol (50 μM) can also induce either evoked or spontaneous burst firing in CA1 pyramidal neurons (three of seven cells in WT and two of seven cells in TRPC5KO; data not shown). Spontaneous burst firing was observed in the presence of 30 μM 1S,3R-ACPD in TRPC1 KO mice (five of eight cells) and TRPC1/4 DKO mice (five of nine cells), however, the 1S,3R-ACPD-induced bursts were retarded, with significantly reduced plateau amplitudes and spike numbers per burst (Fig. 6, A–C). The number of spikes per bursts was 3.5 ± 0.7 for TRPC1 KO (n = 5) and 2.8 ± 0.3 for TRPC1/4 DKO mice (n = 5), and the amplitude of plateau was 6.6 ± 1.1 mV for TRPC1 KO and 5.3 ± 0.6 mV for TRPC1/4 DKO mice. Therefore, TRPC1 and TRPC4, but not TRPC5 contribute significantly to the plateau potential underlying the 1S,3R-ACPD-induced burst firing in CA1 pyramidal neurons. The reduction of pilocarpine-induced seizures and neuronal cell death in TRPC5 KO mice cannot be attributed to a disruption of burst firing induced by activation of mGluRs.
Long-Term Potentiation at the Schaffer Collateral Synapses Is Significantly Reduced in TRPC5 KO Mice, but Is Normal in TRPC1 KO and TRPC1/4 DKO Mice.
A change in the burst firing induced by activation of mGluR has been ruled out as a possible underlying mechanism for the reduction of pilocarpine-induced seizures and neuronal cell death in TRPC5 KO mice. What else could be the alternative mechanism? The generation and propagation of seizures require a reduction in inhibitory synaptic transmission and an increase in excitatory synaptic transmission (Dichter and Ayala, 1987; Dudek et al., 1999). Because TRPC5 is expressed in pyramidal neurons, we first explored whether TRPC5 plays a critical role in synaptic plasticity at a principal excitatory synapse in the hippocampus, the Schaffer collateral synapse.
A previous study reported a change in the paired-pulse facilitation (PPF) in the lateral amygdala in TRPC5 KO mice (Riccio et al., 2009). To determine whether similar changes occur at the Schaffer collateral synapses in TRPC5 KO mice, we determined the PPF at a series of increasing intervals (Fig. 7, A and B). Although there were small increases in the PPF at the shorter intervals, there was no statistical significance when the PPF in TRPC5 KO mice was compared with the PPF in WT mice. We also investigated the PPF at the Schaffer collateral synapses in TRPC1 KO and TPRC1/4 DKO mice and did not detect any significant changes when it was compared with the PPF in WT mice (Fig. 7, A, C, and D). Taken together, our data suggest that TRPC1 and TRPC5 are not critical for the PPF at the Schaffer collateral synapses in adult mice.
Fig. 7.

Normal paired-pulse facilitation in TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice. (A) Representative traces of paired-pulse facilitation (PPF) of Schaffer collateral field excitatory postsynaptic potential (fEPSP) in WT, TRPC5 KO, TRPC1KO, and TRPC1/4 DKO mice. A pair of electric stimuli with increasing intervals (40, 80, 120, 160, 200, 240, 280, and 320 ms) was delivered at 10-second intervals, and the resulting pair of fEPSPs was recorded. (B–D) The averaged PPF ratios (the peak of the second EPSP over the peak of the first EPSP in each pair) and standard errors were plotted (n = 5-15, 5-13, 6, 6 for WT, TRPC5 KO, TRPC1 KO and TRPC1/4DKO, respectively). Note the peak of PPF occurs around 40–50 ms intervals and the subsequent exponential decay at greater intervals. There was no statistically significant difference between WT and TRPC5 KO mice, between WT and TRPC1 KO mice, or between WT and TRPC1/4DKO mice (two-way ANOVA).
Because we did not find significant alteration in the PPF at the Schaffer collateral synapses in the CA1 region, we investigated the LTP induced by high-frequency stimuli (HFS) at the same synapse. The fEPSP slope was potentiated by 42% in WT mice, but the fEPSP slope was potentiated by 6% 30 minutes after the HFS in TRPC5 KO mice (Fig. 8, A and B). In comparison, the fEPSP was still approximately 35% and 31% above the baseline 30 minutes after the HFS in TRPC1 KO mice and TRPC1/4 DKO mice (Fig. 8, A and C). Because TRPC1 KO mice showed normal LTP but TRPC5 KO mice showed reduced LTP, our data indicate that TRPC5 channels play a uniquely critical role in the HFS-induced LTP at the Schaffer collateral synapses in the CA1 region of hippocampus.
Fig. 8.
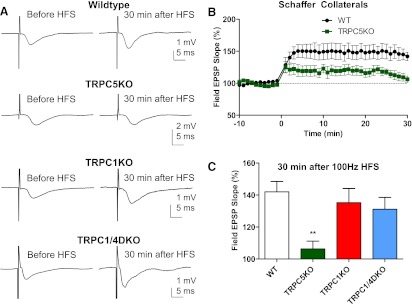
Reduced high-frequency stimuli-induced long-term potentiation (LTP) in the CA1 region in TRPC5 KO, but not in TRPC1 KO and TRPC1/4 DKO mice. (A) Representative traces of Schaffer collateral field excitatory postsynaptic potential (fEPSP) recorded before and 30 minutes after high-frequency stimuli (HFS; 100 Hz, 1 second; repeated three times with a 20-second interval) in WT, TRPC5 KO, TRPC1 KO, and TRPC1/4 DKO mice. Traces shown were the average of 12 consecutive recordings collected at 0.2 Hz. (B) Field EPSP slopes for each minute were determined by averaging 12 consecutive fEPSP recordings in each mouse, and the normalized mean and standard error were plotted (P < 0.01 for genotype effects, two-way ANOVA; n = 14, 9 for WT and TRPC5KO mice, respectively). (C) The averaged fEPSP slope 30 minutes after 100 Hz HFS in WT (n = 14), TRPC5 KO (n = 9), TRPC1 KO (n = 12), and TRPC1/4DKO mice (n = 9). Note the statistically significantly reduced LTP in TRPC5 KO mice; the LTP was normal in TRPC1 KO and TRPC1/4 DKO mice. **P < 0.01, ANOVA and Tukey’s post hoc tests.
TRPC5 KO Mice Exhibited Normal Spatial Learning.
Our data indicated that genetic ablation of TRPC5 resulted in reduced LTP at Schaffer collateral synapses and reduced pilocarpine-induced seizures and neuronal cell death. Is spatial learning also impaired in TRPC5 KO mice? This question is important because a molecular target that can selectively reduce seizure and associated neuronal cell death while not affecting spatial learning would be ideal for new therapy. To answer this question, we tested WT and TRPC5 KO mice in a radial arm water maze using a method described previously by Alamed et al. (2006). TRPC5 KO mice were able to perform as well as WT mice in finding the hidden platform (Fig. 9). The normal spatial learning exhibited by TRPC5 KO suggests that TRPC5 could be a novel molecular target that would allow selective blockade of seizure propagation and excitotoxicity with little impairment of spatial learning.
Fig. 9.

TRPC5 KO mice exhibited normal spatial learning in radial arm water maze. Adult male WT (n = 6) and TRPC5 KO mice (n = 8) were tested in a radial arm water maze using the method described previously by Alamed et al. (2006). There was no statistically significant difference between WT and TRPC5 KO mice (two-way ANOVA).
Discussion
Our data strongly suggest that TRPC1/TRPC4 and TRPC5 serve distinct roles in the brain. TRPC5 plays an important role in pilocarpine-induced seizure. Because pilocarpine-induced seizures were not altered in TRPC1 KO or TRPC1/4 DKO mice (Phelan et al., 2012), this critical role of TRPC5 in pilocarpine-induced seizures is not shared with TRPC1 and TPRC4. Another surprising finding is that TRPC5, but not TRPC1 or TRPC4, plays a critical role in HFS-induced LTP at Schaffer collateral synapses. In contrast, the plateau potential underlying the burst firing induced by mGluR agonists is likely mediated by TRPC1/4 channels. This functional role of TRPC1/TRPC4 is not shared with TRPC5. Given the high sequence homology and similar channel properties between TRPC4 and TRPC5, these findings are surprising.
As the cellular equivalent of hypersynchronous bursts, epileptiform burst firing is the fundamental element of epileptogenesis. There has been a longstanding debate regarding the molecular identity of the ion channels that mediate the plateau potential underlying burst firing induced by mGluR agonists and muscarinic mAChR agonists. Early leading candidates were the persistent Na+ current (Azouz et al., 1996) and the CAN current (Wong and Prince, 1981). Gee et al. (2003) first suggested that the CAN current activated by mGluR1 in pyramidal neurons showed similarity to TRP channels. However, the most often used TRPC channel blocker SKF96365 is also a potent blocker of T-type voltage gated Ca2+ channels (Singh et al., 2010), raising the possibility that the plateau attributed to TRPC channels could be mediated by Cav3 channels. Indeed, a role of T-type voltage-gated Ca2+ channels in epileptogenesis has been reported (Cain & Snutch, 2010). Our data suggest that the plateau potential underlying 1S,3R-ACPD-induced spontaneous burst firing in CA1 pyramidal neurons is mainly mediated by TRPC1/4 channels. Although we cannot completely rule out contributions from the persistent Na+ current or the T-type voltage-gated Ca2+ channel, the near abolishment of the burst in TRPC1/4 DKO mice suggests that the contribution of other channels to the plateau potential is minor in comparison with the contribution of TRPC1/4 channels.
Our data also imply that the epileptiform burst firing induced by mGluR agonists at the cellular level in CA1 pyramidal cells is clearly dissociated from experimental temporal lobe seizures in vivo. TRPC5 KO mice, which show normal burst firing induced by mGluR agonists, actually exhibit reduced pilocarpine-induced seizures whereas TRPC1KO and TRPC1/4DKO mice, which lack this burst firing, show unaltered pilocarpine-induced seizures. This surprising disconnect between the mGluR agonist-induced burst firing at the cellular level and the temporal lobe seizures in vivo raises questions regarding the transition from hypersynchronous bursts to seizures. Our data suggest that increases in the excitatory synaptic drive through LTP which requires TRPC5 are critical for this transition whereas the long-lasting epileptiform bursting is not required.
Because PPF is normal in TRPC5 KO mice, the contribution of TRPC5 to Schaffer collateral LTP is probably mediated through postsynaptic mechanisms. Our data from TRPC5 KO are similar to the data from a selective m1 negative allosteric regulator (Sheffler et al., 2009); both showed a reduction in seizure severity without disruption of spatial learning. This suggests that spatial learning may be more closely linked to a certain type of LTP, such as the N-methyl-d-aspartate receptor-dependent LTP, whereas TRPC5 may contribute mainly to N-methyl-d-aspartate-receptor-independent LTP, which may not be closely associated with spatial learning. Further studies are required to resolve these issues.
There are several possible explanations for the distinct functional roles of TRPC5 versus TRPC1/TRPC4. Although it has been assumed that TRPC1, 4, and 5 can form heteromeric channels, empirical evidence for the existence of such channels in vivo is lacking. One possibility is that TRPC4 and TRPC5 are distributed differently in neurons. We have shown previously that TRPC4 is located on the cell surface at the soma and primary dendrites of lateral septal neurons (Phelan et al., 2012). The surface expression of TRPC5 may be restricted to the distal dendrites and dendritic spines. If this is the case, it seems logical that TRPC5 plays a far more critical role in synaptic plasticity than TRPC4. Another possibility is that TRPC5 may normally remain in intracellular compartments and is only inserted into the membrane when m1AChRs or mGluR1 or mGluR5 receptors are activated. It has been reported that TRPC5 exhibits carbachol-induced membrane insertion whereas surface expression of TRPC1 and TRPC4 was unchanged after carbachol exposure (Tai et al., 2011). However, it should be noted that a dynamic regulation of surface expression of TRPC1 or TRPC4 has also been reported previously (Odell et al., 2005; Wang et al., 2007: Cheng et al., 2010). Either way, additional studies are needed to identify the molecular or cellular mechanisms that confer these unique functional roles to TRPC5 channels.
Finally, our present data suggest that TRPC5 is a novel contributor to epileptogenesis and excitotoxicity in the time frame investigated in this study (2 days after pilocarpine injection). Excitotoxic neuronal cell death, caused by overactivation of glutamate receptors, is a shared mechanism in the pathophysiology of stroke and epilepsy (Choi, 1988; Lipton and Rosenberg, 1994; Fujikawa, 2005). We propose that TRPC channels are critically involved in epileptogenesis and excitotoxicity, with two distinct mechanisms. TRPC1 and TRPC4, likely operating as TRPC1/4 heteromers and located at the soma and primary dendrites, mediate the plateau potential that leads to burst firing; TRPC5 channels, likely operating as homomeric channels and located at the synapses, play a critical role in synaptic plasticity. Both act synergistically to trigger neuronal apoptosis under pathologic conditions such as stroke or epilepsy. Our present data suggest that TRPC5 is a particularly promising novel molecular target for new therapy. In particular, we have shown that TRPC5 is critically involved in seizure and excitotoxicity, but not in normal spatial learning. In contrast, M channels are critically involved in both seizure and spatial learning (Peters et al., 2005). Drugs that can selectively block TRPC5 channels could offer new treatment options for currently untreatable neurologic disorders such as intractable epilepsy or stroke.
Acknowledgments
The authors thank Dr. Stephen Traynelis for critical comments of this manuscript and valuable suggestions.
Abbreviations
- AChR
acetylcholine receptor
- ACSF
artificial cerebrospinal fluid
- CAN
Ca2+-activated nonselective cation
- DKO
double knockout
- EPSP
excitatory postsynaptic potential
- fEPSP
field excitatory postsynaptic potential
- FJC
FluoroJade C
- GFP
green fluorescent protein
- HFS
high-frequency stimuli
- KO
knockout
- LTP
long-term potentiation
- mGluR
metabotropic glutamate receptor
- NMDA
N-methyl-d-aspartate
- PPF
paired-pulse facilitation
- RT-PCR
reverse-transcription polymerase chain reaction
- 1S,3R-ACPD
(1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid
- TM
transmembrane domain
- TRPC
canonical transient receptor channels
- WT
wild type
Authorship Contributions
Participated in research design: Zheng, Phelan, Birnbaumer, Abramowitz, Rhee, Gottschall.
Conducted experiments: Phelan, Zheng, Shwe, Wu, Rhee, Howell.
Contributed new reagents or analytic tools: Freichel, Flockerzi, Birmbaumer, Abramowitz.
Performed data analysis: Zheng, Phelan, Shwe, Wu, Rhee, Howell, Gottschall.
Wrote or contributed to the writing of the manuscript: Zheng, Phelan, Shwe, Birnbaumer, Abramowitz, Flockerzi, Freichel, Rhee, Howell, Gottschall.
Footnotes
This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants NS050381 and NS047546]; the University of Arkansas for Medical Sciences Tobacco Research Fund; and the University of Arkansas for Medical Sciences Hornick Research Award; and also was supported in part by the Intramural Research Program of the National Institutes of Health [Grant Z01-ES 101684].
References
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. (2006) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc 1:1671–1679 [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. (1996) Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol 492:211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ME, Shannon HE. (2005) Behavioral and convulsant effects of the (S) enantiomer of the group I metabotropic glutamate receptor agonist 3,5-DHPG in mice. Neuropharmacology 48:779–787 [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Yildirim E, Abramowitz J. (2003) A comparison of the genes coding for canonical TRP channels and their M, V and P relatives. Cell Calcium 33:419–432 [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. (2003) Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol 182:21–34 [DOI] [PubMed] [Google Scholar]
- Caeser M, Brown DA, Gähwiler BH, Knöpfel T. (1993) Characterization of a calcium-dependent current generating a slow afterdepolarization of CA3 pyramidal cells in rat hippocampal slice cultures. Eur J Neurosci 5:560–569 [DOI] [PubMed] [Google Scholar]
- Cain SM, Snutch TP. (2010) Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 4:475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Su H, Yue C, Remy S, Royeck M, Sochivko D, Opitz T, Beck H, Yaari Y. (2011) An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J Neurophysiol 105:117–129 [DOI] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. (2010) Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol 9:e1001025 DOI: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634 [DOI] [PubMed] [Google Scholar]
- Dichter MA, Ayala GF. (1987) Cellular mechanisms of epilepsy: a status report. Science 237:157–164 [DOI] [PubMed] [Google Scholar]
- Dudek FE, Patrylo PR, Wuarin JP. (1999) Mechanisms of neuronal synchronization during epileptiform activity. Adv Neurol 79:699–708 [PubMed] [Google Scholar]
- Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. (2007) Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS ONE 2:e573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG. (2005) Prolonged seizures and cellular injury: understanding the connection. Epilepsy Behav 7 (Suppl 3):S3–S11 [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U. (2003) Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol 546:655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99:7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Spicher K, Boulay G, Martín-Requero A, Dye CA, Rudolph U, Birnbaumer L. (2002) Mouse gene knockout and knockin strategies in application to alpha subunits of Gi/Go family of G proteins. Methods Enzymol 344:277–298 [DOI] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. (1961) Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol 24:243–259 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. (2003) Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426:285–291 [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176 [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622 [DOI] [PubMed] [Google Scholar]
- Lüthi A, Gähwiler BH, Gerber U. (1997) 1S, 3R-ACPD induces a region of negative slope conductance in the steady-state current-voltage relationship of hippocampal pyramidal cells. J Neurophysiol 77:221–228 [DOI] [PubMed] [Google Scholar]
- Mori Y, Takada N, Okada T, Wakamori M, Imoto K, Wanifuchi H, Oka H, Oba A, Ikenaka K, Kurosaki T. (1998) Differential distribution of TRP Ca2+ channel isoforms in mouse brain. Neuroreport 9:507–515 [PubMed] [Google Scholar]
- Odell AF, Scott JL, Van Helden DF. (2005) Epidermal growth factor induces tyrosine phosphorylation, membrane insertion, and activation of transient receptor potential channel 4. J Biol Chem 280:37974–37987 [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. (2005) Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 8:51–60 [DOI] [PubMed] [Google Scholar]
- Phelan KD, Mock MM, Kretz O, Shwe UT, Kozhemyakin M, Greenfield LJ, Dietrich A, Birnbaumer L, Freichel M, Flockerzi V, Zheng F. (2012) Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol Pharmacol 81:384–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, Gapon S, Yao GL, Tsvetkov E, Rodig SJ, et al. (2009) Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 137:761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacaan AI, Schoepp DD. (1992) Activation of hippocampal metabotropic excitatory amino acid receptors leads to seizures and neuronal damage. Neurosci Lett 139:77–82 [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. (2005) Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31 [DOI] [PubMed] [Google Scholar]
- Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, et al. (2009) A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol Pharmacol 76:356–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Hildebrand ME, Garcia E, Snutch TP. (2010) The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br J Pharmacol 160:1464–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroh O, Freichel M, Kretz O, Birnbaumer L, Hartmann J, Egger V. (2012) NMDA receptor-dependent synaptic activation of TRPC channels in olfactory bulb granule cells. J Neurosci 32:5737–5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. (2001) TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29:645–655 [DOI] [PubMed] [Google Scholar]
- Tai C, Hines DJ, Choi HB, MacVicar BA. (2011) Plasma membrane insertion of TRPC5 channels contributes to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. Hippocampus 21:958–967 [DOI] [PubMed] [Google Scholar]
- Wang M, Bianchi R, Chuang SC, Zhao W, Wong RK. (2007) Group I metabotropic glutamate receptor-dependent TRPC channel trafficking in hippocampal neurons. J Neurochem 101:411–421 [DOI] [PubMed] [Google Scholar]
- Wong RKS, Prince DA. (1981) Afterpotential generation in hippocampal pyramidal cells. J Neurophysiol 45:86–97 [DOI] [PubMed] [Google Scholar]
- Yan HD, Villalobos C, Andrade R. (2009) TRPC Channels Mediate a Muscarinic Receptor-Induced Afterdepolarization in Cerebral Cortex. J Neurosci 29:10038–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reboreda A, Alonso A, Barker PA, Séguéla P. (2011) TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 21:386–397 [DOI] [PubMed] [Google Scholar]
- Zheng F, Gallagher JP. (1991) Trans-ACPD (trans-D,L-1-amino-1,3-cyclopentanedicarboxylic acid) elicited oscillation of membrane potentials in rat dorsolateral septal nucleus neurons recorded intracellularly in vitro. Neurosci Lett 125:147–150 [DOI] [PubMed] [Google Scholar]
- Zheng F, Gallagher JP. (1992) Metabotropic glutamate receptor agonists potentiate a slow afterdepolarization in CNS neurons. Neuroreport 3:622–624 [DOI] [PubMed] [Google Scholar]
- Zheng F, Gallagher JP, Connor JA. (1996) Activation of a metabotropic excitatory amino acid receptor potentiates spike-driven calcium increases in neurons of the dorsolateral septum. J Neurosci 16:6079–6088 [DOI] [PMC free article] [PubMed] [Google Scholar]