

Abstract
B-cell receptor (BCR) associated kinases including spleen tyrosine kinase (SYK) contribute to the pathogenesis of B-cell malignancies. SYK is persistently phosphorylated in a subset of non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL), and SYK inhibition results in abrogation of downstream kinase activity and apoptosis. P505-15 (also known as PRT062607) is a novel, highly selective, and orally bioavailable small molecule SYK inhibitor (SYK IC50 = 1 nM) with anti-SYK activity that is at least 80-fold greater than its affinity for other kinases. We evaluated the preclinical characteristics of P505-15 in models of NHL and CLL. P505-15 successfully inhibited SYK-mediated B-cell receptor signaling and decreased cell viability in NHL and CLL. Oral dosing in mice prevented BCR-mediated splenomegaly and significantly inhibited NHL tumor growth in a xenograft model. In addition, combination treatment of primary CLL cells with P505-15 plus fludarabine produced synergistic enhancement of activity at nanomolar concentrations. Our findings support the ongoing development of P505-15 as a therapeutic agent for B-cell malignancies. A dose finding study in healthy volunteers has been completed.
Introduction
Spleen tyrosine kinase (SYK) is a 72-kDa cytoplasmic tyrosine kinase primarily expressed in hematopoietic cells including B-cells. In B-cells, signal transduction is initiated by BCR activation via Src family kinase Lyn mediated phosphorylation of immune-receptor tyrosine-based activation motifs (ITAMs). This leads to the recruitment, autophosphorylation, and sustained activity of SYK and activation of a number of downstream effectors (Mócsai et al., 2010). Recent evidence suggests that B-cell malignancies including non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL) can be driven by aberrant activity of cellular signaling pathways and by extrinsic factors from the microenvironment that interact with the BCR (Caligaris-Cappio and Chiorazzi, 2010). Increased SYK expression and/or activity has been implicated in a number of NHL histologies (Rinaldi et al., 2006; Tavolaro et al., 2010 Chen et al., 2008; Davis et al., 2010). In CLL, constitutive SYK activity, as well as activation after BCR cross-linking, has been described (Baudot et al., 2009; Gobessi et al., 2009). Increased expression of BCR associated kinases including SYK is associated with a shorter treatment-free interval (Rodriguez et al., 2007), and SYK inhibition results in apoptosis (Baudot et al., 2009; Hoellenriegel et al., 2012) and disruption of chemokine activity (Rodriguez, et al., 2007; Hoellenriegel et al., 2012).
Targeting SYK has been explored using Fostamatinib disodium (R788), the pro-drug of R406. R788/406 is a SYK inhibitor (IC50 = 41 nM) found to have activity in a phase 2 study in NHL and CLL (Friedberg et al., 2010). However, R788/406 is known to have significant off target effects, including FMS-related tyrosine kinase 3 (FLT-3), Lck, Janus kinase 1 and 3, and c-kit (Braselmann et al., 2006), which may be responsible for some of its activity (Davis et al., 2010). No further development of Fostamatinib or of another selective SYK inhibitor has been reported in lymphoid cancers. Therefore, the characterization of novel SYK inhibitors is warranted. Given the therapeutic potential of SYK inhibition and the need to develop SYK inhibitors without off target effects, we evaluated P505-15, a novel and highly selective (IC50 = 1 nM) SYK inhibitor with anti-SYK activity that is 80-fold greater than its affinity for other kinases. P505-15 has been shown to potently inhibit SYK- and BCR-dependent activation of normal B-cells (Coffey, et al., 2012) and has been shown to decrease CLL cell viability (Hoellenriegel et al., 2012). We aimed to further define the preclinical properties of P505-15 in NHL and CLL and its activity as a single agent or in combination with fludarabine in primary CLL samples, including those obtained from patients with poor risk biologic features.
Materials and Methods
Chemical Structure and Kinase Profiling.
Synthesis and characterization of P505-15 [(4-(3-(2H-1,2,3-triazol-2-yl)phenylamino)-2-((1R,2S)-2-aminocyclohexylamino)pyrimidine-5-carboxamide acetate] (PRT062607) as well as its potency and selectivity for SYK have been reported (Coffey et al., 2012).
Cell Lines and Antibodies.
The human non-Hodgkin lymphoma B-cell lines SUDHL-4 (#ACC495), SUDHL-6 (#ACC572), and Karpas-422 (#ACC32) were obtained from DSMZ (Braunschweig, Germany); Toledo (#CRL-2631) and Ramos (#CRL-1596) were obtained from the American Type Culture Collection (ATCC; Manassas, VA). All cells were maintained in RPMI media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (ATCC) and penicillin/streptomycin (Invitrogen), and maintained in a 37°C humidified tissue culture incubator. Antibodies included polyclonal goat F(ab)’2 anti-human IgG and IgM (LifeTechnologies, Grand Island, NY); rabbit anti-human SYK, anti-human phospho-SYK (Y525/526), and alexafluor 488-conjugated anti-human phospho-ERK (Y204) (Cell Signaling Technologies, Inc., Danvers, MA); phycoerythrin-conjugated anti-human phospho-AKT (S473), fluorescein isothiocyanate-conjugated anti-human CD19, allophycocyanin-conjugated anti-human CD5, phycoerythrin-conjugated anti-human phospho-SYK (Y352), fluorescein isothiocyanate-conjugated anti-mouse CD80 and CD86, and allophycocyanin-conjugated anti-mouse CD45R/B220 (BD Biosciences, San Jose, CA).
SYK Autophosphorylation.
Ramos cells (107 cells per experiment) were preincubated for 30 minutes with or without P505-15 and then stimulated (30 minutes at 37°C) with 1 μg/ml anti-IgM. Signaling was terminated by resuspending cells in RIPA lysis buffer (50 mM Tris 7.4, 1% NP40, 0.5% sodium deoxycholate, 150 mM NaCl, 0.5 mM EDTA) containing fresh protease (Roche Pharmaceuticals, Indianapolis, IN) and phosphatase (Sigma Pharmaceuticals, Monticello, IA) inhibitors and incubated on ice for 1 hour. Following centrifugation, protein lysates were pre-cleared with protein A/G sepharose beads (Thermo Scientific, Rockford, IL). Lysates were incubated overnight (4°C) with rabbit anti-SYK antibody and precipitated with protein A/G sepharose beads. Washed beads were denatured and proteins were resolved by 10% SDS-PAGE, immunoblotted with rabbit anti-SYK antibody, stripped, and reblotted with rabbit anti-pSYK Y526/526 antibody.
Intracellular Phospho-flow Cytometry.
Ramos cells (0.5 × 106) were suspended in 200 μl fresh media (RPMI plus 10% fetal calf serum) and pretreated with vehicle or P505-15 (30 minutes at 37°C). Cells were left unstimulated or were stimulated with 1 μg/ml goat F(ab)’2 anti-IgM (Life Technologies) for 10 minutes. Ficoll purified (2 × 106) frozen viable CLL cells obtained from peripheral blood of CLL patients (n = 7) were thawed at 37°C, washed in 10 ml RPMI media plus 10% fetal calf serum by centrifugation, and resuspended at 106 cells/ml in the same media. Aliquots (200 μl) of cells were treated with vehicle or P505-15 for 30 minutes prior to stimulation with 0.3 mM H2O2 (Reth 2002; Irish et al., 2006) (8.8 M stock; Sigma) followed by addition of 6 μg of a 1:1 mixture of goat F(ab’)2 anti-human IgG and anti-human IgM for 10 minutes. Signaling was stopped by addition of 60 μl of a 16% paraformaldehyde solution (Electron Microscopy Sciences, Hatfield, PA) followed by 10-minute incubation at room temperature. Fixed cells were washed twice in ice-cold phosphate buffered saline (PBS) by centrifugation, suspended in a 50% methanol solution in PBS prechilled to −20°C, and stored at 4°C overnight. Cells were stained for intracellular phosphoflow cytometry by washing the permeabilized cells twice in PBS containing 1% BSA, followed by incubation in the same buffer containing various antibodies, as indicated in the results, per manufacturer instructions. Cells were washed once more in PBS containing 1% BSA prior to FACS analysis, in which at least 2,000 events were collected using a BD Biosciences FACS Calibur. Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Apoptosis.
Apoptosis was measured using the PE-conjugated monoclonal active caspase-3 antibody apoptosis kit (BD Biosciences). Cells were suspended in growth media (0.5 × 106 cells/ml) and treated with the indicated concentrations of P505-15 or vehicle control for 72 hours prior to FACS analysis. In some experiments, SU-DHL6 (0.5 × 106 cells) was mixed with 100 µl heparinized human whole blood. Samples were treated with 1 µM P505-15 for 24 hours and then surface stained with anti-human CD19 antibody prior to preparation for FACS analysis of active caspase-3.
In Vitro and In Vivo Stimulation with Anti-IgD.
Spleens were harvested from Balb/c mice and separated into a single cell suspension using a single cell strainer (BD Biosciences). Cells were collected in PBS containing 1% BSA by centrifugation, washed once in the same buffer, and resuspended in RPMI (containing 10% FCS) at 106 cells/ml. Aliquots (190 µl) were then treated with various concentrations of P505-15 for 1 hour prior to stimulating overnight in a 37°C tissue culture incubator with 1 µl anti-mouse immunoglobulin D (IgD) (EBiosciences, San Diego, CA). Following stimulation, cells were stained with anti-CD80/86 FITC and anti-CD45R/B220 APC for 30 minutes at room temperature, washed once in PBS containing 1% BSA, and resuspend in 300 μl of the same buffer for collection of 2000 CD45-positive events by flow cytometry.
Balb/c mice (n = 5 per group) received daily oral BID doses of vehicle (0.5% methylcellulose in water) or P505-15 (15 mg/kg) for a total of 5 days. On study day 1, 1 hour after the first oral dose, mice received a single 200-μl subcutaneous injection of control goat serum (Sigma) or anti-IgD serum (EBiosciences). On study day 5, mice were anesthetized with SC ketamine cocktail and exsanguinated via cardiac puncture. Spleens were weighed and then sectioned and stained with hematoxylin and eosin for histology.
Xenograft Studies.
NOD/SCID mice were acclimated in-house at least 3 days prior to use. Ramos cells (3 × 106) were injected subcutaneously into the hind flank area of conscious mice using a 27-gauge needle in an injection volume of less than 0.5 ml. Following injection, mice were randomized into treatment groups (n = 15) and dosed twice daily by oral gavage with vehicle (0.5% methylcellulose in water) or 10, 15, or 20 mg/kg P505-15. Body weights were obtained at least once per week, and caliper measurements of tumors were determined twice per week beginning when palpable tumors were formed until the end of the study. Tumor volume was assessed by caliper measurement using a formula [maximum length × width × height × pi/6]. Twice daily dosing of vehicle or P505-15 continued until the vehicle or any treatment group exhibited tumors that exceeded 1.5 g in size. At the time of termination (5 weeks post-Ramos inoculation) the mice were anesthetized with a ketamine cocktail. A blood sample via cardiac puncture was obtained for CBC and plasma concentration determination, and then the mice were euthanized via cervical dislocation. Tumors were excised, weighed, and then snap frozen in liquid nitrogen for determination of concentration of P505-15 in the tumor tissue. Statistical comparison of tumor weights across groups was performed using a t-test.
Tumor and Plasma Concentration Analysis.
Tumor-bearing mice were dosed twice daily for a total of 27 days. On the last day of dosing, tumor and plasma samples were collected at predose and 1.5, 4, and 8 hour postdose. Each tumor sample was homogenized in 3 ml of saline per gram of tumor using the Kontes Microtube Pellet Pestle Rods and Motor (Kimble Chase, Vineland, NJ). Plasma and tumor samples were extracted and analyzed for P505-15 concentration using a liquid chromatography tandem mass spectrometer (LC/MS/MS). Pharmacokinetic parameters of P505-15 in plasma and tumor were calculated using WinNonlin program, version 5.3 (Pharsight, Cary, NC).
Patient Samples, Viability Assay, and Synergy Analysis.
Blood samples were obtained after written informed consent approved by the Institutional Review Board. CLL cells (n = 42) were purified using a Ficoll gradient and added to wells (5 × 104 per well) containing 20% serum containing RPMI media plus βME and four serial dilutions of P505-15 (10 nM to 10 μM) with or without fludarabine (10 nM to 10 mM; Hospira Pharmaceuticals, Chicago, IL). After 72 hours of culture, a tetrazolium-based cell viability assay (MTS) (BioRad, Hercules, CA) was performed. The viability data were normalized to untreated controls and used to calculate half-maximal inhibitory concentration (IC50) values. The Loewe surface/synergy was calculated using Combitool (Dressler et al., 1999). Chou median effects analysis and Bliss independence measures analysis were performed as described elsewhere (Bliss 1939; Chou 2010). Statistical significance was determined using the confidence interval.
Results
P505-15 Inhibits SYK Activation and Induces Caspase-dependent Apoptosis in a Subset of NHL Cell Lines.
To validate target inhibition, lysates from NHL cell lines stimulated with anti-IgM and treated with P505-15 (2 µM) were immunoblotted with antibodies specific for the SYK autophosphorylation sites (Y525/526). Significant reduction in SYK activity was observed (Fig. 1A). To demonstrate inhibition of signaling more distal to the BCR/SYK complex, ERK (Y204) and AKT (S473) phosphorylation were monitored with or without SYK inhibition with P505-15 following BCR stimulation. Significant decreases in phosphorylation of both targets were seen (Fig. 1B). Half-maximal inhibition was observed at approximately 50 nM, with complete inhibition of ERK and AKT phosphorylation at 250 nM. As a specificity control, Lyn phosphorylation of SYK at Y352 was also measured. Although inhibition was observed, the effect was not entirely concentration-dependent and complete inhibition could not be achieved (Fig. 1B). Data from a representative experiment is shown in Fig. 1C. These data are consistent with previous observations that P505-15 is a selective and potent inhibitor of Syk, with no appreciable activity against other BCR-related kinases (Coffey et al., 2012).
Fig. 1.
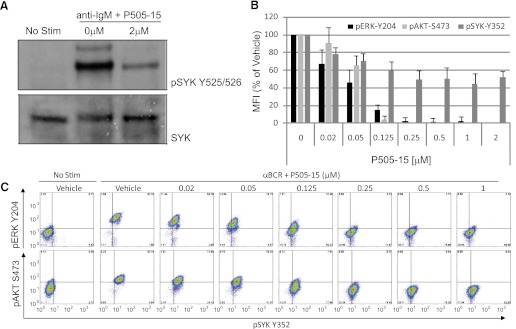
Suppression of BCR-induced SYK signaling. (A) Ramos cells were left unstimulated (No Stim) or stimulated with anti-BCR antibody (anti-IgM) in the presence or absence of 2 µM P505-15. SYK autophosphorylation (pSYK Y525/526) is depicted in the top panel, and total SYK protein (SYK) in the bottom panel. (B) cells stimulated through the BCR and treated with the concentrations of P505-15 shown were evaluated by intracellular phospho-flow cytometry for the induction of ERK Y204 (black bars), AKT S473 (light gray bars), and SYK Y352 (dark gray bars). The mean fluorescent intensity (MFI) normalized to percent of vehicle control is plotted on the y-axis (mean + S.D., n = 3). After treatment of Ramos cells with P505-15, ERK (Y204), and AKT (S473) phosphorylation was inhibited. (C) representative experiment depicting the induction of ERK Y204 (top row y-axis), AKT S473 (bottom row y-axis), and SYK Y352 (x-axis) following treatment with the various concentrations of P505-15 shown.
The effect of SYK inhibition on B-cell survival was measured by induction of caspase 3. The SU-DHL4, SU-DHL6, and Ramos B-cell lines express functional BCR, whereas the Toledo and Karpas-422 lines lack BCR and BLNK expression, respectively (Gabay, et al., 1999; Sprangers et al., 2006). P505-15 induced apoptosis only in the BCR-signaling competent cell lines, consistent with observations made using other SYK inhibitors (Chen et al., 2008, 2011) and highlighting the compound’s specificity against its target kinase (Fig. 2A). To compare the relative sensitivity of primary versus tumor B-cells, we performed mixing experiments in which SU-DHL6 was combined with human whole blood and treated with P505-15. Under these conditions, whereas the tumor B-cell line underwent apoptosis in response to SYK inhibition, primary B-cells did not (Fig. 2B). Caspase 3 cleavage could be induced in primary B-cells upon treatment with the blasticidin (data not shown).
Fig. 2.

NHL cell lines are more reliant on SYK for survival than primary B-cells. (A) NHL cell lines treated with vehicle control, 1 µM or 3 µM P505-15. Histograms (representative of 2–3 experiments) depict induction of apoptosis via caspase 3 cleavage on the x-axis. The three sensitive cell lines are shown on the top row, and the two resistant lines on the bottom row. (B) the side scatter (SSC) by CD19 scatter plots depict primary B-cell and SU-DHL6 gating strategies for human whole blood leukocytes (left plot) and human whole blood mixed with SU-DHL6 (right plot). Second row histograms depict primary B-cell or SU-DHL6 apoptosis following treatment with 1 µM P505-15 for 24 hours. The dotted line represents vehicle control while the shaded gray histogram represents P505-15 treatment.
SYK Inhibition Protects against Tumor Formation in a Mouse Xenograft Model.
The effect of SYK inhibition on Ramos tumor formation in a mouse xenograft model was assessed. Mice were dosed twice daily with 10, 15, or 20 mg/kg P505-15 or vehicle control beginning on the day of tumor cell inoculation. Caliper measurements were initiated when tumors began to form, approximately 3 weeks post-tumor inoculation, and repeated every third day until termination of the study. The study was terminated when tumor weights began reaching approximately 1.5 mg, at which time tumors were excised and weighed.
Tumor and plasma samples were evaluated for P505-15 levels on the last day of dosing. Immediately prior to the last dose on day 27, samples were collected to determine Cmin; following the last dose, samples were collected at 1.5, 4, and 8 hours, data from which were used for a composite PK analysis. More than dose-proportional increases in plasma and tumor Cmax and AUC (0–8 hours) and tumor Cmin were observed as the dose was increased, but a dose-proportional increase in plasma Cmin was noted. Mean Cmax and AUC (0–8 hours) in plasma was at least twofold greater than that in tumor for all doses examined; however, mean nadir concentrations (Cmin) were higher in tumor than in plasma (Table 1), suggesting accumulation of P505-15 in the tumor and slower elimination from this compartment. The difference between plasma and tumor Cmin became more prominent as the dose was increased, as indicated by the increase in tumor/plasma ratios determined from Cmin (Table 2). Tumor/plasma ratios determined from Cmax and AUC (0–8 hours) were similar across the various dose groups. Tumor concentrations of P505-15 at steady state were sustained above 63, 176, and 325 nM over the entire dosing interval following 10, 15, and 20 mg/kg, respectively. The half-life of P505-15 in mice following 1 mg/kg i.v. dose was 3.9 hours.
TABLE 1.
Steady-state plasma and tumor pharmacokinetics following oral dosing of P505-15 in mice
Plasma and tumor composite PK analysis from steady-state Cmin (collected 16 hours following the penultimate dose) and 1.5, 4, and 8 hours post-last dose on day 27.
| Dosing Regimen | Tmax | Cmin | Cmax | AUC (0–8 hours) |
|---|---|---|---|---|
| hours | nM | nM | ng*h/ml | |
| Determined from Plasma | ||||
| 10 mg/kg BID | 1.5 | 45 | 466 | 738 |
| 15 mg/kg BID | 1.5 | 69 | 893 | 1671 |
| 20 mg/kg BID | 4.0 | 102 | 1484 | 3191 |
| Determined from Tumor | ||||
| 10 mg/kg BID | 8.0 | 63 | 143 | 353 |
| 15 mg/kg BID | 4.0 | 176 | 424 | 475 |
| 20 mg/kg BID | 4.0 | 325 | 656 | 1453 |
TABLE 2.
Tumor/plasma ratios
Accumulation of P505-15 within the tumor is evidenced by ratios of greater than 1 at Cmin for each of the dose levels.
| Dosing Regimen |
Tumor/Plasma Ratio |
||
|---|---|---|---|
| AUC Based | Cmax Based | Cmin Based | |
| 10 mg/kg BID | 0.478 | 0.308 | 1.39 |
| 15 mg/kg BID | 0.284 | 0.475 | 2.55 |
| 20 mg/kg BID | 0.455 | 0.442 | 3.15 |
Mice dosed with all three concentrations of P505-15 were protected from Ramos tumor growth in vivo. This was first evident from caliper measurements (Supplemental Fig. 1), which revealed a reduced rate of tumor growth in the presence of P505-15. Upon study completion, mice were euthanized and tumors were excised and weighed. Consistent with caliper measurements, a statistically significant reduction in average tumor weight was achieved in all dosing groups relative to vehicle control. Data are presented in Fig. 3A. These data reveal that submicromolar concentrations of P505-15 can prevent tumor formation by an aggressive NHL cell line in mice. In a separate study (data not shown), treatment with P505-15 was initiated when tumor size was estimated by caliper measurement to be approximately 200 mg. Mice were then randomized into treatment groups and treated with oral vehicle or P505-15 for the next 10 days, after which tumor size approximated 1.5 g and the study was terminated. Under these conditions P505-15 did not affect tumor progression. Cells derived from the extracted tumors did undergo apoptosis in response to in vitro treatment with P505-15, however, indicating that a non-responsive clone was not selected in vivo. Histologic observation revealed that the tumors were minimally vascularized and therefore may not be an appropriate model under these conditions. Irrespective, these data suggest that the efficacy observed under prophylactic treatment (Fig. 3A) may be completely or in part due to inhibition of tumor engraftment.
Fig. 3.

SYK inhibition prevents NHL tumor formation in xenograft mice. Mice were dosed twice daily with 10, 15, or 20 mg/kg P505-15 or vehicle control beginning the day of tumor cell inoculation. (A) individual tumor weights (n = 13–15) were determined at 4 weeks postinoculation for each treatment condition. Statistical differences relative to vehicle control are depicted as P values within the graph. (B) Values shown depict blood cell counts (mean + S.D., n = 13-15) of mice treated with each concentration of P505-15 or vehicle control.
Mice dosed with the P505-15 did not present with reduced numbers in any leukocyte subsets. In fact, the only effect observed was an increase in the number of lymphocytes in mice treated with 15 mg/kg P505-15, which was not repeated in mice dosed with 10 or 20 mg/kg (Fig. 3B). The relative percent of each cell subtype analyzed was also unaffected. (data not shown).
SYK Inhibition Protects against BCR-Induced Splenomegaly in Mice.
Chronic BCR stimulation in secondary lymphoid organs appears to be an important driver of B-cell survival and clonal expansion in certain B-cell malignancies, particularly in CLL. To study this in vivo, we used an anti-mouse IgD that was previously shown to induce BCR signaling (Coffey et al., 2012) and immune cell activation in vivo following subcutaneous administration in mice (Mountz et al., 1987). We report here that P505-15 suppresses mouse B-cell activation following stimulation with this anti-IgD (Fig. 4A). Furthermore, subcutaneous administration of this activating anti-mouse IgD results in rapid B-cell expansion and splenomegaly in the animals. To determine the effect of P505-15 on BCR-induced splenomegaly in vivo, groups of five mice were pretreated with vehicle or 15 mg/kg compound for 1 hour prior to in vivo administration of anti-IgD antibody. Twice daily dosing was then continued for 4 days, with analysis of splenomegaly performed on day 5 post-anti-IgD injection. In vivo stimulation with anti-IgD resulted in an approximate doubling of spleen weights, which was significantly reduced by treatment with P505-15 (Fig. 4B). Spleens were sectioned and H&E stained to assess associated changes in morphology. Examples from mice treated with control goat serum (Fig. 4C, left panel), anti-IgD (Fig. 4C, middle panel), and anti-IgD plus 15 mg/kg P505-15 is presented in Fig. 4C. Expansion of the marginal B-cell zone was observed in all anti-IgD-stimulated mice treated with vehicle control, a phenomenon that was mostly corrected in mice upon treatment with the SYK inhibitor, as shown in Fig. 4C, right panel. These data demonstrate that P505-15 can penetrate the mouse spleen following oral dosing in mice, and inhibit BCR signaling and clonal expansion in this environment.
Fig. 4.

Specific SYK inhibition prevents splenomegaly in a mouse model of chronic BCR stimulation. (A) mouse splenocytes were isolated and treated with various concentrations of P505-15 as shown. Data represent inhibition of anti-IgD mediated B-cell activation (CD80/86 MFI) relative to vehicle control (mean + S.D., n = 4). (B) mice were injected subcutaneously with goat serum (GS) or anti-IgD. Average spleen weight is graphed (mean + S.D., n = 5) from mice treated as indicated. *Statistical difference (P < 0.05) relative to anti-IgD mice treated with vehicle control (Vh). The subset photograph depicts two representative spleens from each group of mice treated with GS and vehicle control (left set), anti-IgD with vehicle (middle set), and anti-IgD with 15 mg/kg P505-15 (right set). (C) representative H&E staining of spleen sections from mice treated with GS (left panel), anti-IgD with vehicle (middle panel), and anti-IgD with 15 mg/kg P505-15 (right panel).
P505-15 Inhibits BCR-Mediated Signaling and Decreases Cell Viability in Primary CLL and Shows Synergy When Combined with Fludarabine.
We next evaluated the ability of P505-15 to inhibit BCR-mediated SYK kinase activity in primary CLL samples (n = 7). Representative flow cytometry data depicting BCR-mediated AKT phosphorylation in CD19/CD5 double positive CLL cells is shown in Fig. 5A. Under these conditions, treatment with P505-15 resulted in a concentration-dependent decrease in AKT phosphorylation, with complete inhibition of this target between 0.3 and 1 μM in all samples tested (Fig. 5B). Despite the potential for increased BCR responsiveness in an unmutated variable region of the immunoglobulin heavy chain (IgVh) samples (Stevenson et al., 2011), decreased AKT phosphorylation after P505-15 treatment did not appear to be related to Zap 70 or IgVh mutational status (data not shown). The ability of P505-15 to inhibit AKT is of particular importance, because agents that disrupt AKT activity have shown therapeutic promise in CLL (Lannutti et al., 2011).
Fig. 5.

P505-15 inhibits BCR mediated signaling, and when combined with fludarabine shows increased cell killing and is fludarabine sparing at nanomolar and low micromolar concentrations. (A) representative BC-mediated AKT phosphorylation in CLL cells as shown by flow cytometry. The left and middle panels depict gating on CD5+/CD19+ CLL cells. The histogram shows a representative (n = 7) induction of AKT S473 following BCR ligation. (B) P505-15 inhibits AKT phosphorylation following BCR ligation of CLL cells in a concentration-dependent manner (mean + S.E.M., n = 7). (C) CLL cells were treated with fludarabine alone, P505-15 alone, or in the combinations indicated. Percent inhibition of viability (mean and S.E.M., n = 13) is plotted on the y-axis. Percent inhibition is P505-15 is dose dependent. The dark gray bars direct attention to cell killing in the presence of 10 µM fludarabine alone (far left bar) relative to 1 µM fludarabine combined with 1 µM or 0.1 µM P505-15 where equivalent cell kill was observed.
Of the 42 primary CLL samples treated with P505-15 alone, activity was seen (defined as IC50 < 3 μM for each sample tested) in 15/42 (36%) samples. P505-15 single agent activity is consistent with prior work showing that P505-15 decreases CLL cell viability in the presence of anti-IgM mediated BCR activation or nurse-like cell coculture (Hoellenriegel et al., 2012). We further characterized the effect of P505-15 across biologic subgroups. Importantly, significant activity was seen in samples obtained from patients with biologic features suggestive of high risk CLL, which is characterized by the presence of having an unmutated variable region of the immunoglobulin heavy chain (IgVh), or deletion of chromosome 17p where p53 resides, or deletion of chromosome 11q (ATM gene). When further evaluating response (IC50 < 3 μM) by poor risk groups, three of seven samples (43%) with 17p deletion, three of eight (38%) with 11q deletion, and four of seventeen (24%) with unmutated IgVh were found to be responsive to P505-15 treatment.
Fludarabine is a purine nuceloside analog that is very active in CLL and continues to serve as the chemotherapy backbone in a number of chemotherapy regimens. However, it is associated with significant and at times long-term myelotoxicty, opportunistic infections, and secondary malignancies. Furthermore, it’s efficacy in poor risk patients is often lacking. Thus, combinations that incorporate drugs with novel mechanisms of action that could potentially decrease fludarabine exposure and toxicity while increasing activity are urgently needed. When primary CLL cells (n = 20) were treated with P505-15 alone or in combination with fludarabine, cell killing was observed for both single agent and combinations (Fig. 5C). When the highest concentration of fludarabine tested (10 µM) was compared with lower concentrations (1 µM) combined with P505-15 (100 nM and 1 µM), equivalent cell killing was seen (Fig. 5C; note dark shaded bars for comparison), indicating a fludarabine sparing effect when inhibiting SYK with P505-15. Furthermore, synergy was detected by employing three independent analysis strategies: Loewe additivity, Bliss independence analysis, and Chou median effect analysis. By all three methods, 1 µM fludarabine in combination with 1 µM P505-15 resulted in synergistic cell kill. The Loewe and Chou analysis also detected synergism at 1 µM fludarabine in combination with 0.1 µM P505-15. The Loewe additivity analysis is represented in Fig. 6.
Fig. 6.

P505-15 is synergistic with fludarabine at nanomolar concentrations in a CLL viability assay. Cell viability data from P505-15 and fludarabine combination studies (n = 18) were analyzed by CombiTool for determination of synergy. The combination effect is calculated by subtraction of the observed effect at certain concentrations of fludarabine and P505-15 from the calculated Loewe surface (Loewe surface in gray). The difference between observed activity and effect predicted from single drug controls (fludarabine or P505-15 alone by Loewe additivity model) are shown on three-dimensional residual plots. Points that fall below the additive zero-interaction plane (z = 0) are synergistic (closed circles), because the observed inhibitory effect was greater than predicted. For example, points corresponding to fludarabine (1 µM) + P505-15 (1 µM) for all 20 points and for fludarabine (1 µM) + P505-15 (100 nM) for 17/18 points are below the Loewe surface, demonstrating synergistic activity. For the point above the zero interaction plane using 0.1 µM concentrations of P505-15 and fludarabine, minimal growth inhibition was seen for either drug and thus no synergy could be appreciated.
Discussion
Several receptors expressed by hematopoietic cells use SYK for signal transduction, including Fc receptors, BCR, integrins, and members of the lectin and selectin families (Turner et al., 2000; Mócsai et al., 2002; Rogers et al., 2005). In B-cells, signaling through the BCR is made possible by its association with Igα and Igβ, each bearing an ITAM domain that is phosphorylated by the Src family member Lyn (Jumaa et al., 2005). SYK engages the diphosphorylated ITAMs, a process that enhances its kinase activity, resulting in SYK autophosphorylation and tyrosine phosphorylation of multiple downstream substrates that ultimately regulate the cell’s activation status, promoting survival and clonal expansion (Rolli et al., 2002). This signaling pathway is active in B-cells beginning at the transition from the pro- to pre-B-cell stage of development, when the newly formed pre-BCR is expressed. In fact, B-cell development arrests at the pro-B-cell stage in SYK knockout mice and corresponds with a loss of mature B cells (Cheng, et al., 1995; Turner et al., 1995). Importantly, inducible loss of Syk for 10 days had no impact on mouse B cell populations (Wex et al., 2011), whereas at 4 weeks postinducible SYK depletion in adult mice a loss of mature B cells was observed, recapitulating the embryonic SYK knockout data (Ozaki et al., 2012). Inducible loss of the BCR (Lam et al., 1997) or Igα (Kraus et al., 2004) also result in loss of peripheral mature B-cells in mice. These data suggest that normal B cells depend upon SYK for signals related to functional response and development, but not necessarily for survival.
However, the oncogenic potential of SYK has been described in a number of different settings. Clinically, SYK overexpression is reported in mantle cell lymphoma (Rinaldi, et al., 2006) and the TEL-SYK fusion protein (translocated ETS leukemia) generated by a chromosomal translocation [t(9;12)(q22;p12)] leads to increased SYK activity and is associated with myelodysplastic syndrome (Kuno et al., 2001). SYK was also reported to mediate mTOR (mammalian target of rapamycin) survival signals in follicular, mantle cell, Burkitt’s, and diffuse large B-cell (DLBCL) NHL (Leseux et al., 2006). Additional studies also suggest that SYK-dependent survival signals may play a role in DLBCL, mantle cell lymphoma, and follicular lymphoma (Gururajan et al., 2006; Irish et al., 2006). In mice, a leukemia-like disease is induced by the adoptive transfer of bone marrow cells that express human TEL-SYK (Wossning et al., 2006), and BCR signaling is implicated in CLL tumor survival (Quiroga et al., 2009).
There is also evidence that primary lymphoma and human cell lines of NHL origin may depend on SYK signaling for survival. This was first reported by Monti et al. ( 2005) who found that of 116 primary NHL tumor specimens analyzed for mRNA expression profiles, 43% clustered into a group characterized as overexpressing SYK and other components of the BCR signaling pathway. A subset of NHL cell lines was also reported to be dependent upon SYK expression in RNA interference studies (Davis et al., 2010). To determine if SYK was required for the survival of certain lymphomas, the protein tyrosine phosphatase PTP-RO, a negative regulator of SYK, was overexpressed in NHL cell lines. This resulted in the suppression of SYK signaling and induction of apoptosis (Chen et al., 2006).
Consistently, pharmacological inhibition of SYK activity induces apoptosis in a subset of NHL cell lines (Chen et al., 2008, 2011). It was previously shown that SYK inhibition with R406 effectively inhibits SYK activity in NHL cell lines and demonstrates single agent activity in CLL including samples taken from patients with high risk features (Quiroga et al., 2009). Clinical activity of this compound in CLL/NHL trial was confirmed in a phase II trial (Friedberg et al., 2010); however, this agent has significant off target effects and phase III trials in oncology have not been pursued as yet in cancer. More recently, similar to other agents that target B-cell signaling, P505-15, a highly selective novel SYK inhibitor (IC50 = 1 nM) with greater than 80-fold selectivity for other kinases (Coffey et al., 2012), was demonstrated to inhibit BCR signaling. The inhibitor was also active in experimental systems that used chemokine and nurse-like cells to mimic the enhanced protection of CLL to cytotoxic chemotherapy or immunotherapy (Hoellenriegel et al., 2012).
Our studies confirm and extend previous observations by demonstrating that the capacity of SYK inhibitors to suppress BCR signaling and induce malignant B-cell apoptosis is not confined to cell lines or to cell cultures. Inhibition of BCR-mediated splenomegaly in mice provides strong evidence that P505-15 suppresses BCR signaling in vivo following oral dosing, and results from the Ramos xenograft model reveal that antitumor activity can be achieved at concentrations that do not affect primary leukocyte survival. These data were confirmed with the observation that tumor B-cells were uniquely sensitive to SYK inhibition in a mixture containing peripheral blood B-cells.
We also report here that P505-15 has modest single agent activity against CLL survival, but is synergistic with fludarabine at nanomolar concentrations. The observation that P505-15 concentrations in the range of 10 to 100 nM enhance the effects of fludarabine (10 nM to 1 µM) demonstrates activity in excess of that expected from a purely additive effect (Tallarida, 2001). These findings are of particular importance given the well known dose limiting toxicity associated with repeated fludarabine treatment. Importantly, activity was seen in samples obtained from poor prognosis patients, including those harboring the 17p deletion, a group in tremendous need of additional therapeutic options. The activity seen in the 17p-deleted samples is consistent with prior work suggesting that SYK mediated apoptosis is p53 independent (Baudot et al., 2009). The reliance on BCR-mediated signaling in CLL with unmutated IgVh suggested that SYK inhibitors, especially in the presence of antigen, would preferentially exhibit increased activity in samples from patients with unmutated IgVh; however, this was not the case after P505-15 treatment. Furthermore, Zap 70 and CD38 expression, which are also markers of increased BCR responsiveness, did not correlate with P505-15 mediated growth inhibition.
Given its preclinical activity and specificity, especially when compared with previously studied SYK inhibitors, P505-15 is an attractive compound for clinical development. Additional studies aimed at defining the biologic characteristics associated with SYK sensitivity are needed. Exploring additional P505-15 combinations including chemotherapy, monoclonal antibodies, or novel combinations of kinase inhibitors targeting multiple branches of signaling pathways, may identify further therapeutic opportunities. A dose finding study using P505-15 in healthy volunteers has been completed and includes single and multiple dosing regimens.
Supplementary Material
Abbreviations
- BCR
B-cell receptor
- CLL
chronic lymphocytic leukemia
- Ig
immunoglobulin
- NHL
non-Hodgkin’s lymphoma
- phospho
phosphorylation
- SYK
spleen tyrosine kinase
Authorship Contributions
Participated in research design: Spurgeon, Tyner, Druker, Coffey, Sinha.
Conducted experiments: Fletcher, Burke, Coffey, DeGuzman, Baker, Hollenbach.
Contributed new reagents or analytic tools: Pandey.
Performed data analysis: Spurgeon, Loriaux, Betz, Coffey, Pak.
Wrote or contributed to the writing of the manuscript: Spurgeon, Loriaux, Coffey, Sinha.
Footnotes
B.J.D. is a Howard Hughes Investigator; J.W.T. is supported by grants from the William Lawrence and Blanche Hughes Fund, the Leukemia & Lymphoma Society, the V Foundation for Cancer Research, and National Institutes of Health National Cancer Institute [4R00CA151457-03]. S.E.S. receives funding from the Medical Foundation of Oregon. R.B. is supported by funding from the Division of Hematology and Medical Oncology at Oregon Health & Science University. M.M.L. is supported by grants from the Leukemia and Lymphoma Society and National Cancer Institute [5R21CA159265]. P505-15 was provided by Portola Pharmaceuticals. G.C., A.B., F.D.,Y.P., D.B., A.P., S.H., U.S. are paid employees of Portola Pharmaceuticals and accordingly received support for portions of this research.
Preliminary data were presented at the Annual Society of Hematology Meeting ASH Annual Meeting Abstracts (2010) Blood 116: 2839.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Baudot AD, Jeandel PY, Mouska X, Maurer U, Tartare-Deckert S, Raynaud SD, Cassuto JP, Ticchioni M, Deckert M. (2009) The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCdelta and proteasome-dependent regulation of Mcl-1 expression. Oncogene 28:3261–3273 [DOI] [PubMed] [Google Scholar]
- Bliss CI. (1939) The toxicity of poisons applied jointly. Ann Appl Biol 26:585–615 [Google Scholar]
- Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, Qu K, Herlaar E, Lau A, Young C, et al. (2006) R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther 319:998–1008 [DOI] [PubMed] [Google Scholar]
- Caligaris-Cappio F, Chiorazzi N. (2010) Where is the biology of CLL leading us? Semin Cancer Biol 20:361–362 [DOI] [PubMed] [Google Scholar]
- Chen L, Juszczynski P, Takeyama K, Aguiar RC, Shipp MA. (2006) Protein tyrosine phosphatase receptor-type O truncated (PTPROt) regulates SYK phosphorylation, proximal B-cell-receptor signaling, and cellular proliferation. Blood 108:3428–3433 [DOI] [PubMed] [Google Scholar]
- Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, Habermann TM, Kutok JL, Shipp MA. (2008) SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood 111:2230–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. (1995) Syk tyrosine kinase required for mouse viability and B-cell development. Nature 378:303–306 [DOI] [PubMed] [Google Scholar]
- Cheng S, Coffey G, Zhang XH, Shaknovich R, Song Z, Lu P, Pandey A, Melnick AM, Sinha U, Wang YL. (2011) SYK inhibition and response prediction in diffuse large B-cell lymphoma. Blood 118:6342–6352 [DOI] [PubMed] [Google Scholar]
- Chou TC. (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446 [DOI] [PubMed] [Google Scholar]
- Coffey G, DeGuzman F, Inagaki M, Pak Y, Delaney SM, Ives D, Betz A, Jia ZJ, Pandey A, Baker D, et al. (2012) Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J Pharmacol Exp Ther 340:350–359 [DOI] [PubMed] [Google Scholar]
- Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al. (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463:88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler V, Müller G, Sühnel J. (1999) CombiTool—a new computer program for analyzing combination experiments with biologically active agents. Comput Biomed Res 32:145–160 [DOI] [PubMed] [Google Scholar]
- Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, Schaefer-Cutillo J, De Vos S, Sinha R, Leonard JP, et al. (2010) Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 115:2578–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Ben-Bassat H, Schlesinger M, Laskov R. (1999) Somatic mutations and intraclonal variations in the rearranged Vkappa genes of B-non-Hodgkin’s lymphoma cell lines. Eur J Haematol 63:180–191 [DOI] [PubMed] [Google Scholar]
- Gobessi S, Laurenti L, Longo PG, Carsetti L, Berno V, Sica S, Leone G, Efremov DG. (2009) Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia 23:686–697 [DOI] [PubMed] [Google Scholar]
- Gururajan M, Jennings CD, Bondada S. (2006) Cutting edge: constitutive B cell receptor signaling is critical for basal growth of B lymphoma. J Immunol 176:5715–5719 [DOI] [PubMed] [Google Scholar]
- Hoellenriegel J, Coffey GP, Sinha U, Pandey A, Sivina M, Ferrajoli A, Ravandi F, Wierda WG, O’Brien S, Keating MJ, et al. (2012) Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia 26:1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Czerwinski DK, Nolan GP, Levy R. (2006) Altered B-cell receptor signaling kinetics distinguish human follicular lymphoma B cells from tumor-infiltrating nonmalignant B cells. Blood 108:3135–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaa H, Hendriks RW, Reth M. (2005) B cell signaling and tumorigenesis. Annu Rev Immunol 23:415–445 [DOI] [PubMed] [Google Scholar]
- Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. (2004) Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117:787–800 [DOI] [PubMed] [Google Scholar]
- Kuno Y, Abe A, Emi N, Iida M, Yokozawa T, Towatari M, Tanimoto M, Saito H. (2001) Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12). Blood 97:1050–1055 [DOI] [PubMed] [Google Scholar]
- Lam KP, Kühn R, Rajewsky K. (1997) In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90:1073–1083 [DOI] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, et al. (2011) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117:591–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leseux L, Hamdi SM, Al Saati T, Capilla F, Recher C, Laurent G, Bezombes C. (2006) Syk-dependent mTOR activation in follicular lymphoma cells. Blood 108:4156–4162 [DOI] [PubMed] [Google Scholar]
- Mócsai A, Ruland J, Tybulewicz VL. (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 10:387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mócsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. (2002) Syk is required for integrin signaling in neutrophils. Immunity 16:547–558 [DOI] [PubMed] [Google Scholar]
- Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, Wu B, Pasqualucci L, Neuberg D, Aguiar RC, et al. (2005) Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 105:1851–1861 [DOI] [PubMed] [Google Scholar]
- Mountz JD, Smith J, Snapper CM, Mushinski JF, Finkelman FD. (1987) Polyclonal activation of the murine immune system by an antibody to IgD. VIII. Stimulation of IgD secretion. J Immunol 139:2172–2178 [PubMed] [Google Scholar]
- Ozaki N, Suzuki S, Ishida M, Harada Y, Tanaka K, Sato Y, Kono T, Kubo M, Kitamura D, Encinas J, et al. (2012) Syk-dependent signaling pathways in neutrophils and macrophages are indispensable in the pathogenesis of anti-collagen antibody-induced arthritis. Int Immunol 24:539–550 [DOI] [PubMed] [Google Scholar]
- Quiroga MP, Balakrishnan K, Kurtova AV, Sivina M, Keating MJ, Wierda WG, Gandhi V, Burger JA. (2009) B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood 114:1029–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reth M. (2002) Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol 3:1129–1134 [DOI] [PubMed] [Google Scholar]
- Rinaldi A, Kwee I, Taborelli M, Largo C, Uccella S, Martin V, Poretti G, Gaidano G, Calabrese G, Martinelli G, et al. (2006) Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 132:303–316 [DOI] [PubMed] [Google Scholar]
- Rodríguez A, Villuendas R, Yáñez L, Gómez ME, Díaz R, Pollán M, Hernández N, de la Cueva P, Marín MC, Swat A, et al. Spanish National Cancer Centre (CNIO) (2007) Molecular heterogeneity in chronic lymphocytic leukemia is dependent on BCR signaling: clinical correlation. Leukemia 21:1984–1991 [DOI] [PubMed] [Google Scholar]
- Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, et al. (2005) Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22:507–517 [DOI] [PubMed] [Google Scholar]
- Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zürn C, Reth M. (2002) Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell 10:1057–1069 [DOI] [PubMed] [Google Scholar]
- Sprangers M, Feldhahn N, Liedtke S, Jumaa H, Siebert R, Müschen M. (2006) SLP65 deficiency results in perpetual V(D)J recombinase activity in pre-B-lymphoblastic leukemia and B-cell lymphoma cells. Oncogene 25:5180–5186 [DOI] [PubMed] [Google Scholar]
- Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. (2011) B-cell receptor signaling in chronic lymphocytic leukemia. Blood 118:4313–4320 [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. (2001) Drug synergism: its detection and applications. J Pharmacol Exp Ther 298:865–872 [PubMed] [Google Scholar]
- Tavolaro S, Chiaretti S, Messina M, Peragine N, Del Giudice I, Marinelli M, Santangelo S, Mauro FR, Guarini A, Foà R. (2010) Gene expression profile of protein kinases reveals a distinctive signature in chronic lymphocytic leukemia and in vitro experiments support a role of second generation protein kinase inhibitors. Leuk Res 34:733–741 [DOI] [PubMed] [Google Scholar]
- Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, Furlong MT, Geahlen RL, Tybulewicz VL. (1995) Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature 378:298–302 [DOI] [PubMed] [Google Scholar]
- Turner M, Schweighoffer E, Colucci F, Di Santo JP, Tybulewicz VL. (2000) Tyrosine kinase SYK: essential functions for immunoreceptor signalling. Immunol Today 21:148–154 [DOI] [PubMed] [Google Scholar]
- Wex E, Bouyssou T, Duechs MJ, Erb KJ, Gantner F, Sanderson MP, Schnapp A, Stierstorfer BE, Wollin L. (2011) Induced Syk deletion leads to suppressed allergic responses but has no effect on neutrophil or monocyte migration in vivo. Eur J Immunol 41:3208–3218 [DOI] [PubMed] [Google Scholar]
- Wossning T, Herzog S, Köhler F, Meixlsperger S, Kulathu Y, Mittler G, Abe A, Fuchs U, Borkhardt A, Jumaa H. (2006) Deregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cells. J Exp Med 203:2829–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.