

Abstract
The intracellular aspect of the sixth transmembrane segment within the ion-permeating pore is a common binding site for many voltage-gated ion channel blockers. However, the exact site(s) at which drugs bind remain controversial. We used extensive site-directed mutagenesis coupled with molecular modeling to examine mechanisms in drug block of the human cardiac potassium channel KCNQ1. A total of 48 amino acid residues in the S6 segment, S4-S5 linker, and the proximal C-terminus of the KCNQ1 channel were mutated individually to alanine; alanines were mutated to cysteines. Residues modulating drug block were identified when mutant channels displayed <50% block on exposure to drug concentrations that inhibited wild-type current by ≥90%. Homology modeling of the KCNQ1 channel based on the Kv1.2 structure unexpectedly predicted that the key residue modulating drug block (F351) faces away from the permeating pore. In the open-state channel model, F351 lines a pocket that also includes residues L251 and V254 in S4-S5 linker. Docking calculations indicated that this pocket is large enough to accommodate quinidine. To test this hypothesis, L251A and V254A mutants were generated that display a reduced sensitivity to blockage with quinidine. Thus, our data support a model in which open state block of this channel occurs not via binding to a site directly in the pore but rather by a novel allosteric mechanism: drug access to a side pocket generated in the open-state channel configuration and lined by S6 and S4-S5 residues.
Introduction
The slowly activating delayed rectifier IKs, a major repolarizing K+ current in heart, is formed by coassembly of the α-subunit KCNQ1 and the β-subunit KCNE1(Barhanin et al., 1996; Sanguinetti et al., 1996). Mutations in either of the subunits cause congenital long QT syndrome. In addition, KCNQ1 and slowly activated potassium current (IKs) channels are targets for multiple drugs with antiarrhythmic or proarrhythmic actions. These include nonselective drugs used to treat arrhythmias, such as quinidine (Balser et al., 1991), amiodarone (Kamiya et al., 2001), azimilide (Busch et al., 1997), and clofilium (Yang et al., 1997), and drugs designed as specific IKs blockers, such as L-7 (Seebohm et al., 2003b), HMR-1556 (Gogelein et al., 2000), and chromanol 293B (Bosch et al., 1998). In addition, drugs not used in antiarrhythmic therapy have been reported to activate cardiac IKs, such as stilbenes-fenamates (Abitbol et al., 1999) and R-L3 (Seebohm et al., 2003c).
The S6 domain helix, lining the pore of many voltage-gated ion channels, has been identified as a key site for high-affinity drug binding and block in the ion channels. This includes the S6 region of domains II and IV in Na+ channels (e.g., local anesthetics and antiarrhythmics) and L-type Ca2+ channels (dihydropyridines). Among K+ channels, the S6 segment has been identified as a key region determining drug block of human ether-a-go-go potassium channel (HERG; KCNH2) and Kv1.5 (KCNA5) channels. In KCNH2, the S6 aromatic residues I647, Y652, and F656 have previously been identified as binding sites for IKr/HERG blockers (Mitcheson et al., 2000;Ishii et al., 2001; Perry et al., 2004). Mutation of these sites to alanine renders the HERG channel resistant to specific IKr inhibitors (such as dofetilide and MK-499) and the nonspecific blocker quinidine. Quinidine interacts with S6 residues in KCNA5 to inhibit the expressed current (Snyders and Yeola, 1995;Yeola et al., 1996). Previous studies (Seebohm et al., 2003a; Seebohm et al., 2003b; Seebohm et al., 2003c) have similarly identified S6 residues required for benzodiazepine IKs agonist (R-L3) or antagonist (L-7) binding. In these studies, a common feature of residues identified for drug block is that they are predicted to interact with the binding drug by facing the permeating pore.
In the present study, we initially used alanine/cysteine scanning mutagenesis of an extensive region of S6 segment and the proximal C-terminus to identify residues important for drug block. However, homology modeling suggested that key residues identified in this way were not oriented toward the channel pore. This suggests that an alternative mechanism could be involved in drug binding and/or block. Accordingly, further studies were conducted that implicate drug binding in a side pocket that is formed by several S6 and S4-S5 linker residues, previously identified as likely interacters with the C-terminal end of S6 segment (Boulet et al., 2007; Choveau et al., 2011; Labro et al., 2011) adjacent to the channel pore as a mechanism of allosteric ion channel block.
Materials and Methods
Site-Directed Mutagenesis.
A total of 48 amino acid residues in the S6 segment (30 residues, V324 to L353), S4-S5 linker (3 residues), and the proximal C-terminus (15 residues, K354 to I368) of KCNQ1 channel were substituted individually with alanine by recombinant polymerase chain reaction (PCR) mutagenesis; five wild-type (WT) alanines (A329, A336, A341, A345, and A352) were individually substituted with cysteines. For each mutation, four primers were used: two were mutagenic primers, and two were flanking primers. The mutagenic primers contained the desired point mutation at their center and were complementary to each other. The flanking primers were complementary to target sequences 5′ and 3′ of the desired mutation. They were chosen to abut convenient restriction sites to allow those sites to be used for reinsertion into the target sequence. The target sequence was denatured, and the mutagenic primers were annealed and extended using Taq polymerase. The two extension products carrying the point mutations close to their 5′ ends were then allowed to anneal and were extended, creating a DNA duplex with the mutation roughly at the center. The mutagenic primers were then removed using a QIAquick PCR Purification Kit (QIAGEN Inc., Chatsworth, CA) according to the manufacturer's instructions. The two flanking primers were then used in a standard PCR with use of the mutated DNA as template. The resulting mutated PCR fragment was then inserted into the target sequence via the restriction sites in the flanking primers. Standard PCR conditions used to accomplish these reactions included 10–100 ng of template DNA; 40 pMol each primer; 10 mM Tris (pH, 9.0; room temperature); 1.5 mM MgCl2; 50 mM KCl; 0.1% Triton X-100‘ 0.2 mM each dATP, dCTP, dGTP, dTTP; and 5 units of Taq DNA polymerase. The temperature profiles used typically were 95°C for 5 minute, at which point the polymerase was added. This was followed by 30 cycles of 95°C for 1 minute, 60°C for 1 minute, and 72°C for 1 minute. A final extension step for 5 minutes at 72°C was then added. To optimize the PCR conditions, Mg2+ concentration ranged from 1 mM to 3 mM, and the annealing temperatures ranged from 53°C to 62°C. Duplicate mutated cDNAs fragments were sequenced to ensure that polymerase or other errors had not been introduced.
Electrophysiology, FuGENE6-Mediated Channel Expression, and Cell Transfection.
cDNAs for WT human KCNQ1 and KCNE1 were provided by Drs. Michael Sanguinetti (University of Utah) and Mark Keating (currently at Novartis Institute for Biomedical Research, Cambridge, MA). Chinese hamster ovary cells were used for transient transfections. The cells lack endogenous outward currents and are thus suitable for K+ current studies. The cells were grown to confluence in F-12 nutrient mixture medium (Invitrogen, Carlsbad, CA) supplemented with 10% horse serum at 37°C. For transfection, 2 µg of the cDNA(s) of interest in the pBK-vector and 2 µg of cDNA encoding green fluorescent protein were mixed with 12 µl FuGENE6 (Roche Berlin, Germany) in 0.5 ml serum free HAM medium for 6–8 hours, after which the standard medium was restored for an additional 48-hour incubation. Green fluorescent protein was cotransfected as a marker to identify successfully transfected cells for the voltage-clamp analysis. The cells were harvested by brief trypsinization and stored in standard medium for the experiments within the next 12 hours.
Whole-Cell Voltage Clamp.
The voltage-clamp studies were performed using the same methods as previously reported (Yang et al., 2003;Kanki et al., 2004; Yang et al., 2009;Abraham et al., 2010). To determine the percentage reduction of IKCQN1 (or IKs) by drugs, activating current was elicited with 1-second (or 5 seconds for IKs) single repetitive depolarizing pulses to +60 mV from the holding potential of −80 mV, and tail current was recorded after return to −40 mV. Pulses were delivered every 15 seconds, and drug block was measured at the end of activating IKCQN1 (or IKs) current. Data were collected when the current reached a steady-state level before and during drug administration.
Solutions and Drugs.
To record ion currents, the internal pipette-filling solution contained 110 mM KCl, 5 mM K4BAPTA, 5 mM K2ATP, 1 mM MgCl2, and 10 mM HEPES. The solution was adjusted to pH of 7.2 with KOH, yielding a final [K+]i of ∼145 mM, as we have described previously. The external solution was normal Tyrode’s, containing 130 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose and was adjusted to pH of 7.35 with NaOH. Drug block was assessed after exposure to drug for ∼20 minutes. Three blocking drugs were used; quinidine and clofilium were purchased from Sigma-Aldrich (St. Louis, MO), and L-7 (Seebohm et al., 2003b) was provided by Merck Research (West Point, PA). To observe the maximal inhibitory effect, drug concentrations used in these experiments were those required to suppress the current by >90%.
Construction of 3D Models for Open- and Closed-State KCNQ1.
Full details about our construction of open- and closed-state 3D homology models for KCNQ1 are reported elsewhere (Smith et al., 2007). The open-state model is based on the crystal structure of the mammalian Kv1.2 potassium channel (Long et al., 2005). This structure provides a template for both the pore domain (S5-P-S6) of the channel and the sensor domain helices (S1-S4) and, key to this study, the S4-S5 linker. Side chain identities were missing for S1 and S3 helices in the template crystal structure, and no coordinates are available for the loops in the sensor. Thus, construction of a complete open-state sensor and pore model for KCNQ1 required use of a hybrid method of comparative modeling and Rosetta-Membrane techniques, as reported by Yarov-Yarovoy et al. (Yarov-Yarovoy et al., 2006). Because there were no experimentally determined closed-state structures of mammalian potassium channels, we used the Kv1.2 closed-state model of Yarov-Yarovoy et al. as the template to build the closed-state KCNQ1.
Quinidine Docking Calculations.
The culled cavities and pockets feature of Pymol software, version 1.4.1 (http://www.pymol.org/pymol), was used to locate empty pockets inside the open- and closed-state models of KCNQ1. This routine identified exactly one unique pocket (four identical pockets per tetramer) that contacted each of the residues that had already been implicated in the drug block mechanism by mutagenesis and patch clamp experiments. We docked one quinidine ligand into one pocket per protein tetramer with use of RosettaLigand (Meiler and Baker, 2006) automated docking software in Rosetta, version 3. As a starting point for the docking calculations, 470 different rotamers were computed for quinidine with MOE software. One of these rotamers was manually placed into the pocket located between subunit A and B of KCNQ1 to provide the docking software with a rough guideline for where to initially place quinidine. Each of the 470 quinidine rotamers was then automatically placed and optimized by the RosettaLigand Monte-Carlo refinement routines in Rosetta, version 3.0 (http://www.rosettacommons.org/software/), with full flexibility for the ligand and protein sidechains. Extra rotamers (-ex1, -ex1aro, -ex2) were used for both buried and surface protein residues. The improve_orientation option was also enabled to place and test 1000 random orientations of the ligand in the pocket to improve its positioning before the Monte Carlo search began. The abbrev2 protocol was used with ligand minimization, harmonic torsions, and protein backbone atom minimization, allowing the Cα atoms to move up to 0.3Å from initial positions. These calculations resulted in 10,000 docked complexes, which were then ranked by the RosettaLigand interface score for further analysis.
Data and Statistical Analysis.
Data are expressed as mean ± S.E.M. For comparisons among means of more than two groups, analysis of variance was used, with post hoc pairwise comparisons by Duncan’s test if statistically significant differences among means were detected. If only two groups were being compared, Student's t test was used. A P value <0.05 was considered to be statistically significant.
Results
Sites of Drug Block.
In a first set of experiments, we assessed the effects of quinidine at a concentration (100 µM) that inhibits WT KCNQ1 current by ≥90% (Fig. 1A). Figure 1B–D shows results with the three mutants most resistant to drug block among the 45 residues tested: F351A, L353A, and K358A. Of note, all three are located in the proximal C-terminus of the protein and not in the intracellular region of S6.
Fig. 1.

Representative current traces for quinidine block of WT (A) and mutant KCNQ1 channels (B-D). The three mutations shown are those in the S6 and proximal C-terminal region of KCNQ1 channel that displayed the greatest resistance to block by quinidine. Raw currents were elicited with single 1-second pulsing protocol to +60 mV from a holding potential of −80 mV before and after quinidine (100 µM; to suppress WT current by ≥ 90%). All current traces shown in all figures are obtained at the steady-state levels.
In these experiments, we noted that some mutations display dramatic gating changes, including F351A, which shows an IKs-like behavior (Fig. 1B), as previously reported (Boulet et al., 2007; Ma et al., 2011). To address the possibility that mutagenesis-induced changes in F351A channel kinetics influence drug block, we examined current-voltage (I-V) relationships before and after quinidine. As shown in Fig. 2, A and B, current amplitudes for F351A with and without quinidine were unaffected, confirming that F351A is insensitive to quinidine block. Compared with the WT KCNQ1 channel (Fig. 2C), however, the F351A channel activates very slowly, with a large positive voltage shift by approximately +40 mV. Furthermore, although we have previously reported that WT KCNQ1 channels are less sensitive to quinidine than IKs (Yang et al., 2003), the IKs-like F351A channels are insensitive to quinidine (Fig. 2D).
Fig. 2.
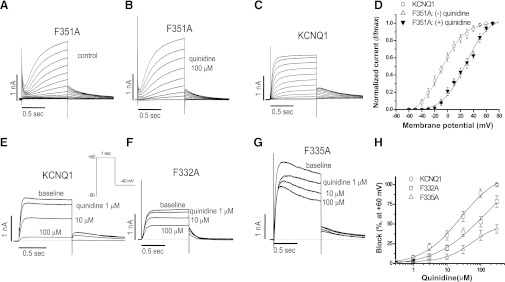
Gating changes with the F351A mutant and concentration-response relations for block of some key mutants by quinidine. (A–D) Gating changes with F351A in the absence and presence of quinidine, compared with WT-KCNQ1 alone. F351A markedly slowed channel activation and positively shifted the voltage of activation by ∼40 mV: V1/2 values were −9.5 ± 1.1 mV for WT-KCNQ1, 28.3 ± 2.2 mV for F351A without quinidine, and 27.6 ± 2.1 mV for F351A with quinidine. (E–H) Two other aromatic residue mutants (F332A and F335A) in S6 segment become quinidine-insensitive, with IC50 values at ∼120 µM for F332A and >300 µM for F335A, compared with block of WT KCNQ1 by quinidine at an IC50 value of 19.4 ± 1.7 µM. The number of cells tested was 4–5 in each group.
We also assessed concentration-response relations for quinidine block in two aromatic residue mutations (F332A and F335A) in the S6 segment. As shown in Fig. 2E–F, although quinidine blocked the WT KCNQ1 channel with an IC50 value of 19.4 ± 1.7 µM, the F332A and F335A channels were much less sensitive, with IC50 values of ∼120 µM and >300 µM, respectively.
Figure 3A summarizes the data for all 45 mutants in the distal S6 and proximal C-terminal regions. Aromatic residues in the pore region displayed variable sensitivity to drug block, greatest with F332 and F335, and near WT sensitivity for F339 and F340. Figure 3B shows that this effect is not quinidine specific and was also seen with a high concentration of clofilium (30 µM).
Fig. 3.
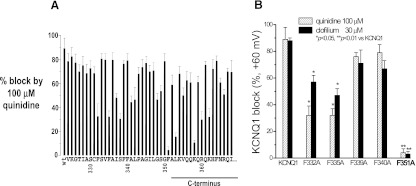
Summarized data for quinidine block of WT and all 45 mutants in the S6 and proximal C-terminal region of KCNQ1 channel. (A) Responses of all 45 mutants in the S6 (V324A-L353A) and proximal C-terminus (K354A-I368A) to block by quinidine (100 µM). F351A in the distal S6 and K358A in the proximal C-terminus were highly resistant to quinidine block (P < 0.01), whereas other mutants (F332A, F335A, I337A, S338A, A341C, L342A, S349A, L353A, K358A, R360A, and K362A) were also less sensitive to block by quinidine (P < 0.05). (B) Block of 5 aromatic residues by both quinidine (100 µM) and clofilium (30 µM). Drug block was assessed using a single 1-second pulse protocol to +60 mV from −80 mV. Steady-state block was measured at the end of activating current (n = 4–8 each).
Because both quinidine and clofilium are nonselective blockers, we also tested the effects of a specific blocker L-7 (10 µM) on WT and F351A mutant channels in the absence and presence of coexpression of the ancillary subunit KCNE1. Regardless of whether KCNE1 was present, L-7 at this concentration was a potent blocker (Fig. 4, A and D), and this effect was nearly completely absent with the F351A mutant (Fig. 4, B and E). Similar effects were observed with the K358A mutant (Fig. 4, C and F). These data are summarized in Fig. 4G. Taken together, these results suggest that the proximal C-terminus of KCNQ1 plays a crucial role in regulating drug access to its binding/blocking site.
Fig. 4.
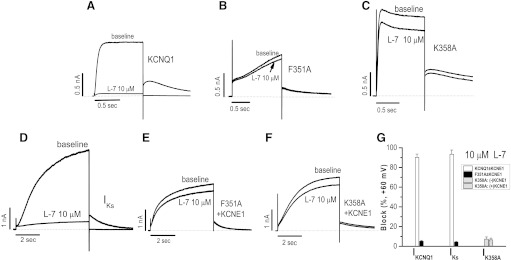
Block of IKCNQ1 and IKs by L-7, a specific IKs blocker. (A–F) Typical L-7 block of the currents expressed with the mutant F351 or K358A ± KCNE1. (G) Summary of data. To elicit the current, a single pulse to +60 mV from −80 mV was given for 1 second (for IKCNQ1) or 5 seconds (for IKs). The number of cells tested was 4–5 in each group.
Structural Modeling at the F351 Site.
Our initial hypothesis, based on analogy to KCNA5 (Kv1.5) and KCNH2 (HERG), was that residue(s) clearly linked to drug block would be oriented into the intracellular mouth of the K+-permeating channel. However, modeling KCNQ1 based on the Kv1.2 structure did not support this idea. Instead, the open-state channel model predicted that the residues (F351, L353, and K358) displaying the greatest insensitivity to drug block were pointing away from the channel and/or making contact with the S4-S5 linker (Fig. 5). Furthermore, the model predicted that three S4-S5 residues (L250, L251, and V254) would form a hydrophobic pocket in which the phenyl ring of residue F351 fits. This model, therefore, suggests that this pocket may, in fact, be a site at which quinidine interacts with the channel protein.
Fig. 5.

Stereo view of KCNQ1-F351 and S4-S5 linker residues associated with drug block. The open-state Kv1.2 structure was used as a template to model F351 and other related residues in the S4-S5 linker of KCNQ1. In this open-state channel model, residue F351 (in orange) sits a largely hydrophobic side pocket (translucent surface) formed by its own subunit’s S4-S5 linker (yellow helix, top left) and the N-terminus of S5 from a neighboring subunit (green helix, bottom left). The side chains (in gray) forming this pocket are T247, L250, L251, V254, T264, and I268. Both T264 and I268 are from the neighboring subunit (green ribbon), and the others are from the same subunit as F351 (yellow ribbon). Side chains for K358 and L353 sites, which are also highly resistant to drug block, are shown in yellow.
Drug Block at Key S4-S5 Linker Residues.
To further examine this prediction, we evaluated the drug sensitivity of the L251A and V254A mutants in the S4-S5 linker. Figure 6 shows that both the L251A and V254A mutants displayed moderately reduced current amplitude at baseline with or without KCNE1 coexpression, with slowed activation of IKCNQ1 with L251A. The most striking finding, however, was that these mutant channels displayed highly reduced sensitivity to drug block. For example, although 100 µM quinidine reduced WT currents by 94% ± 5% (IKCNQ1) and 96% ± 3% (IKs), the corresponding reductions of the currents for L251A were 32% ± 6% and 34% ± 4% and 39% ±7 % and 40% ± 5% for V254A. In our study, the mutant L250A did not generate any current for drug test, in good agreement with another work (Labro et al., 2011).
Fig. 6.
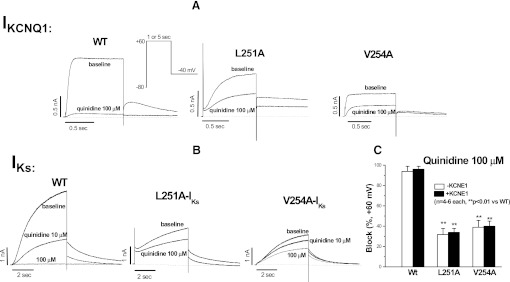
Reduced sensitivities to quinidine block in two mutants (L251A and V254A) in the S4-S5 linker of KCNQ1 channel in the absence (A) and presence (B) of KCNE1 coexpression. Representative traces before and after quinidine are presented in individual panels. The summarized data are shown in (C). Drug block was assessed using a single pulse protocol to +60 mV from −80 mV for 1 second (for IKCNQ1) or 5 seconds (for IKs).
There are three LQT1 mutations (L250H, V254L, and V254M) located in the KCNQ1 S4-S5 linker (Zareba et al., 2003; Napolitano et al., 2005; Kapa et al., 2009). We generated these mutants to determine their biophysical properties and block by quinidine. L250H and V254M expressed very small (or absent) currents; thus, evaluation of drug block was not possible. The V254L mutation also generated a reduced current; even with KCNE1 coexpression, the mutant IKs amplitude was 1846 ± 175 pA, compared with 3683 ± 254 pA (WT-IKs), at +60 mV (P < 0.01, n = 5 each). We found that the mutation was markedly resistant to drug block; 100 µM quinidine reduced V254L-IKs by 36% ± 3%, compared with 97% ± 2% for (WT-IKs) reduction, at +60 mV.
Quinidine Docking in the KCNQ1 Channel.
To further evaluate that quinidine interacts with this side pocket of the protein, we investigated whether our open-state model would be able to accommodate the molecule at this site. To explore that possibility, PyMOL software, version 1.4.1 (http://www.pymol.org/pymol), was used to identify and visualize any internal pockets present in this region of the model. As shown in Fig. 7, we did indeed identify a single pocket in this region. Furthermore, the open-state cavity is buried and qualitatively appears to be the necessary size and shape to accommodate quinidine. This cavity is bounded by the S4-S5 linker, S6 residues 347–351 of the same subunit, S6 residues 335–343 of the neighboring subunit, and S5 residues 268–275 of the same neighboring subunit. In the closed state, however, the corresponding cavity is significantly larger and is exposed to the surface of the protein on two sides. These observations correlate with the experimental observation that quinidine binds to the open state and interacts with residues in this region of the protein, but not in the closed state, because after channel closing, this hypothetical binding site changes shape and appears to have increased exposure to the exterior of the protein. Figure 8 shows top-scoring model of quinidine molecule docked to the KCNQ1 channel. This docking model further supports the idea that quinidine interacts with the KCNQ1 channel protein by binding to a pocket formed by three S4-S5 residues (L250, L251, and V254) in which the phenyl ring of residue F351 fits.
Fig. 7.

A cavity (red volume) was identified between each adjacent KCNQ1 subunit in the open- and closed-state models near the F351 side chain (orange sticks). In the open state (left panel), this cavity is approximately the correct size for quinidine to tightly fill the space and is buried inside the protein. In the closed state (right panel), the cavity is significantly larger and exposed on top and bottom to the outside of the protein. The open-state cavity is bounded by the S4-S5 linker, S6 residues 347-351 of the same subunit, S6 residues 335-343 of the neighboring subunit, and S5 residues 268-275 of the same neighboring subunit.
Fig. 8.

Top-scoring model of quinidine docked to KCNQ1. Quinidine is shown in white/blue/red. KCNQ1 side chains within 4Å of quinidine are shown in orange. Subunit A of KCNQ1 tetramer is shown in green (S4-S5 linker in top right foreground, S5 helix top left foreground, S6 helix lower left/background). Subunit B is cyan (S6 helix lower left, S5 helix lower right).
To explore possible binding modes and interaction surfaces, a model of quinidine was docked into the open-state cavity described above with use of RosettaLigand automated docking routines in Rosetta, version 3.0. Docking calculations resulted in 10,000 docked complexes, which were then ranked by the RosettaLigand interface score. Docking small molecules into a comparative model is intrinsically difficult, given that the binding pocket model is potentially inaccurate at atomic detail. Therefore, we do not expect to necessarily recover the exact binding pose of quinidine. However, we do expect to obtain a qualitative view of its general binding region and contacts it makes with neighboring residues. Thus, we analyzed the top 10 scoring models for quinidine-protein contacts. These contacts are summarized in Table 1, and the complex with the best interface score is shown in Fig. 8, highlighting the locations of these contacts. Close contacts between quinidine and F351/L251/V254 are observed in every one of these top 10 scoring models. This correlates extremely well with the experimental evidence that these three residues are involved with quinidine block and strengthens our hypothesis that the mechanism is an allosteric modulation induced by quinidine binding at the site that we have identified.
TABLE 1.
Defining the proposed quinidine/KCNQ1 interface by counting contacts between quinidine and KCNQ1 side chains across the 10 best-scoring complexes obtained from docking calculations
This table identifies each KCNQ1 residue with side chain atoms that were within 4Å of quinidine atoms in more than 50% of the 10 best-scoring complexes. Quinidine was docked in the cavity between KCNQ1 subunits A and B (The red volume located between the cyan and green subunits in Fig. 7). A “10” in column 4 means that the residue identified in columns 1–3 was in contact with quinidine in every one of the 10 best-scoring complexes. Note that contacts between quinidine and F351/L251/V254 (bold) are observed in every one of these models. This correlates exactly with our experimental evidence that these three residues are involved with quinidine block and strengthens our hypothesis that the mechanism is an allosteric modulation induced by quinidine binding at this site.
| Residue | Helix | Chain | Quinidine contacts |
|---|---|---|---|
| L347 | S6 | A | 10 |
| F351 | S6 | A | 10 |
| L251 | S4-S5 | A | 10 |
| V254 | S4-S5 | A | 10 |
| V255 | S4-S5 | A | 10 |
| F339 | S6 | B | 10 |
| L342 | S6 | B | 10 |
| I268 | S5 | B | 10 |
| P343 | S6 | A | 9 |
| L262 | S5 | A | 7 |
| T264 | S5 | B | 7 |
| F335 | S5 | B | 7 |
Discussion
In this study, we used a combination of alanine/cysteine mutagenesis, patch clamp, and homology modeling to identify a novel allosteric mechanism for drug block of the human cardiac K+ channel KCNQ1. We have found a previously unrecognized role of the S4-S5 linker and the proximal C-terminus in modulating drug block of the KCNQ1 channel. In an open-state channel model based on the Kv1.2 structure, we have strong evidence that block does not occur via binding to a site within the channel pore through which K+ permeates. Instead, the structural model predicts that the key residue, F351, is oriented into a pocket also lined by S4-S5 residues and that mutagenesis at these pocket sites also restricts quinidine block. Taken together, the findings support a model in which quinidine binds to residues within this pocket and, thus, blocks this channel by an allosteric mechanism.
In the studies of drug block, inhibition of ion current through drug binding to sites in the ion-conducting pathway (the inner pore of the channel, especially in S6 segment) has been implicated as a common mechanism for ion channel block. The block becomes more prominent when the channel is activated to open, or the open channel block. Previous studies (Mitcheson et al., 2000; Chen et al., 2002; Perry et al., 2004) with HERG implicated two aromatic residues, I647 and Y652, oriented toward the ion conducting pore, as key sites affecting this type of block. In this study, of interest, mutagenizing along S6 and into the proximal C-terminus of this channel failed to identify these residues as key binding sites, but rather identified residues more distal in S6 and in the C-terminus as key ones. Other studies (Seebohm et al., 2003b;Thomas et al., 2003) have reported that specific IKs blockers (e.g., L-7 and HMR-1556) bind to multiple potential interacting sites in the pore and S6 areas of KCNQ1 to inhibit the KCNQ1 current. In addition, studies have shown that HERG or KCNQ1 channel activators may enhance currents through interaction with residues in the S4-S5 linker of the channel (Perry et al., 2007; Perry et al., 2009). In this study, we have also observed that several KCNQ1 mutations (L251A, V254A, and V254L) in the S4-S5 linker display remarkable resistance to block by a high concentration of quinidine.
Homology structural modeling and docking of a channel-active drug have been broadly used to understand molecular basis of the drug-channel interaction. The HERG channel studies used homology modeling to the crystallized K+ channel KcsA structure to support a model of drug-channel interaction suggested by the electrophysiologic data on alanine-substituted mutants. Therefore, an obvious question is whether our data on KCNQ1 drug block can similarly be supported. To further learn how the residue F351 might limit drug block, we used crystallized K+ channel structures KcsA and KvAP to understand the orientation of this (and other) residue(s) in a potential drug-binding region. KcsA represents a closed state model, KvAP models both closed and open states, and the MthK structure displays the open state channel. Our initial modeling in based on the KcsA template positions F351 in, rather than below, the membrane and suggests that the aromatic residue points toward the channel pore (unpublished data). In addition, F340 appears to be very close to F351 in the adjacent KCNQ1 monomer, raising the possibility that the blocker interacts with dual sites on adjacent subunits; however, this would not explain our finding that F340A displayed drug sensitivity that was not significantly different from WT channel. Our docking study correlates well with these data, because none of the best scoring protein-ligand complex models predict contacts between quinidine and F340; the side-chain for this residue is on the wrong side of the S6 helix in the adjacent subunit and, therefore, faces away from the proposed binding site.
On the basis of the Kv1.2 structure, our homology modeling of KCNQ1 unexpectedly suggested that residue F351 faces away from the permeating pore. This finding argues against pore block as the mechanism for drug inhibition of KCNQ1 current. In the open-state channel model of the Kv1.2 structure, F351 lines a side pocket that is also associated with residue L251 and V254 in the S4-S5 linker. The model allows drug access to this pocket, and alanine substitutions at both sites yielded K+ currents that were resistant to quinidine block (↓34±4% for L251A and ↓40±5% for V254A) (Fig. 6). This further supports the concept that the S4-S5 linker and proximal C-terminus play an important role in modulation of drug block of the channel. Other studies with two HERG channel activators have also indicated importance of interaction between S4-S5 linker and S6 segment and drug binding to these regions (Perry et al., 2007; Perry and Sanguinetti, 2008; Perry et al., 2009).
Previous studies have suggested that the S4-S5 linker interacts with the C-terminal portion of S6 segment, and this interaction provides a mechanism whereby depolarization allows movement of the S4 voltage sensor to be transduced into channel opening (Labro et al., 2011; Choveau et al., 2011). Mutations (L353A, V254A, and V254L) tested here led to disruption of this interaction, and peptides modeled on the S4-S5 linker similarly inhibit KCNQ1 current (Boulet et al., 2007; Labro et al., 2011). Thus, the reduced quinidine block observed in the L353A, V254A, and V254L mutants may also reflect disruption of the interaction between the S4-S5 linker and the S6 domain; a similar mechanism could underlie the effect that we see with the F351A S6 mutant. It is well known that point mutations in the S6 segment and pore regions of K+ channels may alter channel gating, and such changes in turn can modulate drug block; thus, it may be difficult to separate the effects of drug sensitivity and gating that the mutations may confer. Previous structure-function studies of high-affinity drug binding in HERG channels have provided evidence that intact C-type inactivation is important for drug binding, because some mutations with disrupted inactivation markedly attenuated sensitivity to specific IKr blockers. This effect results most likely from allosteric changes in the drug binding site in HERG channels. The KCNQ1 channel alone shows some inactivation that can be removed by the β subunit KCNE1. In fact, two S6 mutants, F339A and F340A, did display ultra-fast inactivation (unpublished data), but did not affect block by quinidine and clofilium in this study. In addition, slowed activation for F351A did not influence the sensitivity to the channel to the blockers. Other researchers have also found that gating alterations in the KCNQ1 channel mutations had no or little interference in determining the effects of drugs on the channel (Seebohm et al., 2003a-c). These data are in good agreement with a report showing that mutations altering KCNQ1 inactivation showed no changes in sensitivity to a potent IKs blocker L-7 (Seebohm et al., 2003b).
Taken together, our data in the present study strongly support a model in which open-state block of this channel occurs not via binding to a site in the pore but rather by a novel allosteric mechanism: drug access to a side pocket generated in the open-state channel configuration and lined by S6 and S4-S5 residues. These findings may form the basis for development of new classes of channel blockers.
Abbreviations
- IKCNQ1
KCNQ1 current
- IKs
slowly activated potassium current
- KCNA5 (Kv1.5)
human cardiac potassium channel underlying a ultra-rapidly activated potassium current
- KCNE1 (minK)
the β-subunit to encode a cardiac slowly-activated potassium channel
- KCNH2 (HERG)
human ether-a-go-go potassium channel
- KCNQ1 (KvLQT1)
the α-subunit to encode a cardiac slowly activated potassium channel
- L-7
a selective IKs blocking compound
- PCR
polymerase chain reaction
- WT
wild type
Authorship Contributions
Participated in research design: Yang, Smith, Meiler, Roden.
Conducted experiments: Yang, Smith, Leake, Meiler.
Contributed new reagents or analytic tools: Smith, Meiler, Sanders.
Performed data analysis: Yang, Smith, Meiler.
Wrote or contributed to the writing of the manuscript: Yang, Smith, Meiler, Sanders, Roden.
Footnotes
This work was supported in part by grants from the United States Public Health Service (HL46681, HL49989, DC007416, GM080403, MH090192) and the American Heart Association (0565306B).
References
- Abitbol I, Peretz A, Lerche C, Busch AE, Attali B. (1999) Stilbenes and fenamates rescue the loss of I(KS) channel function induced by an LQT5 mutation and other IsK mutants. EMBO J 18:4137–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham RL, Yang T, Blair M, Roden DM, Darbar D. (2010) Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol 48:181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR, Bennett PB, Hondeghem LM, Roden DM. (1991) Suppression of time-dependent outward current in guinea pig ventricular myocytes. Actions of quinidine and amiodarone. Circ Res 69:519–529 [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384:78–80 [DOI] [PubMed] [Google Scholar]
- Bosch RF, Gaspo R, Busch AE, Lang HJ, Li GR, Nattel S. (1998) Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res 38:441–450 [DOI] [PubMed] [Google Scholar]
- Boulet IR, Labro AJ, Raes AL, Snyders DJ. (2007) Role of the S6 C-terminus in KCNQ1 channel gating. J Physiol 585:325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Busch GL, Ford E, Suessbrich H, Lang HJ, Greger R, Kunzelmann K, Attali B, Stühmer W. (1997) The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br J Pharmacol 122:187–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. (2002) Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci USA 99:12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choveau FS, Rodriguez N, Abderemane Ali F, Labro AJ, Rose T, Dahimène S, Boudin H, Le Hénaff C, Escande D, Snyders DJ, et al. (2011) KCNQ1 channels voltage dependence through a voltage-dependent binding of the S4-S5 linker to the pore domain. J Biol Chem 286:707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gögelein H, Brüggemann A, Gerlach U, Brendel J, Busch AE. (2000) Inhibition of IKs channels by HMR 1556. Naunyn Schmiedebergs Arch Pharmacol 362:480–488 [DOI] [PubMed] [Google Scholar]
- Ishii K, Kondo K, Takahashi M, Kimura M, Endoh M. (2001) An amino acid residue whose change by mutation affects drug binding to the HERG channel. FEBS Lett 506:191–195 [DOI] [PubMed] [Google Scholar]
- Kamiya K, Nishiyama A, Yasui K, Hojo M, Sanguinetti MC, Kodama I. (2001) Short- and long-term effects of amiodarone on the two components of cardiac delayed rectifier K(+) current. Circulation 103:1317–1324 [DOI] [PubMed] [Google Scholar]
- Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM. (2004) A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem 279:33976–33983 [DOI] [PubMed] [Google Scholar]
- Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. (2009) Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 120:1752–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro AJ, Boulet IR, Choveau FS, Mayeur E, Bruyns T, Loussouarn G, Raes AL, Snyders DJ. (2011) The S4-S5 linker of KCNQ1 channels forms a structural scaffold with the S6 segment controlling gate closure. J Biol Chem 286:717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309:897–903 [DOI] [PubMed] [Google Scholar]
- Ma LJ, Ohmert I, Vardanyan V. (2011) Allosteric features of KCNQ1 gating revealed by alanine scanning mutagenesis. Biophys J 100:885–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Baker D. (2006) ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins 65:538–548 [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. (2000) A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 97:12329–12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. (2005) Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 294:2975–2980 [DOI] [PubMed] [Google Scholar]
- Perry M, de Groot MJ, Helliwell R, Leishman D, Tristani-Firouzi M, Sanguinetti MC, Mitcheson J. (2004) Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol 66:240–249 [DOI] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Sanguinetti MC. (2007) Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc Natl Acad Sci USA 104:13827–13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sanguinetti MC. (2008) A single amino acid difference between ether-a-go-go- related gene channel subtypes determines differential sensitivity to a small molecule activator. Mol Pharmacol 73:1044–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Abbruzzese J, Sanguinetti MC. (2009) PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc Natl Acad Sci USA 106:20075–20080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384:80–83 [DOI] [PubMed] [Google Scholar]
- Seebohm G, Chen J, Culberson C, Sanguinetti MC. (2003 a) Binding site of a KCNQ1 potassium channel activator (Abstract). Biophys J 84:547A–548A [Google Scholar]
- Seebohm G, Chen J, Strutz N, Culberson C, Lerche C, Sanguinetti MC. (2003 b) Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol Pharmacol 64:70–77 [DOI] [PubMed] [Google Scholar]
- Seebohm G, Pusch M, Chen J, Sanguinetti MC. (2003 c) Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circ Res 93:941–947 [DOI] [PubMed] [Google Scholar]
- Smith JA, Vanoye CG, George AL, Jr, Meiler J, Sanders CR. (2007) Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46:14141–14152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyders DJ, Yeola SW. (1995) Determinants of antiarrhythmic drug action. Electrostatic and hydrophobic components of block of the human cardiac hKv1.5 channel. Circ Res 77:575–583 [DOI] [PubMed] [Google Scholar]
- Thomas GP, Gerlach U, Antzelevitch C. (2003) HMR 1556, a potent and selective blocker of slowly activating delayed rectifier potassium current. J Cardiovasc Pharmacol 41:140–147 [DOI] [PubMed] [Google Scholar]
- Yang T, Kanki H, Roden DM. (2003) Phosphorylation of the IKs channel complex inhibits drug block: novel mechanism underlying variable antiarrhythmic drug actions. Circulation 108:132–134 [DOI] [PubMed] [Google Scholar]
- Yang T, Chung SK, Zhang W, Mullins JG, McCulley CH, Crawford J, MacCormick J, Eddy CA, Shelling AN, French JK, et al. (2009) Biophysical properties of 9 KCNQ1 mutations associated with long-QT syndrome. Circ Arrhythm Electrophysiol 2:417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WP, Levesque PC, Little WA, Conder ML, Shalaby FY, Blanar MA. (1997) KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc Natl Acad Sci USA 94:4017–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Baker D, Catterall WA. (2006) Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc Natl Acad Sci USA 103:7292–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. (1996) Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res 78:1105–1114 [DOI] [PubMed] [Google Scholar]
- Zareba W, Moss AJ, Sheu G, Kaufman ES, Priori S, Vincent GM, Towbin JA, Benhorin J, Schwartz PJ, Napolitano C, et al. International LQTS Registry, University of Rochester, Rochester, New York (2003) Location of mutation in the KCNQ1 and phenotypic presentation of long QT syndrome. J Cardiovasc Electrophysiol 14:1149–1153 [DOI] [PubMed] [Google Scholar]