

Abstract
We have previously identified allosteric modulators of the cannabinoid CB1 receptor (Org 27569, PSNCBAM-1) that display a contradictory pharmacological profile: increasing the specific binding of the CB1 receptor agonist [3H]CP55940 but producing a decrease in CB1 receptor agonist efficacy. Here we investigated the effect one or both compounds in a broad range of signaling endpoints linked to CB1 receptor activation. We assessed the effect of these compounds on CB1 receptor agonist–induced [35S]GTPγS binding, inhibition, and stimulation of forskolin-stimulated cAMP production, phosphorylation of extracellular signal-regulated kinases (ERK), and β-arrestin recruitment. We also investigated the effect of these allosteric modulators on CB1 agonist binding kinetics. Both compounds display ligand dependence, being significantly more potent as modulators of CP55940 signaling as compared with WIN55212 and having little effect on [3H]WIN55212 binding. Org 27569 displays biased antagonism whereby it inhibits: agonist-induced guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding, simulation (Gαs-mediated), and inhibition (Gαi-mediated) of cAMP production and β-arrestin recruitment. In contrast, it acts as an enhancer of agonist-induced ERK phosphorylation. Alone, the compound can act also as an allosteric agonist, increasing cAMP production and ERK phosphorylation. We find that in both saturation and kinetic-binding experiments, the Org 27569 and PSNCBAM-1 appeared to influence only orthosteric ligand maximum occupancy rather than affinity. The data indicate that the allosteric modulators share a common mechanism whereby they increase available high-affinity CB1 agonist binding sites. The receptor conformation stabilized by the allosterics appears to induce signaling and also selectively traffics orthosteric agonist signaling via the ERK phosphorylation pathway.
Introduction
The endocannabinoid system encompasses a family of endogenous ligands, prominent examples including anandamide and 2-arachidonoyl glycerol (2-AG), both of which are synthesized on demand and are rapidly hydrolyzed by the enzymes. Within the brain, the distribution of CB1 receptors is heterogeneous; they are found predominantly on nerve terminals where they attenuate neurotransmitter release. CB1 receptor–competitive antagonists-inverse agonists were developed for the treatment of obesity and nicotine addiction but were withdrawn due to associated serious psychiatric side effects (Nathan et al., 2011).
In 2005 we identified the first allosteric modulators of the cannabinoid CB1 receptor (Price et al., 2005; Ross, 2007), followed by a structurally-related compound, 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea (PSNCBAM-1) (Horswill et al., 2007). These compounds modulate electrically evoked contractions in the mouse vas deferens (Price et al., 2005), affect CB1 ligand modulation of synaptic transmission (Wang et al., 2011) and have hypophagic effects in vivo (Horswill et al., 2007). They display a contradictory pharmacological profile: increasing the specific binding of the CB1 receptor agonist [3H]CP55940 but producing a concentration-related decrease in CB1 receptor agonist efficacy. The molecular mechanisms underlying this paradoxical pharmacological profile remain to be fully elucidated.
CB1 receptors are coupled to the Gi/o family of G proteins. Activation of these receptors leads to inhibition of adenylyl cyclases, and to phosphorylation and activation of mitogen-activated protein kinases (MAPK), including extracellular signal-regulated kinases 1/2 (ERK1/2) (Turu and Hunyady, 2010). Following activation, β-arrestin molecules associate with phosphorylated CB1 receptors. It is now accepted that a single receptor may engage different signaling pathways and that various ligands might influence these pathways differentially (Galandrin et al., 2007). This relatively new concept is termed “functional selectivity” (Baker and Hill, 2007; Kenakin, 2007) and has been described for the CB1 receptor (Glass and Northup, 1999; Mukhopadhyay and Howlett, 2001; Anavi-Goffer et al., 2007). This term can also be applied to allosteric modulators. If it is supposed that numerous “active” receptor conformations (leading to specific signaling outcomes) can be triggered by orthosteric agonists, then one might also postulate that any, but not always all, of these activation states may be stabilized by allosteric ligands (Hall, 2000). Allosteric ligands produce a distinctive receptor conformation which will possess a unique profile of pharmacology.
Recently, Ahn et al. (2012) provided evidence that Org 27569 may behave as a CB1 receptor–biased ligand: acting as an allosteric agonist to induce receptor internalization and ERK phosphorylation in a Gα1 protein–independent manner [pertussis toxin (PTX)-insensitive] while also \acting as negative allosteric modulator of CB1 agonist–mediated guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding. Here we investigated the effects of allosteric modulators using a broad range of signaling end points linked to CB1 receptor activation. We assessed the effects on CB1 receptor agonist–induced (1) [35S]GTPγS binding, which measures the level of G protein activation following agonist occupation of a G coupled–protein receptor (GPCR; PTX-sensitive); (2) inhibition of forskolin-stimulated cAMP production (Gαi-mediated, PTX-sensitive) and stimulation of cAMP production (Gαs-mediated; revealed in presence of PTX); (3) phosphorylation of ERK1/2 (PTX-sensitive); and (4) β-arrestin recruitment (PTX-insensitive). We find that Org 27569 inhibits agonist-induced [35S]GTPγS binding and β-arrestin recruitment. It inhibits Gαi-mediated agonist–induced inhibition of cAMP production and Gαs-mediated stimulation of cAMP production. Alone the compound can act as an agonist, increasing cAMP production in untreated and PTX-treated cells. The compound acts as weak agonist alone inducing ERK phosphorylation in a PTX-sensitive manner; it enhances orthosteric agonist–induced ERK phosphorylation.
The level of cooperativity displayed by an allosteric compound is often ligand-dependent. There are well-documented differences in the ligand-binding pocket for CB1 receptor ligands that lead to ligand-specific conformational changes in the receptor (reviewed by Abood, 2005). An example is the W2795.43A mutation of the CB1 receptor which reduces the binding of WIN55212 by 16-fold but does not affect CP55940 binding (McAllister et al., 2003). Together with other residues, W5.43A has an important role in inducing ligand-selective CB1 receptor activation (McAllister et al., 2003; McAllister et al., 2004). Here we find that Org 25769 and PSNCBAM-1 differentially modulate signaling of the CB1 receptor agonists CP55940 and WIN55212, being significantly more potent as modulators of CP55940 signaling. Furthermore, in W5.43A-mutated cells, Org 27569 loses the ability to inhibit CP55950 signaling.
In addition to signaling effects, we have conducted an in-depth characterization of the effect of allosteric modulators on agonist binding, investigating the ability of these compounds to modulate radioligand binding in saturation, competition, and kinetic binding assays. We found that in both saturation and kinetic binding experiments in brain membranes and hCB1-expressing cells, both allosteric modulators appeared to influence only orthosteric ligand maximum occupancy rather than affinity.
Materials and Methods
Materials
WIN55212, CP55940, and Org 27569 [5-chloro-3-ethyl-1H-indole-2-carboxylic acid [2-(4-piperidin-1-yl-phenyl)-ethyl]-amide] were obtained from Tocris (Bristol, UK), and SR141716A [N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride] from the National Institute on Drug Abuse. PSNCBAM-1 was synthesized by Prosidion Limited (Oxford, UK) as described by Bloxham et al., (2006) (patent). Bovine serum albumin (BSA), cell culture media, dithiothreitol (DTT), nonenzymatic cell dissociation solution, GDP, 5-guanylimidodiphosphate (Gpp(NH)p), GTPγS, G418, l-glutamine, Krebs’ salts, penicillin/streptomycin, Tris buffer, and Triton X-100 were all obtained from Sigma-Aldrich (Poole, UK). [3H]CP55940 (128 Ci/mmol), [3H]CP55940 (44 Ci/mmol), and [35S]GTPγS (1250 Ci/mmol) were obtained from PerkinElmer Life Sciences Inc. (Boston, MA). [3H]N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide ([3H]SR141716A; 43 Ci/mmol) was obtained from the National Institute on Drug Abuse.
Chinese Hamster Ovary hCB1R Cells
Chinese hamster ovary (CHO) cells stably transfected with cDNA encoding human cannabinoid CB1 receptors (see Ross et al., 1999) were maintained in Dulbecco’s modified Eagle’s medium (DMEM)–nutrient mixture F-12 Ham, supplemented with 2 mM l-glutamine, 10% fetal bovine serum (FBS), 0.6% penicillin-streptomycin, hygromycin B (300 μg/ml), and geneticin (600 μg/ml). All cells were maintained at 37°C and 5% CO2 in their respective media and were passage twice a week using nonenzymatic cell dissociation solution. The human cannabinoid 1 receptor (hCB1R) transfected cell line was used for cAMP-, pERK1/2-, and [35S]GTPγS-binding experiments.
Human Embryonic Kidney 293–hCB1R Cells
Untransfected human embryonic kidney (HEK293)–Flp-In T-REx (Invitrogen Ltd., Paisley, UK) cells were cultured in the following growth medium: DMEM containing 4.5g/l glucose, an l-glutamine substitute (GlutaMAX-l), and supplemented with 10% fetal bovine serum, 50 IU/ml penicillin, 50 μg/ml streptomycin, 15 μg/ml blasticidin, and 10 μg/ml zeocin. Cells were grown in tissue culture flasks in an incubator at 37°C under 5% CO2. A stable cell line that overexpresses the human CB1 receptor when induced with tetracycline was created using the Flp-In T-REx system according to the manufacturer’s instructions. HEK293 Flp-In T-REx cells were transfected with the plasmid pcDNA5/FRT/TO (which contains a hygromycin resistance gene) into which the hCB1 receptor open reading frame had been inserted. A population of stable transfectants were selected by culturing cells in growth medium containing 100 μg/ml hygromycin and 15 μg/ml blasticidin. The HEK-hCB1R cell line was used for radioligand binding studies.
Membrane Preparation
Mouse Brain Membrane Preparation.
Whole brains from adult male MF1 mice were suspended in centrifugation buffer (320 mM sucrose, 2 mM EDTA, 5 mM MgCl2) and the tissues were homogenized with an Ultra-Turrax homogenizer (Sigma-Aldrich, UK). Tissue homogenates were centrifuged at 1600g for 10 minutes and the resulting supernatant collected. This pellet was resuspended in centrifugation buffer, centrifuged as before, and the supernatant collected. Supernatants were combined before undergoing further centrifugation at 28,000g for 20 minutes. The supernatant was discarded and the pellet resuspended in buffer A (50 mM Tris, 2 mM EDTA, 5 mM MgCl2 at pH 7.0) and incubated at 37°C for 10 minutes. Following the incubation, the suspension was centrifuged for 20 minutes at 23,000g. After resuspending the pellet in buffer A, the suspension was incubated for 40 minutes at room temperature before a final centrifugation for 15 minutes at 11,000g. The final pellet was resuspended in buffer B (50 mM Tris, 1 mM EDTA, 3 mM MgCl2) and the final protein concentration, determined by Bio-Rad Dc kit, was 1 mg/ml. All centrifugation procedures were carried out at 4°C. Prepared brain membranes were stored at −80°C and defrosted on the day of the experiment.
Cell Membrane Preparation.
A large batch of hCB1R cells was prepared by expanding the cell culture to twenty 220-ml flasks. To prepare cell membranes, cells were washed in phosphate-buffered saline and then incubated with phosphate-buffered saline containing 1 mM EDTA for 5 minutes. Cells were then harvested by scraping into the buffer and centrifuged at 400g for 5 minutes. Cell pellets were then resuspended in ice-cold buffer A (320 mM sucrose, 10 mM HEPES, 1 mM EDTA, pH 7.4) and homogenized using a glass dounce homogenizer. Cell homogenates were then centrifuged at 1600g for 10 minutes at 4°C and the supernatant was collected. The pellet was resuspended, homogenized, and centrifuged at 1600g, and the supernatant was collected. Supernatants were pooled before undergoing further centrifugation at 50,000g for 2 hours at 4°C. The supernatant was discarded and the pellet was resuspended in buffer B (50 mM HEPES, 0.5 mM EDTA, 10 mM MgCl2, pH 7.4), aliquoted into 0.5-ml tubes, and stored at −80°C. Protein concentration was determined against a BSA standard curve using BioRad Bradford protein detection reagent.
Signaling Assays
[35S]GTPγS Binding Assay
Mouse brain membranes (5 μg protein) or hCB1R cell membranes (25 μg protein) were preincubated for 30 minutes at 30°C with adenosine deaminase (0.5 IU/ml). The membranes were then incubated with the agonist ± modulator or vehicle for 60 minutes at 30°C in assay buffer (50 mM Tris-HCl; 50 mM Tris-Base; 5 mM MgCl2; 1 mM EDTA; 100 mM NaCl; 1 mM DTT; 0.1% BSA) in the presence of 0.1 nM [35S]GTPγS and 30 μM GDP, in a final volume of 500 μl. Binding was initiated by the addition of [35S]GTPγS. Nonspecific binding was measured in the presence of 30 μM GTPγS. The reaction was terminated by rapid vacuum filtration (50 mM Tris-HCl; 50 mM Tris-Base; 0.1% BSA) using a 24-well sampling manifold (cell harvester; Brandel, Gaithersburg, MD) and GF/B filters (Whatman, Maidstone, UK) that had been soaked in buffer (50 mM Tris-HCl; 50 mM Tris-Base; 0.1% BSA) for at least 24 hours. Each reaction tube was washed five times with a 1.2-ml aliquot of ice-cold wash buffer. The filters were oven-dried for at least 60 minutes and then placed in 4 ml of scintillation fluid (Ultima Gold XR, PerkinElmer, Cambridge, UK). Radioactivity was quantified by liquid scintillation spectrometry.
Data Analysis.
Raw data were presented as cpm. Basal level was defined as zero. Results were calculated as a percentage change from basal level of [35S]GTPγS binding (in the presence of vehicle). Data were analyzed by nonlinear regression analysis of sigmoidal dose-response curves using GraphPad Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis are presented as Emax with 95% confidence interval (CI) and pEC50 (logEC50) ±S.E.M.
PathHunter CB1 β-Arrestin Assays
PathHunter hCB1 β-arrestin cells were plated 48 hours before use and incubated at 37°C, 5% CO2 in a humidified incubator. Compounds were dissolved in dimethylsulfoxide (DMSO) and diluted in OCC media. Five μl of allosteric modulator or vehicle solution was added to each well and incubated for 60 minutes. Five μl of agonist was added to each well followed by a 90-minute incubation. Fifty-five μl of detection reagent was then added followed by a further 90-minute incubation at room temperature. Chemiluminescence, indicated as relative light units (RLU), was measured on a standard luminescence plate reader.
Data Analysis.
Raw data were RLU. Basal level was defined as zero. Results were calculated as the percentage of CP55940 maximum effect. Data were analyzed by nonlinear regression analysis of sigmoidal dose response curves using GraphPad Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis are presented as Emax with 95% CI and pEC50 (logEC50) ±S.E.M.
Cyclic AMP Assays
hCB1R cells (0.6 × 105 cells/ml) were preincubated in PBS containing 1 mg/ml BSA (assay buffer) for 30 minutes at 37°C with rolipram (10 μM). This was followed by further 30 minutes incubation at 37°C with cannabinoid agonist ± Org 27569 or vehicle. A final incubation of 30 minutes with 5 μM forskolin in a total volume of 500 μl then took place. The reaction was terminated by addition 0.1 M HCl and centrifuged to remove cell debris. The pH was brought to 8 or 9 using 1 M NaOH and cyclic AMP content was then measured using a radioimmunoassay kit (The Biotrak; Amersham). Forskolin and rolipram were dissolved in DMSO.
Data Analysis.
Results were calculated as the percentage inhibition of forskolin-stimulated cAMP production (pmol/mg). Data were analyzed by nonlinear regression analysis of sigmoidal dose response curves using GraphPad Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis were presented as Emax with 95% CI and pEC50 (logEC50) ± S.E.M.
AlphaScreen SureFire ERK 1/2 Phosphorylation Assay
ERK1/2 MAP-Kinase Phosphorylation Assay.
For experimental studies of ERK1/2 MAP-kinase phosphorylation, hCB1R cells (40,000 cells/well) were plated onto 96-well plates and serum-starved for 24 hours. Cells were then washed with DMEM before the addition of agonist ± Org 27569 or vehicle at the desired concentration. After a 6-minute incubation at 37°C in a humidified atmosphere, ice-cold lysis buffer (provided with the AlphaScreen SureFire kit) was added to each well and the plate was placed at −80°C for at least 1 hour.
AlphaScreen SureFire ERK Assay.
The assay was performed in 384-well white Proxiplates according to the manufacturer instructions. Briefly, 4-μl samples were incubated with 7 μl of mixture containing 1 part donor beads,1 part acceptor beads, 10 parts activation buffer, and 60 parts reaction buffer. Plates were incubated for 3 hours at 25°C in the dark and read with the Envision system (PerkinElmer) using AlphaScreen settings.
Data Analysis.
Raw data were presented as “Envision units.” Basal level was defined as zero. Results were presented as means and variability as S.E.M. or 95% CI of the percent stimulation of phosphorylated ERK1/2 above the basal level (in the presence of vehicle). Data were analyzed by nonlinear analysis of log agonist versus response curves using GraphPad Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis were presented as Emax with 95% CI and pEC50 (logEC50) ±S.E.M.
Radioligand Binding Experiments
Competition and Saturation Binding Assays
Mouse Brain Membranes.
Binding assays were performed with the CB1 receptor agonist [3H]CP55940 (0.7 nM for equilibrium or 0.1–10 nM for saturation) and CB1 receptor agonist [3H]WIN55212-2 (1.5 nM) in 1 mg/ml BSA and 50 mM Tris buffer, total assay volume 500 μl. Binding was initiated by the addition of mouse brain membranes (30 μg). Assays were carried out at 37°C for 60 minutes before termination by addition of ice-cold wash buffer (50 mM Tris buffer, 1 mg/ml BSA) and vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester, PerkinElmer) and Whatman GF/B glass-fiber filters that had been soaked in wash buffer at 4°C for 24 hours. Each reaction tube was washed five times with a 1.2-ml aliquot of buffer. The filters were oven-dried for 60 minutes and then placed in 4 ml of scintillation fluid (Ultima Gold XR, Packard), and radioactivity quantitated by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 μM of the corresponding unlabeled ligand and was 70–80% of the total binding.
hCB1R Cells.
Saturation and competition binding assays were performed by incubating 5–10 μg/well hCB1R cell membranes in assay buffer (50 mM Tris, 2.5 mM EDTA, 5 mM MgCl2, 1 mg/ml BSA, pH7.4) at 30°C for 90 minutes. Equilibrium binding assays were performed with [3H]CP55940 (0.8 nM) or [3H]WIN55212 (1.5 nM). Reactions were performed in duplicate or triplicate wells of 96-well, round bottom microtiter plates in a final volume of 200 μl. Following incubation, reactions were filtered onto GF/B filter mats presoaked in distilled H2O using a PerkinElmer Filtermate cell harvester. Filters were washed six times with ice-cold 50 mM Tris buffer (pH 7.4) then air dried and the radioactivity counted in a Microbeta Trilux liquid scintillation counter. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 10 μM of unlabeled ligand.
Data analysis.
Saturation studies are generally carried out by measuring binding of a range of radioligand concentrations at equilibrium to a constant amount of receptor in the presence and absence of a high concentration of competing, unlabeled ligand. Specific binding data can then be analyzed using the following model based on the Hill-Langmuir equation:
| (1) |
Here, Y is specific binding, Bmax is the number of binding sites for the radioligand (A), Kd is the equilibrium dissociation constant for A, and [A] is the concentration of this radioligand at half the maximum occupancy.
The saturation binding assay thus provides the affinity of the radioligand for the receptor, which is the concentration of radioligand that produces half of the maximum binding (Kd), and the receptor density in the tissue under investigation, which is the level of maximum specific binding (Bmax). GraphPad Prism 5.0 (GraphPad, San Diego, CA) was used to calculate the Kd ± S.E.M. and Bmax values with 96% CI.
Association and Dissociation Binding Assays.
Association binding experiments were performed by incubating 5–10 μg/well hCB1R cell membranes with a fixed concentration (Kd or higher) of radioligand ([3H]CP55940) in assay buffer (50 mM Tris, 2.5mM EDTA, 5 mM MgCl2, 1 mg/ml BSA, pH 7.4) at 30°C for various incubation times (1 minute to 2 hours) before termination of reactions by rapid filtration. For dissociation kinetic experiments, the radioligand was first incubated with membranes for 60 minutes to allow full association before an unlabeled competing ligand was added to each reaction at various time-points to initiate dissociation of the radioligand until reactions were terminated by filtration. Dissociation was initiated by the addition of 1 μM unlabeled ligand in the presence and absence of test compounds. For both association and dissociation assays, reactions were performed in duplicate or triplicate wells of 96-well, round bottom microtiter plates in a final volume of 200 μl. Following incubation, reactions were filtered onto GF/B filter mats presoaked in distilled H2O using a PerkinElmer Filtermate cell harvester. Filters were washed six times with ice-cold 50 mM Tris buffer, pH 7.4, then air dried and the radioactivity counted in a Microbeta Trilux liquid scintillation counter.
Data analysis.
Kinetic binding assays can be used to calculate the association or dissociation rate constants of a radioligand. Association experimental data can be of use to establish the time of a radioligand to reach equilibrium and therefore provide an optimal incubation time for saturation or competition binding assays. Association experiments also provide an observed association rate, kobs (the association rate at a given concentration of radioligand), and maximum receptor occupancy at equilibrium (Ymax). Dissociation experiments provide a dissociation rate constant for the radioligand, koff, which can be used with kobs to calculate the association rate constant kon using Eq. 2.
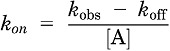 |
(2) |
Data were analyzed using the one-phase association model in GraphPad Prism 5.0 (GraphPad, San Diego, CA) to calculate the kob ± S.E.M., Ymax ± S.E.M., and koff ± S.E.M.
Statistical Analysis.
Values have been expressed as means and variability as S.E.M. or as 95% CI. Mean values have been compared using Student’s unpaired t test, one-sample tests, or analysis of variance (ANOVA) followed by Dunnett’s test or the Newman-Keuls test. P values < 0.05 were considered to be significant.
Results
Effect of Allosteric Modulators on CB1 Receptor Agonist Signaling
[35S]GTPγS Binding Assay in Brain Membranes.
In mouse brain membranes, CP55940 stimulated [35S]GTPγS binding with a pEC50 value of 8.20 ± 0.11 and Emax of 62% (95% CI, confidence limits 56 and 69). The potency of WIN55212 (pEC50 = 7.84 ± 0.08) was not significantly different from that of CP55940, but the efficacy was significantly higher: Emax of 98% (95% CI, confidence limits 90 and 107). Both allosteric modulators Org 27569 and PSNCBAM-1 (Fig. 1) produced a concentration-related reduction in the Emax values for both CP55490 and WIN55212 (Fig. 2; Table 1). However, the compounds were significantly less effective as inhibitors of WIN55212 as compared with CP55940. Neither Org27569 nor PSNCBAM-1 significantly decreased the Emax of WIN55212 until a concentration of 1 μM; in comparison, the compounds inhibited the action of CP55940 in the nM range (Fig. 2; Table 1). Neither Org 27569 nor PSNCBAM-1 significantly altered the pEC50 values of CP55940 or WIN55212 at any of the concentrations tested (Table 1). Org 27569 alone had no significant effect on [35S]GTPγS binding in mouse brain membranes at concentrations of 1 nM to 10 μM (unpublished data).
Fig. 1.

Structures of Org 27569 (Org) and PSNCBAM-1 (PSN).
Fig. 2.

[35S]GTPγS binding to mouse brain membranes. (A) The effect of Org 27569 on CP55940; (B) the effect of Org 27569 on WIN55212; (C) the effect of PSNCBAM-1 on CP55940; (D) the effect of PSNCBAM-1 on WIN55212; (E) the effect of Org 27569 on anandamide. Symbols represent mean values ±S.E.M. from three to four experiments carried out in duplicate.
TABLE 1.
Effect of Org 27569 and PSNCBAM-1 on CP55940- and WIN55212-2-stimulated [35S]GTPγS binding in mouse brain membranes
Data are mean ±S.E.M. or with 95% CI.
| Agonist | Vehicle/Modulator | pEC50 a | Emax b (95% CI) | % Inhibition ±SEM |
|---|---|---|---|---|
| % | ||||
| CP55940 | DMSO | 8.2 ± 0.1 | 62.6 (57–69) | |
| Org 27569 (10 nM) | 7.9 ± 0.3 | 43.5 (31–56)† | 33.9 ± 6.7 | |
| Org 27569 (100 nM) | 8.6 ± 0.4 | 17.98 (12–24)† | 71.8 ± 7.0 | |
| Org 27569 (1 μM) | — | — | 95.3 ± 6.6 | |
| WIN55212 | DMSO | 7.8 ± 0.09 | 98.9 (90–107) | |
| Org 27569 (10 nM) | 7.7 ± 0.2 | 83.1 (68–98) | 13.9 ± 8.4 | |
| Org 27569 (100 nM) | 8.0 ± 0.2 | 90.0 (75–105) | 8.7 ± 13.3 | |
| Org 27569 (1 μM) | 8.4 ± 0.3 | 35.9 (27–45)† | 63.6 ± 4.2 | |
| Anandamide | DMSO | 6.7 ± 0.1 | 61.4 (55–68) | |
| Org 27569 (10 nM) | 6.7 ± 0.4 | 56.6 (33–80) | 1.1 ± 12.5 | |
| Org 27569 (100 nM) | 6.4 ± 0.2 | 39.0 (30–48)† | 38.1 ± 3.8 | |
| Org 27569 (1 μM) | 7.6 ± 0.3 | 17.2 (13–22)† | 71.9 ± 3.4 | |
| CP55940 | DMSO | 7.0 ± 0.2 | 53.7 (47–60) | |
| PSN (10 nM) | 6.5 ± 0.2 | 48.7 (39–58) | 7.1 ± 12.7 | |
| PSN (100 nM) | 6.6 ± 0.4 | 32.6 (24–41)† | 41.8 ± 9.6 | |
| PSN (1 μM) | 6.0 ± 0.5 | 15.6 (6–25)† | 72.8 ± 8.4 | |
| WIN55212 | DMSO | 7.8 ± 0.1 | 75.9 (68–84) | |
| PSN (10 nM) | 7.2 ± 0.3 | 78.4 (55–102) | 0.4 ± 15.0 | |
| PSN (100 nM) | 7.1 ± 0.1 | 66.3 (58–74) | 11.0 ± 3.4 | |
| PSN (1 μM) | 6.8 ± 0.4 | 25.9 (13–39)† | 65.0 ± 10.7 |
Negative logarithm of the agonist EC50 value, determined using nonlinear regression analysis. Values represent the mean ± standard error of the mean (S.E.M.) of four to six experiments.
Maximal agonist effect, determined using nonlinear regression analysis. Values represent the mean with 95% CI of four to six experiments.
Significantly different (nonoverlapping confidence limits) from the DMSO vehicle control.
Org 27569 also significantly decreased the efficacy of the endogenous cannabinoid anandamide (AEA) in this assay (Fig. 2E; Table 1). The endocannabinoid was more susceptible to inhibition by the allosteric modulator than WIN55212, with significant inhibition being observed in the presence of 100 nM Org 27569.
[35S]GTPγS Binding Assay in hCB1R Cell Membranes.
In hCB1-expressing cells, CP55940-stimulated [35S]GTPγS binding with a pEC50 value of 7.35 ± 0.19 and Emax of 65% (95% CI, confidence limits 57 and 72). Neither the potency (pEC50 = 6.70 ± 0.17) nor the efficacy (Emax = 70.4%, 95% CI, confidence limits 61 and 79) of WIN55212 was significantly different from that of CP55940 (Fig. 3). Org 27569 produced a concentration-related reduction in the Emax values for both CP55490 and WIN55212. However, the compound was significantly less effective as an inhibitor of WIN55212 as compared with CP55940 (Fig. 3, A and B; Table 2). In hCB1R cells, Org 27569 behaved as a weak inverse agonist producing a small but significant decrease in basal [35S]GTPγS binding at concentrations of 1 and 10 μM (Fig. 3E). This effect was not observed in wild type CHO cells.
Fig. 3.

[35S]GTPγS binding to hCB1-expressing cells. (A) The effect of Org 27569 on CP55940; (B) the effect of Org 27569 on WIN55212; (C) the effect of PSNCBAM-1 on CP55940; (D) the effect of PSNCBAM-1 on WIN55212; (E) the effect of Org 27569 alone in hCB1 cells and nontransfected cells. Symbols represent mean values ±S.E.M. from three to four experiments carried out in duplicate.
TABLE 2.
Effect of Org 27569 on CP55940 and WIN55212 in [35S]GTPγS binding, cAMP, and pERK assay in hCB1R cells
| Agonist | Vehicle/Modulator | pEC50 a | Emax b (%) (95% CI) | % Inhibition SEM |
|---|---|---|---|---|
| [35S]GTPγS Binding (% Stimulation above Basal) | ||||
| CP55940 | DMSO | 7.3 ± 0.2 | 64.7 (57–72) | |
| Org 27569 (10 nM) | 6.2 ± 0.4 | 76.2 (48–105) | 13.8 ± 31 | |
| Org 27569 (100 nM) | 6.9 ± 0.6 | 35.2 (19–52)† | 46.3 ± 18 | |
| Org 27569 (1 μM) | — | —† | 94.4 ± 6.0 | |
| WIN55212 | DMSO | 6.7 ± 0.2 | 70.4 (62–79) | |
| Org 27569 (100 nM) | 6.5 ± 0.3 | 67.7 (49–87) | 1.2 ± 16 | |
| Org 27569 (1 μM) | 6.9 ± 0.4 | 33.1 (20–47)† | 43.4 ± 18 | |
| cAMP Assay (% Inhibition of Forskolin Stimulation) | ||||
| CP55940 | DMSO | 8.1 ± 0.2 | 86.6 (75–98) | |
| Org 27569 (10 nM) | 7.6 ± 0.4 | 55.0 (37–73)† | 30.3 ± 12.0 | |
| Org 27569 (100 nM) | — | —† | 155 ± 9.6 | |
| WIN55212 | DMSO | 7.1 ± 0.2 | 78.6 (64–93) | |
| Org 27569 (100 nM) | 7.0 ± 0.2 | 64.6 (51–78) | 22.3 ± 4.9 | |
| Org 27569 (1 μM) | 6.9 ± 0.3 | 41.1 (23–59)† | 37.0 ± 5.9 | |
| ERK1/2 Phosphorylation (% Increase above Basal) | ||||
| CP55940 | DMSO | 7.6 ± 0.14 | 50.0 (44–56) | |
| Org 27569 (100 nM) | 8.0 ± 0.16 | 72.0 (64–80)† | — | |
| Org 27569 (1 μM) | 7.8 ± 0.23 | 65.0 (58 -72)† | — | |
| WIN55212 | DMSO | 6.6 ± 0.53 | 40.2 (27–54) | |
| Org 27569 (100 nM) | 7.0 ± 0.67 | 50.9 (35–66) | — | |
| Org 27569 (1 μM) | 6.6 ± 0.48 | 37.0 (24–51) | — | |
Negative logarithm of the agonist EC50 value, determined using nonlinear regression analysis. Values represent the mean ± standard error of the mean (S.E.M.) of four to six experiments.
Maximal agonist effect, determined using nonlinear regression analysis. Values represent the mean with 95% CI of four to six experiments.
Significantly different (nonoverlapping confidence limits) from the DMSO vehicle.
PSNCBAM-1 displayed a similar profile to that observed with Org 27569 (Fig. 3, C and D). The stimulation of [35S]GTPγS binding induced by CP55940 was abolished by 300 nM PSNCBAM-1. This concentration did not significantly affect the stimulation induced by WIN55212, the effect of which was only inhibited by micromolar concentrations of PSNCBAM-1.
Cyclic AMP Assays in hCB1R Cell Membranes.
Next we measured the effect of Org 27569 on CB1 agonist–mediated inhibition of forskolin-stimulated cAMP production in hCB1R cells (Fig. 4, A and B). In the presence of vehicle, CP55940 inhibited forskolin-stimulated cAMP formation with an Emax of 86.6% (95% CI, confidence limits 77 and 98) and pEC50 value of 8.01 ± 0.19. WIN55212 was significantly less potent with a pEC50 value of 7.02 ± 0.20 (Student’s unpaired t test) but had similar efficacy with an Emax of 78.5% (95% CI, confidence limits 64 and 93). Org 27569 significantly reduced the Emax for CP55940 at a concentration of 10 nM, with signaling being abolished in the presence of 100 nM of Org 27569 (P < 0.001, one-sample t test) (Fig. 4B, Table 2). In the presence of Org 27569 and CP55940 the level of cAMP was significantly lower than basal (Fig. 4A). In contrast, Org 27569 was less effective as an inhibitor of WIN55212-mediated inhibition of forskolin-stimulated cAMP production (Fig. 4B, Table 2).
Fig. 4.

Cyclic AMP production in hCB1-expressing cells. (A) Effect of Org 27569 on CP55940-induced inhibition of forskolin-stimulated cAMP production (***P < 0.001, **P < 0.01, significance of difference from basal, one-sample t test); (B) effect of Org 27569 on WIN55212-induced inhibition of forskolin-stimulated cAMP production; (C) effect of CP55940 and Org 27569 alone on forskolin-stimulated cAMP production in cells treated with vehicle or PTX (5 ng/ml, 24 hours); (D) effect of SR141716A alone on forskolin-stimulated cAMP production in cells treated with vehicle or PTX (5 ng/ml, 24 hours); (E) effect of Org 27569 (100nM) on the stimulation of cAMP production (in the presence of forskolin) produced by CP55940 in cells treated with PTX (5 ng/ml, 24 hours); (F) effect of Org 27569 and CP55940 on cAMP production in the absence of forskolin. Symbols represent mean values ±S.E.M. from three to six experiments carried out in duplicate.
It has been previously demonstrated that CB1 receptors couple to both Gs and Gi proteins and can stimulate or inhibit the formation of cAMP; thus in the presence of PTX, a simulation of cAMP is revealed (Glass and Felder, 1997; Bonhous et al., 1998). In line with this, we find that, after overnight treatment of hCB1R cells with PTX (5 ng/ml), CP55940 no longer inhibits but rather stimulates the production of cAMP; the Emax for stimulation being 174% (95% CI, confidence limits 106 and 242) and the pEC50 being (5.87 ± 0.35). On the other hand, Org 27569 alone in the absence of PTX pretreatment stimulates cAMP production, an effect that is maintained following PTX treatment (Fig. 4C). The Emax for Org 27569 was 106% (95% CI, confidence limits 61 and 152) in the absence and 141% (95% CI, confidence limits 110 and 173) in the presence of PTX; the pEC50 values for Org 27569 were 5.87 ± 0.28 in the absence and 6.44 ± 0.23 in the presence of PTX. Neither Org 27569 nor CP55940 had any effect on forskolin-stimulated cAMP production in untransfected CHO cells (Fig. 4C). Notably, the CB1 receptor orthosteric inverse agonist SR141 also produced an increase in cAMP levels (Emax of 50%, 95% CI, confidence limits 32–68); an effect that was abolished after PTX pretreatment, indicating the effect was due to constitutive activation of Gi (Fig. 4D).
The stimulation of cAMP produced by CP55940 in PTX pretreated cells was abolished by 100 nM Org 27569, a concentration which alone did not stimulate cAMP production (Fig. 4E).
In the absence of forskolin, CP55940 produced a very small stimulation of cAMP production reaching an Emax of 11% (95% CI, confidence limits 7 and 14). Org 27569 did not significantly affect levels of cAMP in the absence of forskolin (Fig. 4F).
ERK 1/2 Phosphorylation Assay in hCB1R Cell Membranes.
Using an AlphaScreen SureFire ERK 1/2 phosphorylation assay kit, we measured the effect of Org 27569 on activation of ERK 1/2 phosphorylation by CB1 agonists in hCB1R cells (Fig. 5, A and B). CP55940 induced rapid, transient ERK phosphorylation, which peaked at 6 minutes and rapidly decayed by 15–20 minutes (data not shown); this is similar to data obtained by others (Daigle et al., 2008). Subsequent analysis was conducted at the 6-minute time point. In the presence of vehicle, CP55940 induced ERK1/2 phosphorylation above basal with an Emax of 50.0% (95% CI, confidence limits 44 and 56) and pEC50 value of 7.69 ± 0.14. WIN55212 was significantly less potent with a pEC50 value of 6.95 ± 0.42, but its efficacy did not differ from that of CP55940: Emax value of 40.2 (95% CI, confidence limits 27 and 54).
Fig. 5.

ERK phosphorylation in hCB1-expressing cells. (A) Effect of Org 27569 on CP55940-induced ERK phosphorylation; (B) effect of Org 27569 on WIN55212-induced ERK phosphorylation; (C) effect of CP55940 and Org 27569 alone on ERK phosphorylation in cells treated with vehicle or PTX (5 ng/ml, 24 hours).
In contrast to the inhibitory effects observed with Org 27569 in the other signaling assays, at 100 nM and 1 μM this compound significantly increased the Emax for CP55940-induced ERK 1/2 phosphorylation (Fig. 5A; Table 2). In the presence of 1 μM Org 27569, the basal level of the CP55940 log concentration-response curve was 15% (95% CI, confidence limits 6 and 24). Org 27569 had no significant effect on ERK 1/2 phosphorylation induced by WIN55212 (Fig. 5B, Table 2).
CP55940 did not induce ERK1/2 phosphorylation following pretreatment of the cells for 24 hours with PTX (5 ng/ml) (Fig. 5C). Org 27569 induced a small but significant level of ERK1/2 phosphorylation with an Emax of 19% (95% CI, confidence limits 11 and 26) and pEC50 value of 8.55 ± 0.99; the effect was abolished following PTX pretreatment (Fig. 5C).
PathHunter β-Arrestin Assays.
In the PathHunter β-arrestin CB1 assay (Fig. 6, Table 3), CP55940-stimulated β-arrestin recruitment with a pEC50 value of 7.89 ± 0.06 and Emax of 99% (95% CI, confidence limits 95 and 104). The potency of WIN55212 (pEC50 = 6.93 ± 0.14) was significantly lower than that of CP55940 (Student’s unpaired t test), but the efficacy was not significantly different; Emax of 83% (95% CI, confidence limits 70 and 97). The allosteric modulators, Org 27569 and PSNCBAM-1 produced a concentration-related reduction in the Emax values of both CP55490 and WIN55212 (Fig. 6; Table 3). Neither Org 27569 nor PSNCBAM-1 significantly altered the pEC50 values of CP55940 or WIN55212 at any of the concentrations tested (Table 3). Org 25769 also significantly decreased the efficacy of the endogenous cannabinoid anandamide (AEA) in this assay (Fig. 6E; Table 3). Org 27569 alone had no significant effect on β-arrestin recruitment (Fig. 6F).
Fig. 6.

PathHunter β-arrestin assay performed with hCB1 cells. (A) The effect of Org 27569 on CP55940; (B) the effect of Org 27569 on WIN55212; (C) the effect of PSNCBAM-1 on CP55940; (D) the effect of PSNCBAM-1 on WIN55212; (E) the effect of Org 27569 on anandamide; (F) the effect of Org 27569 alone. Symbols represent mean values ±S.E.M. from two to four experiments carried out in duplicate.
TABLE 3.
Effect of Org 27569 and PSNCBAM-1 on CP55940 and WIN55212-2 in the PathHunter β-arrestin assay
Increase in luminescence (relative light units, RLU) are presented as a percentage of the maximal CP55940 stimulation.
| Agonist | Vehicle/Modulator | pEC50 a | Emaxb (%) (95% CI) | % Inhibition ±SEM |
|---|---|---|---|---|
| CP55940 | DMSO | 7.9 ± 0.06 | 99.5 (95–104) | |
| Org 27569 (1 nM) | 8.1 ± 0.1 | 66.7 (60–73)† | 32.9 ± 5.2 | |
| Org 27569 (10 nM) | 8.0 ± 0.1 | 15.3 (13.5–17)† | 84.3 ± 1.1 | |
| Org 27569 (100 nM) | — | —† | 98.9 ± 0.2 | |
| WIN55212 | DMSO | 6.9 ± 0.1 | 83.5 (70–97) | |
| Org 27569 (1 nM) | 7.1 ± 0.07 | 76.4 (70–83) | 8.5 ± 2.7 | |
| Org 27569 (10 nM) | 7.0 ± 0.1 | 37.9 (33–43)† | 54.2 ± 0.7 | |
| Org 27569 (100 nM) | 7.3 ± 0.7 | 3.4 (0.3–7)† | 94.6 ± 1.7 | |
| Anandamide | DMSO | 7.4 ± 0.2 | 100.0 (83– 117) | |
| Org 27569 (1 nM) | 7.0 ± 0.1 | 110.9 (96–126) | −10.92 ± 0.6 | |
| Org 27569 (10 nM) | 6.0 ± 0.6 | —† | 80.2 ± 3.3 | |
| Org 27569 (100 nM) | 8.8 ± 1.6 | —† | 135 ± 1.8 | |
| CP55940 | DMSO | 7.9 ± 0.06 | 102.3 (98–107) | |
| PSNCBAM (1 nM) | 7.9 ± 0.1 | 76.8 (70–83)† | 24.5 ± 4.3 | |
| PSNCBAM (10 nM) | 8.6 ± 0.3 | 18.3 (15–21)† | 82.1 ± 1.2 | |
| PSNCBAM (100 nM) | — | —† | 96.9 ± 2.1 | |
| WIN55212 | DMSO | 7.6 ± 0.1 | 101.3 (90–112) | |
| PSNCBAM (1 nM) | 7.5 ± 0.1 | 90.2 (82–98) | 10.0 ± 6.6 | |
| PSNCBAM (10 nM) | 7.8 ± 0.2 | 46.1 (39–53)† | 53.8 ± 3.7 | |
| PSNCBAM (100 nM) | 8.6 ± 1.5 | 8.9 (5–13)† | 90.5 ± 2.6 |
Negative logarithm of the agonist EC50 value, determined using nonlinear regression analysis. Values represent the mean ± standard error of the mean (S.E.M.) of four to six experiments.
Maximal agonist effect, determined using nonlinear regression analysis. Values represent the mean with 95% CI of four to six experiments.
Significantly different (nonoverlapping confidence limits) from the DMSO vehicle.
As observed in the [35S]GTPγS binding assay, the compounds were significantly less potent as inhibitors of WIN55212 as compared with CP55940. The IC50 values for the inhibitors were calculated from concentration-response curves of the inhibitor concentration versus the percentage reduction in the agonist Emax value. The IC50 values for Org 27569 against CP55940 and WIN55212 (Fig. 7) were 2.03 ± 0.37 nM and 10.17 ± 0.96 nM, respectively (P < 0.001, Student’s unpaired t test). Similarly, the IC50 values for PSNCBAM-1 against CP55940 and WIN55212 (Fig. 7) were 2.72 ± 0.33 nM and 8.74 ± 0.78 nM, respectively (P < 0.001, Student’s unpaired t test).
Fig. 7.

Comparison of IC50 values for Org 27569 and PSNCBAM-1 as inhibitors of CP55940, WIN55212, and anandamide in the β-arrestin assay performed with hCB1 cells. Columns represent mean values ±S.E.M. from three to four experiments carried out in duplicate. The IC50 values (nM) were obtained using Prism 5 to construct concentration-response curves of the inhibitor concentration versus the percentage reduction in each agonist Emax value.
Single-Point Mutation (W5.43A) of the CB1 Receptor.
In view of the significant divergence of the effects of Org 27569 on CP55940 as compared with WIN55212, we investigated its effects in cells expressing the point mutation, W5.43A. McAllister et al. (2003) have demonstrated that WIN55212 is affected by W5.43A mutations, suggesting that these residues are part of the binding site for this ligand. In contrast, CP55940 is unaffected by this mutation. In wild type cells, 1 μM Org 27569 abolished CP55950 stimulation of [35S]GTPγS binding. However, in the cells expressing the W5.43A point mutation of CB1, Org 27569 (1 μM) did not significantly alter the Emax for CP55940-induced stimulation of [35S]GTPγS binding (Supplemental Fig. 1). These data suggest that there may be an overlap in the binding pocket for WIN55212 and Org 27569.
Effect of Allosteric Modulators on CB1 Receptor Agonist Binding
Equilibrium Binding Assays.
In line with our previous studies (Price et al., 2005), we find that Org 27569 causes a significant and concentration-dependent increase in the specific binding of [3H]CP55940 to mouse brain membranes with an Emax of 212 ± 11% and a pEC50 of 6.38 ± 0.21 (Fig. 8A). Here we find that in mouse brain membranes PSNCBAM-1 similarly caused a 141 ± 7.9% increase in the specific binding of [3H]CP55940 with a pEC50 of 6.87 ± 0.46 (Fig. 8A). We have also previously reported (Price et al., 2005) that, in contrast to the increase in specific binding of [3H]CP55940, Org 27569 caused a significant and concentration-related decrease in the specific binding of [3H]SR141716A to mouse brain membranes; the displacement was incomplete and not consistent with a simple model of competitive displacement. We have also previously reported that, in hCB1R cells, PSNCBAM-1 caused a significant and concentration-dependent, increase in [3H]CP55940 binding of 159 ± 9% with a pEC50 of 8.08 ± 0.20 (Horswill et al., 2007). In hCB1R cells PSNCBAM-1 caused a significant and concentration-related decrease in the specific binding of [3H]SR141716A with a pEC50 of 5.65 ± 0.07; the displacement was incomplete and not consistent with a simple model of competitive displacement (Horswill et al., 2007).
Fig. 8.

Effect of allosteric modulators on the equilibrium binding. (A) Effect of Org 27569 and PSNCBAM-1 on [3H]CP55940 binding to mouse brain membranes; (B) effect of Org 27569 and PSNCBAM-1 on [3H]WIN55212 binding to mouse brain membranes; and (C) effect of PSNCBAM-1 on [3H]WIN55212 binding to hCB1R cell membranes. Symbols represent mean values ±S.E.M. from three independent experiments. Data were best fitted by a one-site competition binding model.
Here we have investigated the effect of both compounds on the specific binding of [3H]WIN55212 for the first time. In mouse brain membranes preparations, Org 27569 had no significant effect on the specific binding of [3H]WIN55212 at concentrations of up to 10 μM (Fig. 8B). Unlabeled WIN55212 displaced [3H]WIN55212 binding by 98 ± 4% with a pEC50 of 8.21 ± 0.11 (Fig. 8B). PSNCBAM-1, at concentrations ranging from 1 nM to 10 μM, had a small but significant effect on the specific binding of [3H]WIN55212 in mouse brain membranes (Fig. 8B). In the presence of PSNCBAM-1 [3H]WIN55212 binding was increased with an Emax of 124% (95% CI, confidence limits 110 and 137), which is significantly different from 100%.
In hCB1R cells PSNCBAM-1, at concentrations ranging from 1 nM to 10 μM, had a small but significant effect on the specific binding of [3H]WIN55212. In the presence of PSNCBAM-1 [3H]WIN55212 binding was increased to 109% (95% CI, confidence limits 107 and 111), which is significantly different from 100% (Fig. 8C). SR141716A fully inhibited [3H]WIN55212 binding with a pKi of 8.02 ± 0.08 (Fig. 7C). This pKi value was consistent with that reported for SR141 against [3H]CP55940 (pKi of 8.35 ± 0.11) (Horswill et al., 2007).
These data demonstrate that PSNCBAM-1 and Org 27569 display ligand dependence exhibited in the level of binding cooperativity exhibited with [3H]CP55940 and [3H]WIN55212.
Saturation Binding Assays.
Saturation binding experiments involved investigating the equilibrium dissociation constant (Kd) and maximum occupancy (Bmax) of the CB1 receptor agonist radioligand [3H]CP55940. [3H]CP55940 bound in a saturable manner to mouse brain membranes (Fig. 9A; Table 4). Org 27569 (1 μM and 10 μM) significantly increased the Bmax of [3H]CP55940 (Fig. 8A; Table 4). However, the pKd of [3H]CP55940 was unaffected by Org 27569 at either concentration. Similarly, PSNCBAM-1 significantly increased the Bmax value of [3H]CP55940; neither 1 μM nor 10 μM PSNCBAM-1 significantly affected the pKd of [3H]CP55940 in mouse brain membranes, however there was a trend toward an increase in Kd (Fig. 8B; Table 4). Similarly, in saturation experiments using hCB1R membranes (Fig. 8C; Table 4), the presence of 1 μM PSNCBAM-1 positively modulated binding of [3H]CP55940 by increasing Bmax (Table 4). However, the pKd of [3H]CP55940 was unaffected by PSNCBAM-1 (P > 0.05, Student’s t test).
Fig. 9.

Effect of: (A) Org 27569 on saturation binding of [3H]CP55940 in mouse brain membranes; (B) PSNCBAM-1 on saturation binding of [3H]CP55940 in mouse brain membranes. Data shown are mean ±S.E.M. for five independent experiments. Data were best fitted by a one-binding site saturation model. (C) Effect of PSNCBAM-1 on saturation binding of [3H]CP55940 in hCB1 cell membranes. Data shown are mean ±S.E.M. of triplicate wells from a representative experiment that was performed three times. Data were best fitted by a one-binding site saturation model. The Bmax and Kd from three independent experiments are shown in Table 4. (D) Effect of PSNCBAM-1 on [3H]CP55940 (0.5 nM) association kinetics in hCB1 cell membranes. Data were best fitted by a one-phase association model. Data shown are mean values ± S.E.M. of triplicate wells from a single representative experiment that was performed 3 three times. The kob and Ymax parameters from three independent experiments are presented in Table 5. (E) Effect of PSNCBAM-1 on [3H]CP55940 (0.5 nM) dissociation kinetics in hCB1 cell membranes. Data shown are mean values ± S.E.M. from three3 independent experiments. Data were best fitted by a two-phase dissociation model. Data for 2 μM PSNCBAM-1 were best fitted by a one-phase dissociation model. Phase proportions and koff values for three independent experiments are displayed in Table 6.
TABLE 4.
Effect of allosteric modulators on saturation binding of [3H]CP55940 in mouse brain membranes and hCB1 cell membranes
Data shown are mean with 95% CI of three to six independent experiments.
| Kd | Bmax | |
|---|---|---|
| (95% CI) | (95% CI) | |
|
nM |
pmol/mg |
|
| Mouse Brain Membranes | ||
| [3H]CP55 + Vehicle | 2.67 (1.5–3.8) | 1.59 (1.3–1.8) |
| [3H]CP55 + Org (1 μM) | 2.16 (1.5–2.8) | 2.43 (2.2–2.7)† |
| [3H]CP55 + Org (10 μM) | 2.76 (1.7–3.8) | 3.25 (2.8–3.7)† |
| [3H]CP55 + PSN (1 μM) | 6.00 (3.1–8.9) | 3.64 (2.7–4.5)† |
| [3H]CP55 + PSN (10 μM) | 5.66 (3.6–7.7) | 2.98 (2.4–3.5)† |
| hCB1 | ||
| [3H]CP55 + Vehicle | 0.45 (0.26–0.63) | 6.4 (5.5–8.4) |
| [3H]CP55 + PSN (1 μM) | 0.56 (0.45–0.66) | 11 (10.8–12.2)† |
Indicates the values are significantly different from vehicle as indicated by non-overlapping confidence limits.
This increase in Bmax can be interpreted as an increase in the number of available binding sites for [3H]CP55940. In parallel experiments, PSNCBAM-1 at 1 μM caused no change to the level of nonspecific binding of [3H]CP55940 (data not shown). Thus the effect on Bmax would appear not to be a result of an increase in the level of nonspecific binding.
Association and Dissociation Kinetics.
Competition and saturation experiments at equilibrium showed Org 27569 and PSNCBAM-1 to increase the binding of radiolabeled agonists. To gain greater understanding of the mechanisms underlying this effect, further detailed analysis of the effects of one of these modulators (PSNCBAM-1) on agonist association and dissociation was carried out with [3H]CP55940 in hCB1R cells.
[3H]CP55940 (0.5 nM) bound to hCB1R membranes with an observed association rate constant (kob) of 0.060 ± 0.008/minute and a maximum receptor occupancy at equilibrium (Ymax) of 1.59 ± 0.10 pmol/mg (Fig. 9C). When association of [3H]CP55940 was measured in the presence of PSNCBAM-1 (2 μM), the compound had no effect on kob, values being 6.0 ± 0.8/min × 10−2 and 5.7 ± 0.4/min × 10−2 in DMSO- and PSN-treated, respectively (P > 0.05, Student’s t test), but significantly elevated Ymax of [3H]CP55940; values being 1.59 ± 0.1 pmol/mg and 2.46 ± 0.21 pmol/mg in DMSO- and PSN-treated, respectively (P < 0.05, Student’s t test); an increase of 55%, which corresponds closely to previously observed effects for this compound in competition binding assays.
[3H]CP55940 dissociated from CB1 receptors in a biphasic manner in the absence of PSNCBAM-1 (data were best fitted to a two-phase dissociation curve) (Fig. 9D). This was characterized by a fast phase during which 40.3 ± 1.9% of [3H]CP55940 became dissociated, followed by a slow phase during which the remaining 59.7 ± 1.9% became dissociated. Approximately 20% of radioligand was not dissociated within the time of the experiment (120 minutes) but was predicted to become so from the trend of the curve. Such a biphasic relationship indicates that high- and low-affinity binding sites at the CB1 receptor for [3H]CP55940 are present in these membranes. A common explanation for this is that the fast phase corresponds to low–affinity binding and thus the radioligand dissociates from the receptor quickly, whereas the slow phase corresponds to high-affinity binding and thus the radioligand dissociates more gradually.
When PSNCBAM-1 was present during the dissociation phase at 0.1 μM and 2 μM, it caused a significant concentration-dependent increase in the proportion of slow phase dissociation (Fig. 9D; Table 5), such that its maximal effect produced 100% slow-phase dissociation (data were fitted best to a one-phase dissociation curve) (P < 0.01, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison). In the presence of 2 μM there was a slight alteration of the slow phase koff for [3H]CP55940. When present, the fast phase koff was not significantly altered by 0.03 μM or 0.1 μM PSNCBAM-1 (P > 0.05, one-way ANOVA followed by Dunnett’s multiple comparison) but was decreased marginally by 2 μM PSNCBAM-1 (P < 0.05, one-way ANOVA followed by Dunnett’s multiple comparison). Thus, PSNCBAM-1 appeared to increase the proportion of high-affinity [3H]CP55940 binding while not greatly affecting its dissociation rate.
TABLE 5.
Effect of PSNCBAM-1 on [3H]CP55940 (0.5 nM) dissociation from hCB1 cell membranes
Data shown are mean ±S.E.M. of 3 independent experiments. *P < 0.05 **P < 0.01 one-way ANOVA followed by Dunnett’s multiple comparison.
| Slow Phase | Koff (Slow) | Koff (Fast) | |
|---|---|---|---|
| % | min–1 ×10−2 | min–1 × 10−2 | |
| [3H]CP55 +Vehicle | 59.7 ± 1.9 | 1.1 ± 0.2 | 32 ± 20 |
| [3H]CP55 + 30 nM PSN | 64.4 ± 3.9 | 1.2 ± 0.1 | 63 ± 22 |
| [3H]CP55 + 100 nM PSN | 79.0 ± 4.0** | 0.9 ± 0.1 | 34 ± 9 |
| [3H]CP55 + 2 μM PSN | 100 ± 0** | 0.4 ± 0.04* | — |
Effects of Gpp(NH)p.
Gpp(NH)p is a nonhydrolyzable analog of guanosine triphosphate and is known to cause uncoupling of G proteins from GPCRs. Such uncoupling of G proteins generally leads to a decrease in numbers of receptors in the high-affinity agonist binding conformation, thus reducing agonist binding. To establish whether the ability of PSNCBAM-1 to increase [3H]CP55940 binding was dependent on G proteins being associated with CB1 receptors, binding experiments in the presence of Gpp(NH)p were performed. Gpp(NH)p caused a concentration-dependent decrease in [3H]CP55940 binding to hCB1R membranes. Gpp(NH)p at 100-μM concentration decreased the pKd of [3H]CP55940 while apparently having little effect on Bmax (Fig. 10A); the pKd values were 0.89 nM (95% CI, confidence limits 0.54 and 1.23) and 3.11 nM (95% CI, confidence limits 1.30 and 4.90) in the presence and absence of Gpp(NH)p, respectively; the Bmax values were 3.84 pmol/mg (95% CI, confidence limits 3.32 and 4.35) and 4.09 nM (95% CI, confidence limits 2.82 and 5.36) in the presence and absence of Gpp(NH)p, respectively.
Fig. 10.

Effect of Gpp(NH)p and/or PSNCBAM-1 (1 μM) in hCB1R cell membranes on: (A) saturation binding of [3H]CP55940; (B) [3H]CP55940 equilibrium binding; (C) [3H]CP55940 dissociation. Data shown in (A) are mean values ±S.E.M. of triplicate wells from a single experiment that was performed twice. Data were best fitted using a one-site saturation binding model. Data shown in (B) are mean values ±S.E.M. from three independent experiments. **P < 0.01, #P > 0.05; one-way ANOVA followed by Bonferroni’s multiple comparison. Data shown in (C) are mean values ±S.E.M. of triplicate wells; the experiment was performed twice. Data for all groups were best fitted using a two-phase dissociation model.
We also demonstrated that PSNCBAM-1 increases the Bmax of [3H]CP55940 in both brain membranes and hCB1R cells (Fig. 9, A and B). The mechanism underlying this increase in Bmax remains to be established. However, one possible explanation for this increase is that it resulted from an increase in the number of receptors that are coupled to G protein—a condition that can give receptors higher affinity for agonist binding. If PSNCBAM-1 did indeed exert its effects through increased G protein association with receptors, then it might be expected to lose this property in the presence of Gpp(NH)p, in which receptors are uncoupled from G proteins. As displayed in Fig. 10B, 100 μM Gpp(NH)p significantly reduced 0.5 nM [3H]CP55940 binding by 46 ± 0.4% as would be predicted from the data displayed in Fig. 10A (P < 0.01, one-way ANOVA followed by Bonferroni’s multiple comparison). Similar to results obtain in previous equilibrium binding experiments, PSNCBAM-1 significantly increased [3H]CP55940 binding by 59 ± 8% (P < 0.01, one-way ANOVA followed by Bonferroni’s multiple comparison). Surprisingly, when Gpp(NH)p was present, PSNCBAM-1 not only blocked the reduction in agonist binding caused by this agent, but also significantly increased the binding of [3H]CP55940 by 35 ± 0.1% above its level of binding in the absence of Gpp(NH)p (P < 0.01, one-way ANOVA followed by Bonferroni’s multiple comparison). The difference between the effect of 1μM PSNCBAM-1 treatment in the presence and absence of Gpp(NH)p was not statistically significant indicating that Gpp(NH)p has little effect on agonist binding in the presence of PSNCBAM-1 (P > 0.05, one-way ANOVA followed by Bonferroni’s multiple comparison). Hence PSNCBAM-1 apparently retained its ability to increase [3H]CP55940 binding even under conditions when CB1 receptors would be expected to be uncoupled from G proteins.
As shown in Fig. 10C, Gpp(NH)p (100 μM) affected [3H]CP55940 dissociation by decreasing the slow phase of dissociation from 57.4 (95% CI, confidence limits 54.6 and 59.8), to 29% (95% CI, confidence limits 25 and 33). This effect was the opposite of that produced by PSNCBAM-1 which increased the slow phase of dissociation (Fig. 9C). Like PSNCBAM-1, Gpp(NH)p caused only a small change in either the rapid or slow koff values (Table 6). Hence Gpp(NH)p produced effects which are consistent with an decrease in the proportion of high-affinity [3H]CP55940 binding sites. As also shown in Fig. 10C, in the presence of both PSNCBAM-1 (2 µM) and Gpp(NH)p (100 μM) the slow phase of [3H]CP55940 dissociation was 58% (95% CI, confidence limits 57 and 59) of total binding, a proportion which is very close to that in the absence of these agents. Again there was little change in the rapid or slow koff values when both PSNCBAM-1 and Gpp(NH)p were present (Table 6). Thus, in dissociation experiments, PSNCBAM-1 appeared completely to reverse the effects of Gpp(NH)p (as was previously observed in equilibrium binding experiments).
TABLE 6.
Effect of Gpp(NH)p and PSNCBAM-1 on [3H]CP55940 dissociation from hCB1 cell membranes
Control data are mean ±S.E.M. of three experiments. Data in the presence of Gpp(NH)p are mean with 95% CI of two independent experiments.
| Slow Phase | Koff (Slow) | Koff (Fast) | |
|---|---|---|---|
| Control | 57.4 (54.6–59.8) | 1.1 (0.7–1.7) | 32 (8.9–111.5) |
| 100 μM Gpp(NH)p | 29 (25–33)† | 2.2 (2.4–2.1)† | 44.3 (44.2–44.4) |
| 100 μM Gpp(NH)p + 2 μM PSN | 58 (57–59) | 0.9 (0.8–1.0) | 49.6 (47.1–52.2) |
Significantly different (nonoverlapping confidence limits) from the DMSO vehicle.
Discussion
This study further highlights the unique pharmacological profile displayed by CB1 receptor allosteric modulators. Here we present a comprehensive characterization of the effects of two allosteric modulators on a diverse range of CB1 receptor–coupled signaling pathways and on agonist-binding kinetics. Three major findings are presented here: These allosteric modulators display (1) ligand-dependent effects, (2) signaling pathway–dependent effects, and (3) effects on the maximum occupancy of CB1 receptor agonists.
Studies have highlighted differences in the ligand recognition site for WIN55212 (and other aminoalkylindoles) and that of structurally-distinct agonists, including CP55940 and anandamide (McAllister et al., 2003; Abood, 2005; Kapur et al., 2007). In particular, mutation of W5.43A results in a significant loss of affinity of WIN55212 and SR141716A, while the affinity of CP55940 was unaffected by this mutation (for review see Abood, 2005). In line with previous findings (Wang et al., 2011), here we show that the allosteric inhibitors display a marked ligand-selectivity in a broad range of CB1 receptor agonist signaling assays; both compounds are significantly less potent as inhibitors of WIN55212 signaling compared with CP55940, as measured in the [35S]GTPγS, β-arrestin, and cAMP assays. We find that neither compound affects the specific binding of [3H]WIN55212 at concentrations up to 10 μM. Furthermore, in cells expressing the CB1 mutation W5.43A, Org 27569 no longer inhibits CP55940-mediated signaling. Pharmacological and mutation studies also suggest a separation of the binding sites for the aminoalkylindoles from those of the other classes of cannabinoid ligands. For example the K3.28192 mutation has no effect on the affinity or efficacy of WIN55212 but causes a significant loss of affinity and efficacy of the other three classes of cannabinoid. In line with this, we find that the endocannabinoid anandamide and CP55940 are equally susceptible to antagonism by Org 27569, while the effect on WIN55212 is divergent. Taken together, the data suggest some commonality in the binding pocket for WIN55212 and that of the allosteric modulators, the consequence being that WIN55212 is less susceptible to antagonism by these compounds. It is important to note that it is possible that this mutation may not affect the affinity of Org 27569, but might rather affect the ability of the CB1R to transmit cooperativity between CP55940 and Org27569. Further radioligand binding studies and studies directed at identifying the location and function of the allosteric binding pocket are in progress.
We also found that Org 27569 displays an intriguing profile of signaling pathway specificity. The compound produces a marked inhibition of CP55940-induced stimulation of [35S]GTPγS binding in mouse brain membranes and hCB1R cells. Furthermore, it potently inhibit β-arrestin recruitment, which is PTX-insensitive. Org 27569 also apparently inhibits Gαi-mediated CP55940-induced inhibition of forskolin-stimulated cAMP production. As previously shown by others (Glass and Felder, 1997; Bonhaus et al., 1998; Chen et al., 2010), pretreatment of CB1R expressing cells with PTX unmasks CB1 receptor-mediated, Gαs-coupled stimulation of cAMP production; Org 27569 (100nM) also abolished CP55940-induced increases in cAMP production. However, Org 27569 alone (at concentrations above 100nM) stimulated cAMP production; an effect that was unaffected by PTX pretreatment. Org 27569 had no effect on cAMP levels in the absence of forskolin. It is known that forkolin and Gαs synergize to activate adenylyl cylase (Barovsky and Brooker, 1985; Glass and Felder, 1997). Taken together, the data suggest that Org 27569 can act both as a CB1 receptor allosteric agonist of Gαs-coupled CB1 receptor signaling and (at lower concentrations) as an allosteric inhibitor of orthosteric agonist–induced activation of Gαs signaling. It is unlikely that the increase in cAMP observed with Org 27569 is inverse agonism of Gαi-mediated signaling because the effect is unaffected by PTX treatment. In contrast, the increase in cAMP observed with the CB1 receptor inverse agonist SR141716A is abolished by PTX pretreatment. Because the Org 27569 increases cAMP alone, it is not possible to conclude if the inhibition by this compound of CPP55940-induced inhibition of cAMP is due to inhibition Gαi- signaling or simply “physiologic antagonism” due to concomitant activation of Gαs.
In contrast to the inhibition of orthosteric agonist signaling observed in other assays, Org 27569 enhances CP55940-induced ERK phosphorylation. As observed for Gαs-mediated increases in cAMP, Org 27569 can also act as an allosteric agonist (low-efficacy) to induce ERK phosphorylation; an effect which is Gαi-mediated because this effect is lost in the presence of PTX.
Ahn et al. (2012) have recently reported that Org 27569 acts as an inhibitor of both basal and agonist-induced G protein coupling in a [35S]GTPγS assay. However, Ahn et al. (2012) found that this compound alone, at the high concentration of 10 μM, promotes receptor internalization and increases ERK phosphorylation. Furthermore, the effects on pERK seemed to be G protein independent (PTX-insensitive). We also find that Org 27569 alone acts as a weak inverse agonist in the [35S]GTPγS assay but apparently as an agonist of Gαs signaling and a weak partial agonist of Gαi signaling in pERK assays, respectively. Furthermore we find that this compound alone has no effect on β-arrestin recruitment, which is known to be PTX-insensitive (van der Lee et al., 2009), but does act as an inhibitor of CB1 agonist–induced β-arrestin recruitment. In line with findings of Ahn et al. (2012), we also identify marked divergence of the effect of Org 27569 on CB1 receptor agonist–induced ERK phosphorylation, whereby Org 27569 (100 nM and 1 μM) significantly increases the efficacy (Emax) of CP55940 as compared with inhibition observed in all other assays. Notably the ERK phosphorylation induced by CP55940 and Org 27569 is abolished after PTX treatment. Others have previously demonstrated that CB1-mediated ERK phosphorylation is Gαi-mediated (Chen et al., 2010); our results present a fascinating pharmacological profile for Org 27569, which apparently traffics CB1-mediated signaling, potentially inhibiting orthosteric agonist–mediated cellular events, mediated by cAMP and β-arrestin, while enhancing CB1-pERK–mediated cellular events.
There are well-documented examples of agonist-selective CB1 receptor coupling (reviewed by Bosier et al., 2010). For example, in the mouse tetrad, WIN55212 is more potent in reducing mobility than in producing antinociception, whereas CP 55940 is significantly more potent in reducing motor activity than producing catalepsy. Agonist-selective signaling or pharmacodynamic variations may underlie these ligand differences. Reports have identified the CB1-activated ERK signaling cascade as a key mediator of several forms of cocaine-induced synaptic plasticity, thereby implicating this cascade in addiction (Pan et al., 2011). It is conceivable that a CB1 receptor allosteric modulator may be designed that will selectively modulates pERK, thus providing a more targeted treatment of addiction. To our knowledge, the differential effects of Org 27569 described in this paper provide the first example of signal transduction–related biased antagonism of CB1 receptor signaling.
In an effort to understand the nature of the receptor modulation induced by Org 27569 and PSNCBAM-1, we carried out saturation binding studies. In these experiments both allosteric modulators surprisingly caused an elevation in Bmax of [3H]CP55940, indicating an increase in the number of available binding sites in the presence of these compounds. We observed no significant change in agonist affinity (Kd). This contrasts with the Org 27569-induced increase in [3H]CP55940 affinity with no change in maximum occupancy reported recently by Ahn et al. (2012), although notably, they did observe a trend toward an increase in Bmax in the presence of Org 27569.
In kinetic binding assays with [3H]CP55940 in CB1R cells, it appears that 60% of the CB1 receptors occupied by CP55940 are G protein-coupled (slow dissociation phase, high-affinity), whereas the remaining 40% are uncoupled (fast dissociation phase, low-affinity). The fact that the G protein uncoupling agent Gpp(NH)p reduced the slow phase of binding, and thus the fraction of receptors coupled to G proteins, supports this hypothesis. In hCB1R membranes, PSNCBAM-1 did not significantly affect either the observed association rate constant (kob) or dissociation rate constant (koff) of [3H]CP55940, but instead exerted a substantial effect on the proportions of fast and slow phases of dissociation. The koff of a ligand is highly reflective of its binding affinity and therefore any alteration of koff by a modulator may indicate a change in ligand affinity. The lack of substantial PSNCBAM-1 effects on CP55940 kob and koff implies that this compound causes very little alteration in CP55940 affinity. These results are consistent with saturation binding experiments at equilibrium where neither modulator altered the Kd of [3H]CP55940. In contrast, the slow phase of CP55940 dissociation was greatly augmented from 60% in the absence of PSNCBAM-1 to 100% at the maximum PSNCBAM-1 concentration of 2 μM. Thus, consistent with the increase in Bmax observed in saturation binding assays, the presence of PSNCBAM-1 in kinetic assays appears to cause an increase in the proportion of high-affinity agonist binding sites.
Strikingly, PSNCBAM-1 retained its ability to augment CP55940 binding, even in the presence of Gpp(NH)p and blocked Gpp(NH)p effects on CP55940 dissociation. Hence, while Gpp(NH)p produced the expected effect of reducing agonist binding by uncoupling G proteins, PSNCBAM-1 effectively opposed this action in both equilibrium and kinetic binding experiments. This property of PSNCBAM-1 is consistent with its ability to increase the maximal number of binding sites (Bmax). Thus, even under conditions when G proteins would not be expected to be associated with CB1 receptors, PSNCBAM-1 apparently induces a receptor conformation that displays a high affinity toward agonist compounds.
Previous investigations have demonstrated that structurally distinct ligands regulate CB1 receptor–G protein complexes with Gαi1, Gαi2, and Gαi3 such that multiple conformations of the receptor can be evoked by ligands to regulate individual G proteins (Mukhopadhyay and Howlett, 2001, 2005; Anavi-Goffer et al., 2007). Mukhopadhyay and Howlett, (2005) found that the nonhydrolyzable GTP analog, GTPγS, promoted complete dissociation of all CB1 receptor-Gαi complexes; the CB1 agonist desacetyllevonantradol precluded GTPγS-induced dissociation of Gαi3, while leaving Gαi1 dissociation unaffected. In contrast, WIN55212 did not affect GTPγS-induced dissociation of any of the Gαi subtypes, which is consistent with the nonselectivity of this cannabinoid for Gαi subtypes. It is conceivable that the biased antagonism observed here reflects the fact that allosteric modulators facilitate interactions with specific Gα subtypes while impeding interactions with others. The increase in Bmax may be indicative of a high-affinity coupled complex that is coupled to a unique Gα pool that displays biased signaling. Differential effects of allosteric modulators on CP55940 and WIN55212 may also reflect differential Gα coupling of the agonist-allosteric bound receptor and warrant future investigation. Georgieva et al. (2008) found that CP55940 and WIN55212-bound CB1 receptor conformations have similar affinities for Giα1 but are profoundly different in their ability to activate this G protein type, WIN55212 being significantly more active. The finding presented here indicted that the modulators may promote the binding of a G protein subtype that binds to the CP-bound, but not the WIN-bound receptor
Overall radioligand binding experiments indicate that the allosteric modulators Org 27569 and PSNCBAM-1 may be able to make available a population of CB1 receptors that retains the ability to bind agonists with high affinity but is not active in terms of its capacity to trigger certain CB1 agonist signaling responses. This hypothesis, that Org 27569 induces a high-affinity nonsignaling state, is also supported by data obtained by Ahn et al. (2012). Our data prompt an extension to this hypothesis, which is that in some functional assays the formation of the same complex will result in a loss of coupling to certain signaling pathways (cAMP, β-arrestin) but simultaneously enhance signaling mediated by ERK phosphorylation. In addition, we find that Org 27569 alone can act as an allosteric agonist, activating both Gαi- and Gαs-mediated cellular responses. Taken together, the data suggest a model in which the binding of the modulators to the allosteric site stabilizes a CB1 receptor conformation that is capable of inducing Gα-dependent signaling. It may be that this conformation mimics the GTP-bound conformation and this precludes receptor uncoupling by Gpp(NH)p. As expected, this conformation has high affinity for the orthosteric agonist but inhibits orthosteric agonist signaling via certain Gα-mediated pathways (see Fig. 11).
Fig. 11.

Summary of the complex pharmacology of the CB1 receptor allosteric modulators. Left, the effect of Org 27569 alone, and right, the effect of Org 27569 on binding and signaling of the CB1 receptor orthosteric ligand CP55940.
Supplementary Material
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- CB
cannabinoid
- CB1R
CB1 receptor
- CI
confidence interval
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethyl sulphoxide
- DTT
dithiothreitol
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- GPCR
G protein-coupled receptor
- hCB1R
human cannabinoid 1 receptor
- [3H]SR141716A
[3H]N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- PSNCBAM-1
1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea
- PTX
pertussis toxin
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)triphosphate
Authorship Contributions
Participated in the research design: Baillie, Horswill, Reggio, Abood, Anavi-Goffer, Strange, Stephens, Pertwee, Ross.
Conducted experiments: Baille, Horswill, Bolognini.
Contributed new reagents or analytical tools: Reggio, Abood, McAllister.
Performed data analysis: Baillie, Horswill, Ross.
Wrote or contributed to writing manuscript: Baillie, Horswill, Reggio, Abood, Anavi-Goffer, Stephens, Pertwee, Ross.
Footnotes
The work was funded by Prosidion Inc.; and the National Institutes of Health [Grants DA003934 and DA023204].
G.L.B. and J.G.H. contributed equally to this work.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Abood ME. (2005) Molecular biology of cannabinoid receptors. Handb Exp Pharmacol 168:81–115 [DOI] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Kendall DA. (2012) Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem 287:12070–12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anavi-Goffer S, Fleischer D, Hurst DP, Lynch DL, Barnett-Norris J, Shi S, Lewis DL, Mukhopadhyay S, Howlett AC, Reggio PH, Abood ME (2007) Helix 8 Leu in the CB1 cannabinoid receptor contributes to selective signal transduction mechanisms. J Biol Chem 282:25100–25113 [DOI] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. (2007) Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci 28:374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barovsky K, Brooker G. (1985) Forskolin potentiation of cholera toxin-stimulated cyclic AMP accumulation in intact C6-2B cells. Evidence for enhanced Gs-C coupling. Mol Pharmacol 28:502–507 [PubMed] [Google Scholar]
- Bloxham, et al. (2007). Bicyclic aryl and heteroaryl receptor modulators. European Patent Application number EP2197835.
- Bonhaus DW, Chang LK, Kwan J, Martin GR. (1998) Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther 287:884–888 [PubMed] [Google Scholar]
- Bosier B, Muccioli GG, Hermans E, Lambert DM. (2010) Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol 80:1–12 [DOI] [PubMed] [Google Scholar]
- Chen XP, Yang W, Fan Y, et al. (2010) Structural determinants in the second intracellular loop of the human cannabinoid CB1 receptor mediate selective coupling to G(s) and G(i). Br J Pharmacol 161:1817–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K. (2008) Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology 54:36–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bouvier M. (2007) The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci 28:423–430 [DOI] [PubMed] [Google Scholar]
- Georgieva T, Devanathan S, Stropova D, Park CK, Salamon Z, Tollin G, Hruby VJ, Roeske WR, Yamamura HI, Varga E. (2008) Unique agonist-bound cannabinoid CB1 receptor conformations indicate agonist specificity in signaling. Eur J Pharmacol 581:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Felder CC. (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Northup JK. (1999) Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol Pharmacol 56:1362–1369 [DOI] [PubMed] [Google Scholar]
- Hall DA. (2000) Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol 58:1412–1423 [DOI] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. (2007) PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol 152:805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Hurst DP, Fleischer D, Whitnell R, Thakur GA, Makriyannis A, Reggio PH, Abood ME. (2007) Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol Pharmacol 71:1512–1524 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2007) Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci 28:407–415 [DOI] [PubMed] [Google Scholar]
- McAllister SD, Rizvi G, Anavi-Goffer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Abood ME. (2003) An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region. J Med Chem 46:5139–5152 [DOI] [PubMed] [Google Scholar]
- McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. (2004) Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem 279:48024–48037 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Howlett AC. (2001) CB1 receptor-G protein association. Subtype selectivity is determined by distinct intracellular domains. Eur J Biochem 268:499–505 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Howlett AC. (2005) Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol. Pharmacol 67: 2016–2024 [DOI] [PubMed] [Google Scholar]
- Nathan PJ, O’Neill BV, Napolitano A, Bullmore ET. (2011) Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS Neurosci Ther 17:490–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D, Liu QS. (2011) Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J Neurosci 31:11244–11255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, et al. (2005) Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol 68:1484–1495 [DOI] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. (1999) Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol 126:665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA. (2007) Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol Sci 28:567–572 [DOI] [PubMed] [Google Scholar]
- Turu G, Hunyady L. (2010) Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol 44:75–85 [DOI] [PubMed] [Google Scholar]
- van der Lee MMC, Blomenröhr M, van der Doelen AA, Wat JWY, Smits N, Hanson BJ, van Koppen CJ, Zaman GJR. (2009) Pharmacological Characterization of Receptor Redistribution and β-Arrestin Recruitment Assays for the Cannabinoid Receptor 1. J Biomol Screen 14:811–823 [DOI] [PubMed] [Google Scholar]
- Wang X, Horswill JG, Whalley BJ, Stephens GJ. (2011) Effects of the allosteric antagonist 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea (PSNCBAM-1) on CB1 receptor modulation in the cerebellum. Mol Pharmacol 79:758–767 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.