

Abstract
A major challenge in organophosphate (OP) research has been the identification and utilization of reliable biomarkers for the rapid, sensitive, and efficient detection of OP exposure. Although Tyr 411 OP adducts to human serum albumin (HSA) have been suggested to be one of the most robust biomarkers in the detection of OP exposure, the analysis of HSA-OP adduct detection has been limited to techniques using mass spectrometry. Herein, we describe the procurement of two monoclonal antibodies (mAb-HSA-GD and mAb-HSA-VX) that recognized the HSA Tyr 411 adduct of soman (GD) or S-[2-(diisopropylamino)ethyl]-O-ethyl methylphosphonothioate (VX), respectively, but did not recognize nonphosphonylated HSA. We showed that mAb-HSA-GD was able to detect the HSA Tyr 411 OP adduct at a low level (i.e., human blood plasma treated with 180 nM GD) that could not be detected by mass spectrometry. mAb-HSA-GD and mAb-HSA-VX showed an extremely low-level detection of GD adducted to HSA (on the order of picograms). mAb-HSA-GD could also detect serum albumin OP adducts in blood plasma samples from different animals administered GD, including rats, guinea pigs, and monkeys. The ability of the two antibodies to selectively recognize nerve agents adducted to serum albumin suggests that these antibodies could be used to identify biomarkers of OP exposure and provide a new biologic approach to detect OP exposure in animals.
Introduction
Organophosphates (OPs) constitute a diverse class of chemicals that include insecticides, protease inhibitors, and chemical warfare agents. The use of OP toxicants as chemical warfare weapons is a threat to US military personnel as well as civilians. In addition, agricultural workers who handle OP pesticides are at risk of exposure because there are more than 100 different OP pesticides used worldwide, and it is estimated that globally there are 750,000 to 3 million human intoxications by OPs annually (Kwong, 2002). The worldwide level of OP exposure and the possible toxic consequences underscore the need to develop an efficient, portable, and inexpensive way to detect OPs (Chen and Mulchandani, 1998; Sogorb and Vilanova, 2002).
The acute toxic effects of OPs arise from the inhibition of acetylcholinesterase (AChE) to form a relatively stable OP-AChE adduct. AChE inhibition results in accumulation of excess acetylcholine that leads to stimulation of acetylcholine receptors, which can produce convulsions and other central nervous system toxicity (Marrs et al., 2007). In addition to AChE inhibition, OPs also covalently modify other proteins (Schopfer et al., 2005; Wieseler et al., 2006; Thompson et al., 2010), including butyrylcholinesterase (BChE) and serum albumin.
Analysis of biomedical samples (e.g., urine and blood) can provide not only qualitative but also quantitative information about OP exposure (Marrs et al., 2007). Numerous approaches have been used in the detection of OP exposure (Worek et al., 2005; Thompson et al., 2010), including determination of inhibition of serum AChE and BChE functional activities with biochemical assays and detection of unbound OPs in plasma or their decomposition products with mass spectrometry (MS) methods, including gas chromatography–MS (GC-MS) or liquid chromatography–tandem MS (LC-MS/MS). Other approaches include GC-MS detection of fluoride-induced reactivated AChE or BChE and detection of AChE-, BChE-, or albumin-OP adducts with capillary electrophoresis–MS or LC-MS-MS after proteolytic digestion of those proteins. Among these approaches, determination of inhibition of AChE and BChE functional activity in the blood is still the mainstay for quick initial screening, although determination of relative cholinesterase activity lacks sensitivity and specificity. MS approaches require sophisticated equipment and well trained operators (Worek et al., 2005).
Detection of protein-OP adducts (i.e., AChE, BChE, or serum albumin) as biomarkers for OP exposure has provided insight into the structural biology of OP action (Read et al., 2010; Thompson et al., 2010). Generally, the approach to detect protein-OP adducts has relied on MS (Thompson et al., 2010), although one report showed that antibodies could be raised against phosphorylated AChE (George et al., 2003). However, in that report, the antibodies reported to recognize the inhibited form of AChE did not distinguish OP-AChE adducts arising from different OPs (George et al., 2003). An attempt to develop antibodies against phosphonylated BChE showed that the chemical instability of phosphonylated decapeptide haptens designed to mimic phosphonylated BChE precluded using this approach (MacDonald et al., 2010, and unpublished observations). In addition, despite the fact that AChE and BChE are prominent targets of OP exposure, these enzymes are present at low concentrations (e.g., BChE is present at 4 μg/ml in plasma) and the OP-BChE adduct is labile (Li et al., 2007). In contrast, serum albumin is the most abundant protein in blood plasma (40,000 μg/ml), and after OP exposure, OPs form a stable adduct (e.g., with human serum albumin [HSA] at Tyr 411) on the basis of LC-MS analysis of proteolytic digests (Li et al., 2008). Thus, albumin is a functional scavenger of OPs, resulting in a stable OP adduct after exposure. It has been estimated that 1–2% of HSA forms a stable OP adduct after OP exposure (Ding et al., 2008). Based on the instability of phosphonylated adducts to AChE and BChE, the detection of HSA-OP adducts represent a more feasible strategy to detect in vivo biomarkers of OP exposure because the abundance of serum albumin is 10,000 times more than either AChE or BChE and the Tyr 411 adduct is extremely stable (Ding et al., 2008). Tyr 411 OP adducts of HSA have been reported in a number of cases of OP exposure (Peeples et al., 2005; Li et al., 2010; Lockridge and Schopfer, 2010; Marsillach et al., 2011).
Protein sequence alignment of serum albumin indicates that across different species, including humans, rats, and guinea pigs, the region surrounding Tyr 411 is highly conserved (Fig. 1). We therefore chose this region (i.e., amino acids 408–417, LVRYTKKVPQ, in HSA) as the epitope to synthesize phosphonylated antigens and haptens. Based on the three-dimensional structure of HSA, Tyr 411 lies on the surface of the protein (Ding et al., 2008) and Tyr 411 is apparently readily accessible and efficiently attacked by OPs to form stable covalent adducts of HSA. This may explain why Tyr 411 of HSA is preferentially phosphonylated if treated with OPs (Li et al., 2008).
Fig. 1.

A comparison of the peptide sequences of serum albumin in the epitope region surrounding Tyr 411 for four species: humans (HSA), monkeys (MSA), guinea pigs (GSA), and rats (RSA).
In the present report, we discuss the rationale and the successful generation of five monoclonal antibodies (mAbs) (in which two were studied in detail) that were raised against four different phosphonylated decapeptides corresponding to the region surrounding Tyr 411 in HSA. Two of the mAbs selectively recognized soman (GD) or S-[2-(diisopropylamino)ethyl]-O-ethyl methylphosphonothioate (VX) adducts, respectively, at Tyr 411 on HSA. The two antibodies were shown to possess great sensitivity (i.e., as little as 10−12 g of antigen detected in blood plasma samples) and detected OP-adducted serum albumin from various biologic samples prepared from in vitro and in vivo OP exposure experiments. The antibodies also stereoselectively recognized OP adducted to HSA. Because the OPs used in these studies possess two centers of chirality, four possible OP adducts could form. In the case of GD adducted to HSA, one of the adducts was selectively recognized over the other one. Two mAbs (i.e., mAb-HSA-GD and mAb-HSA-VX) were successfully used in studies to detect OP adducts of serum albumin in different species including humans, rats, guinea pigs, and monkeys. The availability of the two mAbs not only provides a powerful tool in basic research, but also could be used to identify biomarkers of OP exposure and provide a new approach to detect OP exposure in animals.
Materials and Methods
Biologic and Chemical Reagents.
Buffers, reagents, and solvents were purchased from VWR Scientific, Inc. (San Diego, CA) in the highest purity commercially available. The nerve agent model compounds and their corresponding Sp and Rp isomers were synthesized as previously described (Berman and Leonard, 1989; Barakat et al., 2009). The nerve agent model compounds are toxic and should be handled with extreme care. Chemical wastes containing nerve agent model compounds were hydrolyzed by overnight incubation with 2.5 M NaOH and 10% ethanol before disposal. Goat anti-mouse antibodies conjugated with horseradish peroxidase (HRP) and SuperSignal West Pico Chemiluminescent Substrate was purchased from Pierce (Rockford, IL). Molecular biology reagents were purchased from Life Technologies (Carlsbad, CA) unless otherwise specified.
Synthesis of para-Nitrophenyl Esters of Nerve Agent Model Compounds.
The synthesis of para-nitrophenyl esters of nerve agent model compounds was accomplished by combining the corresponding nerve agent monochloridates (50 mg, 0.25 mmol) with triethylamine (150 mg, 0.9 mmol) and dimethylaminopyridine (5 mg, 0.04 mmol) at 4°C in CH2CCl2. After addition of para-nitrophenol (150 mg, 1.1 mmol) and stirring at room temperature for 5 hours, the mixture was cooled to 4°C and made basic, and the separated organic fraction was evaporated to dryness and chromatographed to afford the desired products in 78%–91% yield. Each product was fully characterized by NMR.
Phosphonylation of HSA in the Presence of para-Nitrophenyl Esters of Sarin, GD, and VX.
The incubation comprised 200 nM para-nitrophenyl esters of sarin (GB), GD, and VX (made up in a stock solution of CH3CN) and 10 nM HSA in Tris-HCl buffer (10 mM, pH 8.0) in a total incubation volume of 0.6 ml. The incubation was initiated after thorough mixing and continuously monitored at 400 nm with a Cary UV-Vis spectrophotometer (Agilent Technologies, Santa Clara, CA). The progress curves were plotted as a function of absorbance increase versus time. The half-life and rate constants were determined (n = 3).
Characterization of Phosphonylated Peptide–Amino Caproic Acid Hapten Purity and Aqueous Stability
The purity of phosphonylated peptides was determined by high-performance LC (HPLC). Phosphonylated peptides were run on a Beckman Gold HPLC column using 0.1% trifluoroacetic acid in water-acetonitrile and a reverse-phase C18 column using a Hitachi 2000 HPLC (Hitachi Inc., Dallas, TX). HPLC data were reported for final, purified peptides. Final purities ranged from 92–99%. Peptide purity for each peptide is shown in Table 1. The stability of the phosphonylated peptides was determined by MS. Each phosphonylated peptide was prepared to a final concentration of 1 mg/ml and an internal standard (IS) was included to assist MS quantification (pH 7.0, 25°C). At the appropriate time point, an aliquot of each sample was injected into the MS (Hitachi M-8000; Hitachi Inc.) using electrospray ionization in the positive mode. Signal intensities for each phosphonylated peptide were tabulated and compared with the signal intensities of the IS peptide for each run (Peptide/IS). Peptide/IS for each day was normalized to the value obtained at day 0, and this value was multiplied by 100 to get percent remaining phosphonylated peptide. Plots of percent remaining versus time (days) were made in Excel. Half-lives were calculated for each phosphonylated peptide shown in Table 1.
TABLE 1.
Characterization of phosphonylated peptide-AcA hapten purity and aqueous stability
| Name | Half-lifea | Mol. Wt. | Purityb |
|---|---|---|---|
| days | Da | % | |
| HSA-peptide-AcA-GB | 12.0 | 1465.7 | >92.4 |
| HSA-peptide-AcA-GD | 16.3 | 1507.4 | >99.6 |
| HSA-peptide-AcA-GF | 19.3 | 1505.3 | >99.2 |
| HSA-peptide-AcA-VX | 8.5 | 1451.3 | >98.4 |
AcA, amino caproic acid; GB, sarin; GD, soman; GF, cyclosarin; HSA, human serum albumin; VX, S-[2-(diisopropylamino)ethyl]-O-ethyl methylphosphonothioate.
Peptide half-life was determined by mass spectrometric analysis (deionized H2O, 25°C).
Organophosphate-adducted peptide purity was determined by high-performance liquid chromatography.
Preparation of Antigens and Conjugation of Antigens to Proteins.
Phosphonylated peptide antigens (408LVRY*TKKVPQ417-AcA, where AcA stands for ε-amino caproic acid and * stands for a phosphonyl group, were synthesized as described previously (MacDonald et al., 2010). The purity of each phosphonylated peptide-AcA was >95% as judged by HPLC, and the structure was confirmed by HPLC-MS. Each phosphonylated peptide-AcA was conjugated to keyhole limpet hemocyanin (KLH; Pierce) and chicken egg white ovalbumin (OVAL; Pierce) using ethyl-diisopropyl-carbodiimide and sulfo-N-hydroxysuccinimide (1:1). Highly purified conjugated phosphonylated peptide-AcA carrier protein was obtained after removing excess peptides and reagents by filtration and dialysis against phosphate-buffered saline (PBS) buffer (pH 7.4) through repeated filtration with Amicon Ultra 4 centrifugation tubes (Millipore, Temecula, CA). Protein concentration was determined by a BCA assay (Pierce). The chemical stability of each antigen was tested by incubating the antigen at pH 7.4, room temperature, and analyzing aliquots of the incubation taken over time by MS. All of the antigens examined possessed half-life values >8.5 days (Table 1). In the MS experiment, we did not observe any new signals that may have indicated the loss of phosphonate or related groups from the antigens over the course of the stability tests.
Mouse Immunizations.
The procedures and care of animals used in this work conformed to the Guiding Principles in the Care and Use of Animals provided by the American Physiological Society, as well as all federal and California regulations. Human BioMolecular Research Institute has an approved Assurance from the National Institutes of Health's Office for Protection from Research Risks. Female Swiss Webster mice (5 weeks, 20 g) were purchased from Taconic Farms (Oxnard, CA) and used in immunization studies with the conjugated haptens. After a 1-week stabilization period, 10 mice were immunized with each KLH-conjugated hapten (i.e., HSA-VX-KLH, HSA-GD-KLH, HSA-GF-KLH, HSA-GB-KLH, and nonphosphonylated HSA-KLH). The immunization protocol consisted of an initial i.p. injection of 150 μg of conjugated hapten in 200 μl of an oil-in-water emulsion (PBS-containing Sigma Adjuvant System; Sigma, St. Louis, MO) followed by another boost 4 weeks later. After an additional 3 weeks, each animal was boosted with 100 μg OVAL-conjugated phosphonylated HSA decapeptide (i.e., HSA-GD-OVAL, HSA-VX-OVAL, HSA-GF-OVAL, and HSA-GB-OVAL) in 200 μl PBS-containing alum (Pierce) as an i.p. injection. Mouse blood (50–100 μl) was collected from each animal from the tail vein 7–10 days after each boost. Heparin (Sigma), 1–3 IU, was added to each blood sample and mixed thoroughly, and plasma was separated from blood cells through centrifugation at 5000 rpm for 10 minutes at 4°C. Each plasma sample was tested for the presence of anti–phosphonylated peptide antibodies by enzyme-linked immunosorbent assay (ELISA) (see below). When the titer reached >6400 units, the mice were allowed to rest for at least 1 month. A final tail vein injection was made with KLH-conjugated phosphonylated peptides (25–50 μg in PBS) or nonphosphonylated HSA peptide–conjugated KLH hapten 3 days before the mice were taken for splenectomy and cell fusion.
Cell Culture and Fusions.
Mouse myeloma cells NS-1 (American Type Culture Collection, Manassas, VA) were seeded at 50–100 × 103 cells/ml and grown in Medium A (Stemcell Technologies, Vancouver, BC, Canada). Three days after the final tail vein immunization, mice were sacrificed and 1–2 × 108 isolated spleen cells were fused with 1–2 × 107 myeloma cells using polyethylene glycol (Stemcell Technologies). After cell fusions, five 100-mm dishes were plated with 2–4 × 106 cells/ml in methycellulose medium (Medium D; Stemcell Technologies) for 7–14 days. Single colonies of hybridoma cells were picked from methylcellulose medium and cultured in Medium E (Stemcell Technologies) in 96-well plates. Cell culture supernatants were tested for the presence of antibodies to OVAL-conjugated HSA-OP peptides (i.e., HSA-GB-OVAL, HSA-GD-OVAL, HSA-GF-OVAL, and HSA-VX-OVAL) or nonphosphonylated HSA-OVAL using ELISA. Positive cultures were expanded and supernatants were tested against a panel of antigens to verify antibody selectivity before secondary cloning in methylcellulose Medium D or by limited dilution in Medium E. After recloning of hybridoma cells for 2–3 rounds, the monoclonal cells afforded single heavy-chain and single light-chain clones and were deemed clones that generated mAbs and isotyped with isotyping kit (Pierce).
Purification of Antibodies.
For hybridoma cells expressing the desired anti-HSA-OP mAbs, the cell culture was expanded. Briefly, culture medium of the hybridoma cells was centrifuged at 1000 rpm to separate cells from the supernatant, and the supernatant was filtered to remove residual debris. To the culture medium supernatant was added 0.01% NaN3, and it was mixed thoroughly at room temperature prior to purification. For mAb purification, the protein was dialyzed against PBS (pH 7.4) with 0.01% NaN3 and mixed 1:1 with binding buffer (Pierce) and then chromatographed on Protein G Sepharose 4B (Sigma) equilibrated with PBS (pH 7.4) containing 0.01% NaN3. Culture medium was loaded onto the column at 1 ml/min. The Protein G column was washed extensively with PBS (pH 7.4) containing 0.01% NaN3. Bound antibodies were eluted with low-pH elution buffer (pH 2.0) (Pierce) and immediately neutralized to pH ∼7.5 with 1.0 M Tris. Protein-containing fractions were pooled based on optical density at wavelength of 280 nm (OD280) absorbance and dialyzed against PBS (pH 7.4) containing 0.01% NaN3 for routine application. Purified antibodies were quantified by OD280 absorbance (i.e., OD280 1.4 for 1 mg/ml IgG) and analyzed on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for purity.
Western Blot.
Western blot analysis was conducted to determine levels of phosphonylated and nonphosphonylated OP-adducted serum albumin. The OP-adducted serum albumin was quantified in highly purified protein samples (or plasma protein samples) after removing cells by centrifugation at 5000 rpm at room temperature for 10 minutes. Blood plasma protein samples were prepared by addition to the plasma of a protease inhibitor cocktail (Sigma) in a 1:100 dilution containing 1 mM each of phenylmethylsulfonyl fluoride and Na3VO4 and 1 μg/ml each of aprotinin, leupeptin, and pepstatin. Samples were run on 10% SDS-polyacrylamide gels. The primary antibodies were obtained from hybridoma culture supernatant or highly purified mAbs. Secondary antibodies used for Western blotting included HRP-conjugated goat anti-mouse antibodies (HRP-GAM) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and blots were developed using either SuperSignal West Pico Chemiluminescent (Thermo Fisher, Waltham, MA) for regular detection or Immobilon Western Chemiluminescent HRP Substrate (WBKLS0500; Millipore) for high-sensitivity detection. Polyvinylidene difluoride (PVDF) membranes were used in the experiment (IPVH00010; Millipore). For quantification, Western blots were scanned and densitometry was determined using Scion Image software (Scion Corporation, Frederick, MD).
ELISA.
For quantification of immunoreactivity of serum from immunized mice or analysis of hapten binding of purified antibodies, ELISA was conducted. The wells of 96-well ELISA plates (Greiner, Neuburg, Germany) were coated with 0.1–2.0 μg of HSA-OP peptides or HSA-GB-OVAL, HSA-GD-OVAL, HSA-GF-OVAL, and HSA-VX-OVAL for 1 hour at room temperature in 50 μl of PBS (without calcium or magnesium) at pH 7.4. After rinsing with PBST (PBS with 0.1% Tween-20), the 96-well plates were blocked for 1 hour with 230 μl of blocking buffer [PBS containing 0.05% (v/v) Tween-20 (PBST) and 1% (w/v) bovine casein (Sigma) (PBSTC)]. After removing the blocking buffer, 50–100 μl of hybridoma culture medium or purified antibody in PBSTC was applied to each well and allowed to incubate at 37°C in a CO2 incubator for 1 hour. After rinsing off the primary antibody, a 50-μl aliquot of HRP-GAM secondary antibody (1:4000 dilution in PBSTC) (Jackson Laboratories) was added to each well and incubated at room temperature for 1 hour. After three rinses with PBST, bound antibody was detected by adding 100 μl of Pierce chemiluminescent substrate (Thermo Scientific, Brookfield, WI) to each well and reading the luminosity using a Wallac Victor2 plate reader (Perkin Elmer, Waltham, MA). For evaluation of serum titers, plasma samples were diluted in PBSTC 1:100-fold and subsequently in twofold series. The diluted plasma assayed ranged generally from 1:100- to 1:128,000-fold dilutions depending on the titer.
Preparation of Rat Blood Plasma Samples.
Male Sprague-Dawley rats [Crl:CD(SD); 250–300 g] were obtained from Charles River Laboratories (Wilmington, MA). Rats were housed individually in polycarbonate cages in a temperature-controlled (21 ± 2°C) and humidity-controlled (50% ± 10%) colony room and maintained on a reversed 12-hour light/dark cycle with lights off at 0900 hours. Food and water were available ad libitum in home cages. Blood samples were obtained from nerve agent–exposed animals as part of sample sharing from other studies conducted at the US Army Medical Research Institute of Chemical Defense. VX and GD were obtained from the US Army Edgewood Chemical Biologic Center (Aberdeen Proving Ground, MD) and diluted in sterile saline. For VX, 0.8 × LD50 (LD50 = 16 μg/kg) was administered s.c. into the right flank at a volume of 0.5 ml/kg body weight. For GD, 132 μg/kg (1.2 × LD50) was administered s.c. (0.5 ml/kg), followed 1 minute later with coadministration i.m. (0.5 ml/kg) of the oxime HI-6 (93.6 mg/kg) and atropine sulfate (2 mg/kg). Diazepam (10 mg/kg) was administered s.c. 30 minutes after seizure onset. Standard therapy of an oxime, atropine, and diazepam was given to maximize survival to a lethal dose of agent. Twenty-four hours after GD intoxication, the rats were given a wet mash of food pellets and sugar. S.c. injections of saline (3 ml) were administered as needed to prevent dehydration.
Effect of Sp-GD on Plasma Cholinesterase Functional Activity in Rats.
From six 240-g male Sprague-Dawley rats (Harlan, Milpitas, CA) administered 250 μg/kg of Sp-GD, tail vein blood was collected at different times, including at 0, 2, 4, 6, 8, 24, 48, 72, 96, 144, and 240 hours, and immediately cooled to 4°C. Plasma was obtained from whole blood after separation by centrifugation at 2000g for 10 minutes. Cholinesterase functional activity was detected by the Ellman assay (Ellman et al., 1961). Typically, 20 μl of plasma was used in each assay [1 mM acetylthiocholine, 1 mM 5,5′-dithiobis-(2-nitrobenzoic acid) in 50 mM PBS buffer, pH 7.4]. Absorbance change of 5,5′-dithiobis-(2-nitrobenzoic acid) was monitored at 405 nm using a Cary UV-Vis spectrophotometer. Absorbance at 405 nm versus time was plotted to obtain initial rate measurements. Initial rate measurements were normalized to zero time (i.e., before Sp-GD administration, the change in absorbance at 405 nm was taken as 100% functional activity) and a plot of relative cholinesterase activity versus hours or days posttreatment was done with GraphPad Prism (GraphPad Software, La Jolla, CA).
Data Analysis.
In the kinetic studies of antibody and ligand binding, the data analyses and statistical calculations were conducted with GraphPad Prism version 5.01. (GraphPad Software).
Results
Design and Synthesis of Antigens.
Although there are numerous reports concerning the synthesis of phosphorylated peptides (McMurray et al., 2001), before our publication (MacDonald et al., 2010), there were very few reports in the literature concerning the synthesis of phosphonylated peptides (Fidder et al., 2002). As previously described (MacDonald et al., 2010), four phosphonylated peptides were used in the synthesis of the required haptens (Fig. 2), including peptide conjugates of GB 1, GD 2, cyclosarin (GF) 3, and VX 4. The chemical synthesis of the antigens and conjugated immunogens followed the general strategy described previously (MacDonald et al., 2010). Briefly, Fmoc-protected tyrosine, 5, was treated with monochloridates of nerve agent model compounds 1–4 to afford phosphonylated benzyl-protected Fmoc tyrosine 10–13, respectively (Fig. 3). The monochloridates 1–4 were prepared as described before (MacDonald et al., 2010). After purification by chromatography on silica gel, the benzyl group was removed by hydrogenolysis. All of the compounds were fully characterized by 1H NMR and 31P NMR, MS, and HPLC. The phosphonylated products 10–13 were incorporated into solid-phase peptide synthesis to afford the desired phosphonylated peptide 14–17 containing an AcA at the carboxyl terminus. These products were >95% pure on the basis of HPLC and MS. The carrier protein KLH was conjugated through the AcA to produce the desired conjugated hapten-KLHs that were used to immunize mice (Fig. 3). Alternatively, 14–17 was used to conjugate to OVAL or HSA, which were used in various bioassays. In total, three different phosphonylated peptide conjugates were synthesized (i.e., hapten-KLH, hapten-HSA, and hapten-OVAL) for GB, GD, GF, and VX. In addition, nonphosphonylated peptide conjugates (i.e., peptide-KLH, peptide-HSA, and peptide-OVAL) were prepared and characterized. Characterization of the HSA–phosphonylated peptide–AcAs is summarized in Table 1. Because of the potential chemical lability of haptens 14–17, chemical stability studies were conducted. As shown in Table 1, on the basis of MS studies of the kinetics of loss in the presence of potassium phosphate buffer (pH 7.4), the haptens possessed considerable chemical stability. Loss of material may have been to adherence to plasticware or other surfaces because no detectable degradation products were observed as judged by MS.
Fig. 2.

Chemical structures of the monochloridates of nerve agent model compounds used in the synthesis of haptens. (1) GB; (2) GD; (3) GF; (4) VX.
Fig. 3.

Chemical synthesis of phosphonylated peptides conjugated to KLH via AcA used to generate mAbs that recognize nerve agents adducted to serum albumin. (A) The monochloridates of model compounds of sarin, soman, cyclosarin, and VX (i.e., compounds 1–4) were treated with with Fmoc- and benzyl-protected tyrosine (compound 1) to afford 6–9. (B) After benzyl deprotection, the resulting carboxylic acids (i.e., compounds 10–13) were used in solid-phase peptide synthesis to make the phosphonylated peptide-AcA. (C) Solid-phase peptide synthesis using Fmoc-protected phosphonylated tyrosine. (D) The resulting haptens (14–17) were conjugated to KLH to form the antigen. (E) The conjugated antigen was used to immunize mice to generate antibodies.
Procurement of Antibodies.
Tail vein blood was obtained from the immunized mice, and plasma obtained after centrifugation was tested for the presence of anti–phosphonylated HSA–peptide (i.e., anti-GB-, GD-, GF-, and VX-HSA antibodies) by ELISA. When the titer reached >6400 units, the mice were allowed to rest at least 1 month. A final tail vein injection was made with KLH-conjugated phosphonylated peptides before splenectomy and cell fusion. Spleens from immunized mice were obtained and finely minced for fusion with myeloma cells to give rise to primary hybridoma cells. Fusion of spleens from mice immunized against GB and GF yielded very few primary hybridomas, and thus, we focused our efforts on hybridoma cells from F41 and F46 (i.e., hybridomas that produced mAbs that selectively recognized HSA-GD and HSA-VX, respectively, named mAb41.5D5A2 and mAb46.5H10G7). After screening >200,000 primary cells, 40 clones were obtained. Recloning of these 40 positive hybridoma cells two or three more times provided homogenous cell lines expressing two lines of mAbs that selectively recognized HSA-GD or HSA-VX. Thus, mAb-HSA-GD and mAb-HSA-VX were obtained after cloning and recloning several generations of hydridoma cells. The mAbs were purified to homogeneity and characterized by SDS-PAGE and isotyping (Fig. 4). The isotypes for both mAb-HSA-GD and mAb-HSA-VX were determined to be IgG1b and their light chains were both κ isotype.
Fig. 4.
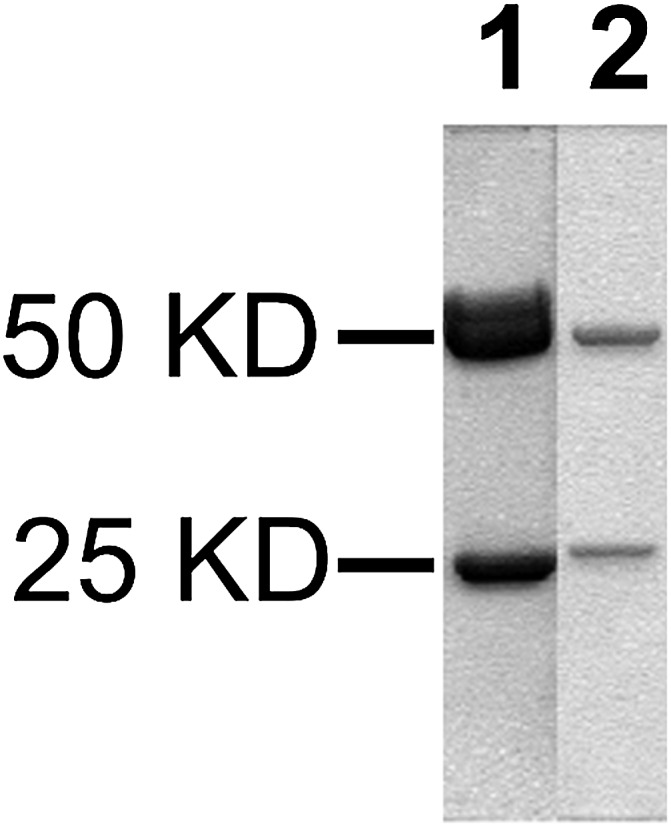
SDS-PAGE of mAbs. Lane 1, mAb-HSA-GD; lane 2, mAb-HSA-VX.
In Vitro Characterization of mAb-HSA-GD and mAb-HSA-VX.
Characterization of the selectivity of mAb-HSA-GD and mAb-HSA-VX was done using four different peptide haptens (i.e., HSA-GD, HSA-VX, HSA-GB, and HSA-GF) that were chemically synthesized to afford the same phosphonyl adduct as the actual nerve agents (Fig. 3). The nonphosphonylated decapeptide (i.e., HSA-NP) was also prepared and used for comparison. As shown in Fig. 5, ELISA data showed that mAb-HSA-GD had the greatest affinity for HSA-GD, while mAb-HSA-VX had the greatest affinity for HSA-VX. Compared with other mAbs obtained, mAb-HSA-GD showed selective recognition of HSA-GD and HSA-GF and to a much lesser extent the other HSA-OP adducts (i.e., GD ∼ GF >> GB > VX). mAb-HSA-GD did not recognize the HSA peptide that did not contain a phosphonyl group on Tyr 411 (i.e., HSA-NP). mAb-HSA-VX selectively recognized HSA-VX compared with the other HSA-OP peptide adducts (i.e., VX >> GB > GF > GD). mAb-HSA-VX did not recognize the HSA peptide that did not contain a phosphonyl group (i.e., HSA-NP). The Kd values for mAb-HSA-GD and mAb-HSA-VX were 30 nM and 0.4 nM, respectively (Fig. 5). The affinity of mAb-HSA-GD for HSA-GD or mAb-HSA-VX for HSA-VX, respectively, was confirmed with Western blot studies. As shown in Fig. 6, the immunoreactivity of mAb-HSA-GD for HSA-GD > HSA-GB ∼ HSA-GF >> HSA-VX. No significant immunoreactive signal was observed for mAb-HSA-GD against HSA-GA (HSA-tabun) or HSA-NP at low immunogen levels.
Fig. 5.

Characterization of antibodies by competitive ELISA with synthetic phosphonylated and nonphosphonylated peptides attached to ovalbumin (i.e., HSA-GB-OVAL, HSA-GD-OVAL, HSA-GF-OVAL, HSA-VX-OVAL, and nonphosphonylated HSA-NP-OVAL). HSA-NP is the nonphosphonylated peptide (NP). (A) mAb-HSA-GD. (B) mAb-HSA-VX. ▼, HSA-VX; □, HSA-GD; △, HSA-GF; ○, HSA-GB; ◇, HSA-NP; RU, relative luminescence units. Experiments were carried out with 1:100-diluted hydridoma cell culture medium. mAb was preincubated separately with each phosphonylated or nonphosphonylated HSA-peptide as described in Materials and Methods. The bound antibody was measured indirectly using an HRP-conjugated secondary antibody.
Fig. 6.

Western blot analysis of haptens conjugated to ovalbumin (i.e., HSA-GB-OVAL, HSA-GD-OVAL, HSA-GA-OVAL, HSA-GF-OVAL, HSA-VX-OVAL, and nonphosphonylated HSA-NP-OVAL). HSA-NP is the nonphosphonylated peptide (NP). mAb-HSA-GD (A) or mAb-HSA-VX (B) were tested for their preferential binding to the haptens (above) by Western blot analysis.
In good agreement with the ELISA data, on the basis of Western blot analysis, the immunoreactivity of mAb-HSA-VX was HSA-VX >> HSA-GB > HSA-GF. No significant immunoreactive signal was observed for mAb-HSA-VX against HSA-GD, HSA-GA, or HSA-NP at low immunogen levels. The affinities of mAb-HSA-VX and mAb-HSA-GD suggested that the antibodies could have utility in the selective analysis of exposure to different nerve agent OPs because the two antibodies readily detected GD and VX, respectively, and to a lesser extent, GB and GF.
Because the antibodies were designed to be used in detection and analysis of OP exposure in vivo, we sought to increase the sensitivity of detection and develop a robust method to detect OPs at very low concentrations. To test the lowest level of detection of the antibodies, we conducted a titration experiment by serially diluting an authentic HSA-GD protein standard. Previously, the amount of phosphonylated HSA Tyr 411 adducted with GD had been determined by tryptic digestion and LC-MS studies and was available as a standard (Li et al., 2007; O. Lockridge, personal communication). An HSA-VX standard protein was not available, so the study was largely conducted with mAb-HSA-GD. As shown in Fig. 7B, the lowest level of detection of the GD adduct on Tyr 411 of HSA was on the order of 10−12 g with a femtogram-sensitive HRP substrate in a Western blot analysis format.
Fig. 7.

Determination of limit of detection by mAb-HSA-GD for HSA-GD protein adduct. (A) Detection of HSA-GD protein adduct with a low-sensitivity chemiluminescent HRP substrate. (B) Detection of HSA-GD protein adduct with a high-sensitivity HRP substrate. (C) Ponceau staining of membranes used in (A) and (B) shows the relative protein content using a Western blot on a PVDF membrane. In the experiment, 1 μg of HSA-GD protein adduct standard (containing 21% of the Tyr 411 amino acid adducted as HSA-GD, independently predetermined by MS) was serially diluted (as indicated by the fold-dilution on the top of the blot) in either PBS or HSA solution (containing 1 mg/ml of unmodified HSA in each lane).
Because mAb-HSA-GD showed superb sensitivity in detecting GD-adducted HSA on the basis of serially diluted standard protein samples in PBS, we extended the studies to detection of OP-adducted serum albumin isolated from animal blood samples. Because it was estimated that only 1–2% of serum albumin would form Tyr 411 adducts during OP exposure (Ding et al., 2008), to recognize the serum albumin–GD adduct in blood plasma, it was required that the antibody not only have the ability to detect low levels of target protein as shown in Fig. 7, but also have the ability to distinguish serum albumin–GD adduct in the presence of large quantities of unmodified serum albumin. We therefore conducted experiments to simulate these conditions in blood plasma before embarking on more complex detection in blood samples from animals exposed to OPs. In these experiments, OP-adducted HSA standards previously quantified by LC-MS (O. Lockridge, personal communication) were serially diluted in the presence of unmodified HSA. The total protein in each dilution experiment was 1 μg, as shown by Ponceau staining in Fig. 7C. When an HSA solution was used in a serial dilution experiment, compared with dilution in PBS, the intensity of the signal was not significantly decreased (e.g., Fig. 7, B and C), and the antibody recognized far less than 1% of a theoretical amount of HSA-GD adduct in solution. For example, in an experiment with 1.6% of the serum albumin adducted with GD (determined by MS after tryptic digestion) at a typical blood plasma concentration, mAb-HSA-GD readily recognized HSA-GD adduct (i.e., the second lane of Fig. 7B represented a typical blood plasma that had 1–2% of HSA-GD adduct). Thus, using the methodology developed, detection of very low levels of HSA-adducted OPs was feasible even in the presence of large amounts of unmodified HSA.
Phosphonylation of HSA in the Presence of para-Nitrophenyl Esters of Nerve Agents.
To study the kinetics of OP adduction of HSA in vitro, we synthesized para-nitrophenyl esters of nerve agent model compounds 18, 19, and 20 (Fig. 8). The para-nitrophenol (pNP) leaving group of these OP esters liberated when the para-nitrophenyl esters covalently modified HSA was continuously monitored at 400 nm. From the time course studies the half-life of adduction of HSA by GB, GD, and VX model compounds 18–20 were 6.1 ± 4, 10.9 ± 5, and 16.4 ± 9 hours, respectively (Table 2).
Fig. 8.

Chemical structures of para-nitrophenyl esters of nerve agent as photometric model compounds. (A) Structure of para-nitrophenyl-GB. (B) Structure of para-nitrophenyl-GD. (C) Structure of para-nitrophenyl-VX.
TABLE 2.
Half-life values and kinetic constants for the pseudo first-order reaction between nerve agent model compound para-nitrophenyl esters 18–20 and human serum albumin
Experiments were carried out at pH 7.4, room temperature.
| Nerve Agent Model Compound | Half-life (hr) | k |
|---|---|---|
| h | h-1 | |
| para-Nitrophenyl-GB 18 | 10.9 ± 5 | 0.06 ± 0.02 |
| para-Nitrophenyl-GD 19 | 6.1 ± 4 | 0.11 ± 0.07 |
| para-Nitrophenyl-VX 20 | 16.4 ± 9 | 0.04 ± 0.02 |
GB, sarin; GD, soman; HSA, human serum albumin; VX, (S-[2-(diisopropylamino)ethyl]-O-ethyl methylphosphonothioate).
Stereoselectivity of Antibody mAb-HSA-GD.
Certain nerve agents and other OPs have a center of chirality about phosphorus. Generally, authentic nerve agents are present as racemic mixtures. However, work has been done with enantiomerically pure nerve agents (Benschop and De Jong, 1988) and nerve agent model compounds (Zheng et al., 2010). It has been observed that nerve agent or nerve agent model compound enantiomers inhibit cholinesterase with considerable stereoselectivity (i.e., for cholinesterase inhibition, Sp nerve agent > Rp nerve agent) (Benschop and De Jong, 1988; Zheng et al., 2010). The observations from in vitro cholinesterase inhibition studies are relevant to in vivo toxicity because it has been shown that nerve agents or nerve agent model compounds with Sp chirality are much more toxic than nerve agents with Rp chirality (Benschop and De Jong, 1988; Kalisiak et al., 2011, 2012).
As a model system to test the ability of mAb-HSA-GD to recognize different optical isomers of HSA-GD adducted to HSA, we examined the stereoselectivity of mAb-HSA-GD recognition of the adduct of Rp- and Sp-GD model compound enantiomers in vitro. Previously, we showed that several nerve agent model compounds recapitulated the stereoselectivity and mechanism of action of actual nerve agents in all respects except that the model compounds were less toxic (Gilley et al., 2009). Accordingly, we incubated Rp-GD methylthiocholine or Sp-GD methylthiocholine (0.2 mg/ml) in the presence of HSA (60 mg/ml) over a 20-day time course to examine whether GD adducts could form and whether the adduct formed was stable. We also examined the stereoselectivity of the adduction. Over the course of a 20-day incubation, based on the area of the product formation curves, Sp-GD methylthiocholine showed approximately a 1.3-fold greater stereoselectivity in HSA adduct formation recognition by mAb-HSA-GD compared with the amount of adduct formed from Rp-GD in the in vitro system (Fig. 9). Although the total amount of OP adduct recognized by the mAb-HSA-GD was significantly different for each OP enantiomer, the stability of the adducts appeared to be similar.
Fig. 9.

A plot of the amount of GD nerve agent model compound stereoisomers adducted to HSA as a function of time based on quantification of immunoreactivity with mAb-HSA-GD. The amount of HSA-GD protein adduct was determined by Western blot analysis. After addition of nerve agent model compound Sp-GD or Rp-GD methylthiocholine to HSA, the incubation was sampled over time and compared with an HSA-GD-adducted protein standard. Quantification was done by densitometry. ○, Sp-GD methylthiocholine; □, Rp-GD methylthiocholine.
Immunodetection of GD in Human Blood Plasma Samples.
As described above, we characterized mAb-HSA-GD and mAb-HSA-VX with highly purified OP-adducted HSA samples in vitro. mAb-HSA-GD was characterized for its ability to recognize nerve agent–adducted HSA in the presence of HSA. We next examined whether mAb-HSA-GD could recognize authentic HSA-adducted GD standards in the presence of human blood plasma. As shown in Fig. 10, mAb-HSA-GD detected extremely low levels of HSA-GD in the presence of human plasma that was treated with 0.18 μM GD for 72 hours. Under similar conditions, no detectable amount of OP adduct could be detected by LC-MS under standard tryptic digestion and analysis conditions (O. Lockridge, personal communication). After analysis by densitometry, the level of HSA-GD detected in human plasma treated with 0.18 μM GD was determined to be 0.1 ng/ml using an HSA-GD protein standard as a positive control.
Fig. 10.
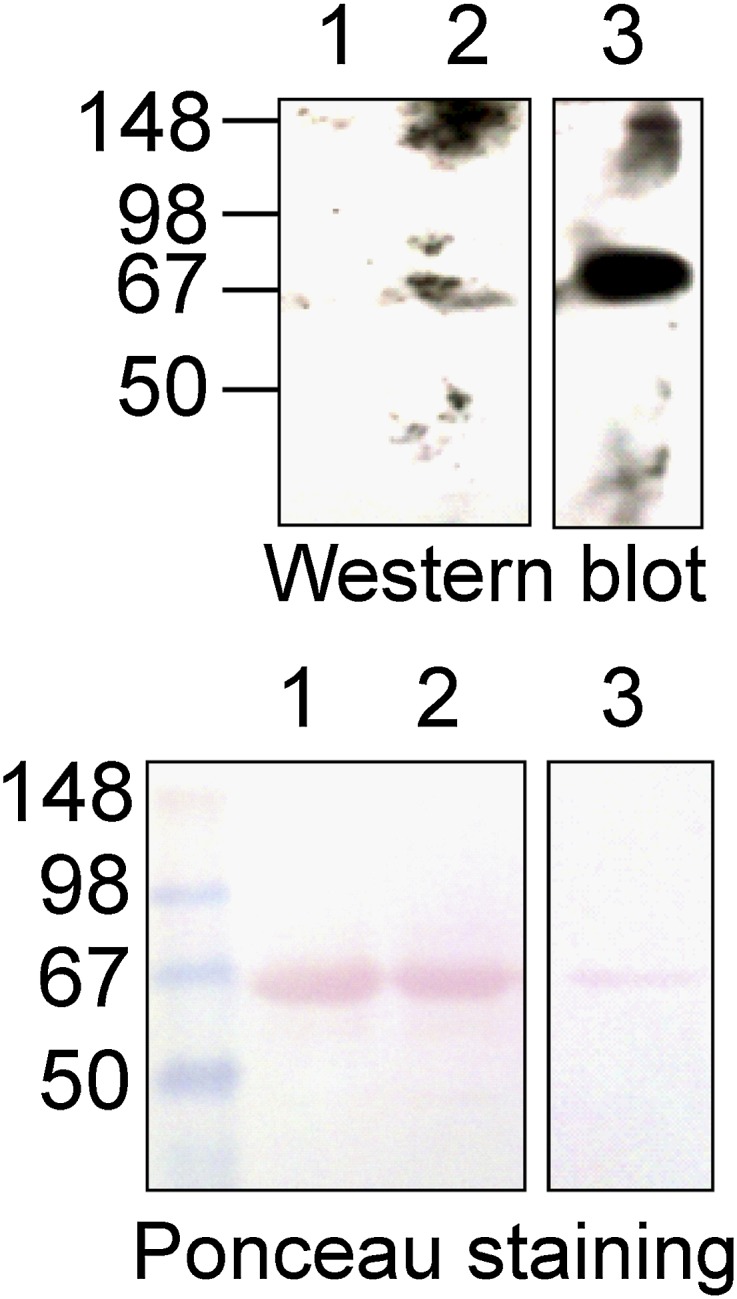
Western blot analysis of human blood plasma samples treated with GD. (Top) Western blot of human blood plasma samples treated with GD (0.18 μM) for 72 hours and probed with mAb-HSA-GD. (Bottom) Ponceau staining of the PVDF membrane used in the Western blot shows an equal amount of serum albumin. Lane 1, untreated human plasma (1 μl); lane 2, human plasma (1 μl) treated with 0.18 μM GD; lane 3, positive control protein HSA-GD-adducted protein standard (1 μg).
Immunodetection of GD Adducts in Rat Serum Albumin from Plasma of Rats Administered GD.
We examined whether mAb-HSA-GD could detect GD-adducted albumin in the presence of blood samples from animals treated with authentic nerve agents. Rat blood plasma samples were prepared from rats 72 hours after the animals had been administered GD s.c. at 1.2-fold the LD50 for GD. As shown in Fig. 11, four different samples from plasma isolated from the blood of rats treated with GD (lanes 2, 3, 5, and 6) showed immunoreactive bands for GD–rat serum albumin (RSA-GD) with mAb-HSA-GD on the basis of Western blot analysis. Samples that showed no OP-adducted RSA signal (lanes 1 and 4) were derived from rats that were treated with PBS. Purified RSA and HSA were used as negative control samples in the same Western blot analysis (lanes 7 and 8). The positive control samples used in characterization of mAb-HSA-GD in vitro (Fig. 7) were also used as positive controls in the Western blot and a quantification standard (lane 9). The results showed that mAb-HSA-GD was suitable for low-level detection and sensitive analysis of authentic biologic samples of GD-adducted blood from rats. The RSA-GD detected was quantified by densitometry using HSA-GD as a standard to be 5.4 ± 1.1 ng/ml plasma.
Fig. 11.

Western blot using mAb-HSA-GD of plasma samples from rats treated with GD (1.2 × LD50) for 72 hours. (A) Lanes 1 and 4, plasma (1 μl) from untreated rats; lanes 2, 3, 5, and 6, plasma from rats treated with GD; lane 7, untreated RSA (1 μg); lane 8, HSA (1 μg); lane 9, a standard of 1 μg of GD-adducted HSA (independently characterized by MS. (B) Coomassie blue staining of an identical gel as blot A run in parallel that showed equal protein loading of serum albumin.
Immunodetection of VX Adducted to RSA from Plasma of Rats Administered VX.
We examined the plasma isolated from rats administered authentic VX with mAb-HSA-VX to detect VX-adducted serum albumin. In a similar fashion to the detection of GD-adducted RSA from rats administered GD described above, VX-adducted RSA was detected in plasma samples from rats treated with authentic VX. As shown in Fig. 12, blood plasma samples isolated from rats 72 hours after administration of VX s.c. at 0.8-fold the LD50 showed immunoreactive RSA-VX adducts. The selective immunodetection showed that VX adducted on Tyr 411 of RSA was detectable even at sublethal doses. The RSA-VX detected was quantified by densitometry using HSA-GD as a standard and was observed to be 5–50 ng/ml (quantification was done by comparing the VX band intensity in the Western blot with the HSA-GD band on the same blot because no independently verified HSA-VX was available).
Fig. 12.
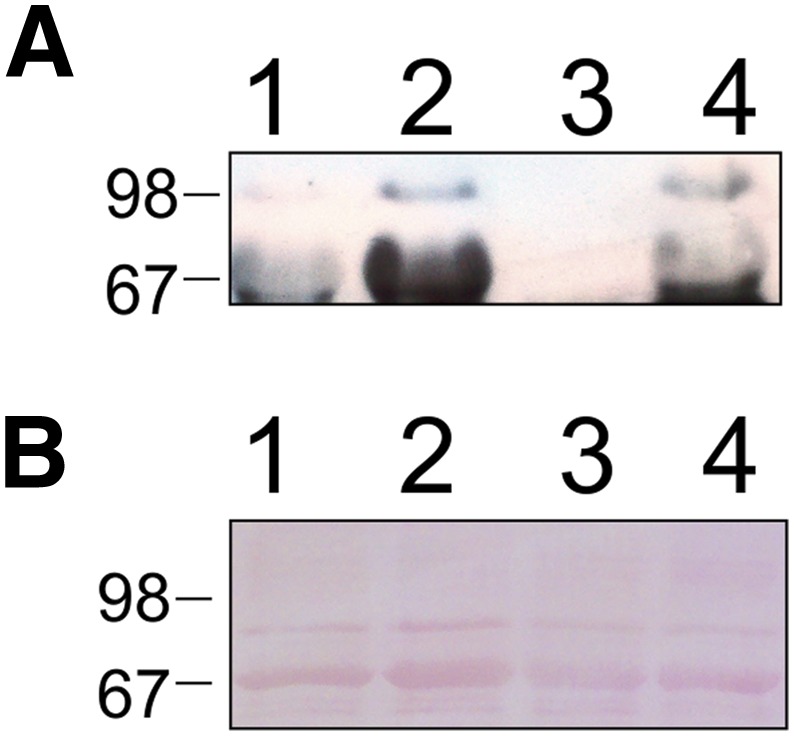
Western blot using mAb-HSA-VX of plasma samples from rats treated with VX (0.8 × LD50) for 72 hours. (A) Lanes 1 and 3, plasma (1 μl) from untreated rats; lanes 2 and 4, plasma (1 μl) from treated rats. (B) Ponceau staining of an identical gel as blot A run in parallel that showed equal protein loading of serum albumin.
In addition to GD and VX detected in RSA from rats administered the actual nerve agents, mAb-HSA-GD was able to detect GD-adducted serum albumin in the presence of plasma 30 minutes after African green monkeys were administered authentic GD (15 μg/kg). In addition, mAb-HSA-VX immunodetected VX-adducted serum albumin from the plasma of guinea pigs 72 hours after administration of VX s.c. at 0.6–0.8 × LD50 (Supplemental Fig. 1). The immunodetection of GD adducted to monkey serum albumin under these conditions was determined to be 1.8 ± 0.4 ng/ml plasma.
Cholinesterase Activity Inhibition by GD Model Compound.
One of the acute toxic effects of nerve agents is the cholinesterase inhibition in the blood. We therefore investigated the time course of cholinesterase inhibition with a GD model compound Rp-GD tertiary amine (Barakat et al., 2009) and compared it to one reported for GD . As shown in Fig. 13A, 80% of the functional activity of cholinesterase in rat blood plasma was inhibited by Sp-GD model compound at the 4-hour time point, but fully recovered to a normal level after 48 hours. At the 72-hour point, the cholinesterase activity was completely recovered to a normal level (Fig. 13B) while RSA-GD was still readily detectable (Fig. 11). Comparison of data for GD [i.e., 0–24 hours (Geller et al., 1987) and up to 10 days (Jovic, 1974)] with the Rp-GD model compound showed similar results in inhibition of cholinesterase functional activity (Fig. 13).
Fig. 13.

Time course for inhibition of rat plasma cholinesterase functional activity by nerve agent model compound Sp-GD tertiary amine (Barakat et al., 2009) or GD after administration to rats. (A) Comparison of inhibition of rat plasma cholinesterase by nerve agent model compound Sp-GD (□) (250 μg/kg, i.p.) and GD (○) (88 μg/kg, i.p) (modified from Geller et al., 1987). (B) Comparison of time course (0–10 days) for inhibition of rat plasma cholinesterase activity by nerve agent model compound Sp-GD tertiary amine (□) (250 μg/kg, i.p.) (Barakat et al., 2009) and GD (◯) (25.5 μg/kg, i.p.) (modified from Jovic, 1974).
Discussion
Herein we describe the characterization of nerve agent– or nerve agent model compound–adducted serum albumin from large and small animals using mAbs raised against nerve agent–phosphonylated peptides of HSA. As shown in Fig. 13, nerve agent model compounds showed a similar time course of action compared with the actual nerve agent (i.e., Sp-GD versus GD itself), albeit at different doses. Consequently, data using nerve agent model compounds provide a useful approach in advance of studies using the actual nerve agents. The mAbs obtained showed remarkable sensitivity and selectivity in detecting phosphonylated serum albumin from both in vitro and in vivo samples. As shown in Fig. 7, A and B, in the case of in vitro samples, mAb-HSA-GD was able to recognize as little as 100 pg of HSA-GD. In contrast, detection of tryptic digests of a similar sample has a limit of detection of 250 ng (Ding et al., 2008). The mAbs described herein are remarkably selective and can distinguish phosphonylated HSA from nonphosphonylated HSA. Even minor structural changes of nerve agent–HSA adducts could be distinguished. As shown in Fig. 7B, mAb-HSA-GD was able to recognize HSA-GD in the presence of nonphosphonylated HSA at as low as a 213:1 molar ratio dilution without losing any sensitivity (i.e., compared with identical fold-dilutions in the presence of PBS or HSA solution). In the case of in vivo samples, mAb-HSA-GD was capable of detecting exposure to nerve agent model compounds or actual nerve agents from biologic samples at extremely low levels that were not detectable by MS methods (Fig. 10) (O. Lockridge, personal communication). Animals treated with nerve agents (i.e., GD or VX) at sublethal doses afforded plasma that showed immunoreactivity to the corresponding adduct at Tyr 411 of serum albumin.
Currently available approaches for the detection of OP exposure including functional enzyme assays and MS-based approaches have certain advantages and drawbacks (Worek et al., 2005; Thompson et al., 2010). For example, assays of functional activity of AChE and BChE are quick and efficient but do not provide information about the type of nerve agent that is present and inhibiting the enzyme. MS-based approaches to detect protein adducts of OPs are quite sensitive but require expensive equipment and well trained operators (Worek et al., 2005). The approach described herein employs a biologic method using immunodetection that is efficient and robust.
Nerve agent adducts of serum albumin were chosen to develop immunodetection because previous studies of BChE showed that the phosphonylated peptide surrounding the active-site serine of BChE is prone to elimination and affords the corresponding dehydroalanine-containing peptide (Masson and Lockridge, 2010). The phosphonylated peptide that corresponds to the region surrounding Tyr 411 of HSA shows significant stability, and the adduct is not prone to elimination (Masson and Lockridge, 2010). The antibodies described herein, mAb-HSA-GD and mAb-HSA-VX, can distinguish phosphonylated Tyr 411 peptides from nonphosphonylated peptides. In particular, the antibodies selectively recognized phosphonylated Tyr 411 on serum albumin, (i.e., anti-HSA-GD recognizes GD-adducted HSA at Tyr 411 and anti-HSA-VX selectively recognizes VX-adducted HSA at Tyr 411) in Western blot analysis. To our knowledge, these antibodies are the first antibodies that can selectively recognize defined phosphonylated peptides of protein samples from in vivo serum samples.
The specificity of the antibodies reported herein was achieved using a defined epitope approach to raise antibodies that could distinguish native and phosphonylated peptide. Previously, a report showed that antibodies could distinguish phosphorylated AChE and inhibited AChE using this strategy (George et al., 2003), but despite their utility, these polyclonal anti–phosphorylated AChE antibodies did not have the ability to distinguish different OP adducts. The results of that report did suggest that it was possible to raise antibodies to individually recognize specific modifications of the dianionic phosphorus of OP-adducted proteins (George et al., 2003).
Another strategy reported in the literature to raise antibodies to selectively recognize OPs is to raise antibodies against different epitopes of the same hapten (Johnson et al., 2005). Although antibodies raised against OPs following this strategy did bind to different OP adducts with some affinity, their binding to the desired OP-adducted protein was reportedly relatively weak and not selective because most of the antibodies described had IC50 values to their desired antigen in the range of 10−4 to 10−6 M (Hunter and Lenz, 1982; Hunter et al., 1982, 1985, 1990; Brimfield et al., 1993; Johnson et al., 2005). For example, antibodies raised against GD that was conjugated to the carrier protein bovine serum albumin had an IC50 value of 10−4 M to its antigen (Lenz et al., 1992). Antibodies raised against GD that were conjugated to carrier protein KLH had a reported IC50 value of 10−6 M to its antigen (Hunter and Lenz, 1982). Compared with the mAbs described herein, the previously reported antibodies to phosphonylated haptens may be useful in qualitative analysis of biomedical samples, but may have less utility in quantitative studies.
Using a defined epitope approach, mAb-HSA-GD and mAb-HSA-VX, described herein, showed excellent detection of phosphonylated serum albumin using Western blot analysis across different species. The cross-species detection is undoubtedly due to the conserved nature of the region in the serum albumin protein sequence used in the hapten.
After exposure to nerve agents, numerous protein-OP adducts are formed in the blood, including cholinesterases and serum albumin. As shown in Fig. 13, following nerve agent model compound or actual nerve agent administration, rat cholinesterase functional activity is largely abrogated at 24 hours but functional activity is recovered to normal levels after 48 hours. Therefore, cholinesterase functional activity can be used as a biomarker in OP poisoning in the short term, but cholinesterase functional activity cannot be reliably used as a biomarker for OP exposure much beyond 48 hours. In contrast, serum albumin–GD adducts from animals exposed to nerve agents were still readily detectable by mAb-HSA-GD 72 hours after exposure, well after cholinesterase functional activity recovered back to normal levels. These data show that the detection of HSA-GD is a more robust biomarker than cholinesterase functional activity at longer time points postadministration. The availability of mAbs described herein also affords detection of nerve agent–adducted serum albumin and provides structural information to identify the chemical nature of the agent possibly long after the subject has been exposed to a nerve agent. In fact, some of the samples used in the studies described herein had been stored at −80°C for 3 years.
It is known that GD contains two centers of chirality. Although it has been reported that stereoisomers of GD have differential effects on their toxic potency in vivo, the potency of each isomer has largely been studied in detail for stereoselectivity of cholinesterase inhibition (Benschop and De Jong, 1988). Little has been reported about the reaction of GD stereoisomers with serum albumin. As shown in Fig. 9, mAb-HSA-GD stereoselectively recognized the Sp-GD isomer in preference to the Rp-GD isomer adducted to HSA. The result may suggest that Sp-GD more avidly binds to HSA compared with the Rp-GD isomer or it may suggest that the mAb-HSA-GD stereoselectively distinguishes GD–serum albumin adducts. If the nerve agent model compounds recapitulate the action of actual nerve agents (albeit at greater doses), the observation regarding stereoselective GD–serum albumin adduction may have some consequences for deconvoluting the mechanism of toxicity of GD (and other nerve agents). In conclusion, using a defined epitope approach, we obtained two mAbs that selectively recognize GD- or VX-adducted HSA at Tyr 411. The two mAbs showed unprecedented sensitivity and selectivity in detection of nerve agent biomarkers both in vitro and in vivo. The mAb approach presented herein provides a powerful tool in the detection of biomarkers for nerve agent OP detection.
Supplementary Material
Acknowledgments
The authors thank Dr. Oksana Lockridge (University of Nebraska Medical School) for providing the HSA-GD adducts that were used as independently verified protein standards. The authors also thank Dr. John McDonaugh (US Army Medical Research Institute of Chemical Defense, Aberdeen Proving Ground) for providing monkey blood plasma samples. The authors also thank Don Kaiser for the work on hybridoma cells and mAb screening. Additionally, the authors thank Drs. Mary MacDonald and Marion Lanier for chemical synthesis of the haptens and immunogens.
Abbreviations
- AcA
amino caproic acid
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- ELISA
enzyme-linked immunosorbent assay
- GA
tabun
- GB
sarin
- GC-MS
gas chromatography–mass spectrometry
- GD
soman
- GF
cyclosarin
- HPLC
high-performance liquid chromatography
- HRP
horseradish peroxidase
- HSA
human serum albumin
- HSA-GB
decapeptide hapten that has a GB phosphonylated tyrosine
- HSA-GD
decapeptide hapten that has a GD phosphonylated tyrosine
- HSA-GF
decapeptide hapten that has a GF phosphonylated tyrosine
- HSA-soman protein adduct
human serum albumin that has conjugated soman
- HSA-VX
decapeptide hapten that has a VX phosphonylated tyrosine
- IS
internal standard
- KLH
keyhole limpet hemocyanin
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- mAb
monoclonal antibody
- mAb-HSA-GD
monoclonal antibody that recognizes HSA tyrosine 411 adduct of GD
- mAb-HSA-VX
monoclonal antibody that recognizes HSA tyrosine 411 adduct of VX
- MS
mass spectrometry
- OP
organophosphate
- PBS
phosphate-buffered saline
- PVDF
polyvinylidene difluoride
- RSA
rat serum albumin
- OVAL
ovalbumin
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- VX
S-[2-(diisopropylamino)ethyl]-O-ethyl methylphosphonothioate
Authorship Contributions
Participated in research design: Chen, Zhang, Cashman.
Conducted experiments: Chen, Zhang, Cashman.
Contributed new reagents or analytic tools: Lumley.
Performed data analysis: Chen, Zhang, Cashman.
Wrote or contributed to the writing of the manuscript: Chen, Zhang, Lumley, Cashman.
Footnotes
This work was supported by the CounterACT Program, National Institutes of Health Office of the Director, and National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant UO1 NS058038] (J.R.C). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the federal government.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Barakat NH, Zheng X, Gilley CB, MacDonald M, Okolotowicz K, Cashman JR, Vyas S, Beck JM, Hadad CM, Zhang J. (2009) Chemical synthesis of two series of nerve agent model compounds and their stereoselective interaction with human acetylcholinesterase and human butyrylcholinesterase. Chem Res Toxicol 22:1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop HP, De Jong LPA. (1988) Nerve agent stereoisomers: analysis, isolation and toxicology. Acc Chem Res 21:368–374 [Google Scholar]
- Berman HA, Leonard K. (1989) Chiral reactions of acetylcholinesterase probed with enantiomeric methylphosphonothioates. Noncovalent determinants of enzyme chirality. J Biol Chem 264:3942–3950 [PubMed] [Google Scholar]
- Brimfield AA, Lenz DE, Maxwell DM, Broomfield CA. (1993) Catalytic antibodies hydrolysing organophosphorus esters. Chem Biol Interact 87:95–102 [DOI] [PubMed] [Google Scholar]
- Chen W, Mulchandani A. (1998) The use of live biocatalysts for pesticide detoxification. Trends Biotechnol 16:71–76 [DOI] [PubMed] [Google Scholar]
- Ding SJ, Carr J, Carlson JE, Tong L, Xue W, Li Y, Schopfer LM, Li B, Nachon F, Asojo Oet al. (2008) Five tyrosines and two serines in human albumin are labeled by the organophosphorus agent FP-biotin. Chem Res Toxicol 21:1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95 [DOI] [PubMed] [Google Scholar]
- Fidder A, Hulst AG, Noort D, de Ruiter R, van der Schans MJ, Benschop HP, Langenberg JP. (2002) Retrospective detection of exposure to organophosphorus anti-cholinesterases: Mass spectrometric analysis of phosphylated human butyrylcholinesterase. Chem Res Toxicol 15:582–590 [DOI] [PubMed] [Google Scholar]
- Geller I, Sawa A, Stavinoha WB. (1987) Effects of subchronic soman on avoidance-escape behavior and cholinesterase activities. Neurotoxicol Teratol 9:377–386 [DOI] [PubMed] [Google Scholar]
- George KM, Schule T, Sandoval LE, Jennings LL, Taylor P, Thompson CM. (2003) Differentiation between acetylcholinesterase and the organophosphate-inhibited form using antibodies and the correlation of antibody recognition with reactivation mechanism and rate. J Biol Chem 278:45512–45518 [DOI] [PubMed] [Google Scholar]
- Gilley C, MacDonald M, Nachon F, Schopfer LM, Zhang J, Cashman JR, Lockridge O. (2009) Nerve agent analogues that produce authentic soman, sarin, tabun, and cyclohexyl methylphosphonate-modified human butyrylcholinesterase. Chem Res Toxicol 22:1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter KW, Jr, Brimfield AA, Knower AT, Powell JA, Feuerstein GZ. (1990) Reversal of intracellular toxicity of the trichothecene mycotoxin T-2 with monoclonal antibody. J Pharmacol Exp Ther 255:1183–1187 [PubMed] [Google Scholar]
- Hunter KW, Jr, Brimfield AA, Miller M, Finkelman FD, Chu SF. (1985) Preparation and characterization of monoclonal antibodies to the trichothecene mycotoxin T-2. Appl Environ Microbiol 49:168–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter KW, Jr, Lenz DE. (1982) Detection and quantification of the organophosphate insecticide paraoxon by competitive inhibition enzyme immunoassay. Life Sci 30:355–361 [DOI] [PubMed] [Google Scholar]
- Hunter KW, Jr, Lenz DE, Brimfield AA, Naylor JA. (1982) Quantification of the organophosphorus nerve agent soman by competitive inhibition enzyme immunoassay using monoclonal antibody. FEBS Lett 149:147–151 [DOI] [PubMed] [Google Scholar]
- Johnson JK, Cerasoli DM, Lenz DE. (2005) Role of immunogen design in induction of soman-specific monoclonal antibodies. Immunol Lett 96:121–127 [DOI] [PubMed] [Google Scholar]
- Jović RC. (1974) Correlation between signs of toxicity and some biochemical changes in rats poisoned by soman. Eur J Pharmacol 25:159–164 [DOI] [PubMed] [Google Scholar]
- Kalisiak J, Ralph EC, Cashman JR. (2012) Nonquaternary reactivators for organophosphate-inhibited cholinesterases. J Med Chem 55:465–474 [DOI] [PubMed] [Google Scholar]
- Kalisiak J, Ralph EC, Zhang J, Cashman JR. (2011) Amidine-oximes: Reactivators for organophosphate exposure. J Med Chem 54:3319–3330 [DOI] [PubMed] [Google Scholar]
- Kwong TC. (2002) Organophosphate pesticides: Biochemistry and clinical toxicology. Ther Drug Monit 24:144–149 [DOI] [PubMed] [Google Scholar]
- Lenz DE, Yourick JJ, Dawson JS, Scott J. (1992) Monoclonal antibodies against soman: Characterization of soman stereoisomers. Immunol Lett 31:131–135 [DOI] [PubMed] [Google Scholar]
- Li B, Nachon F, Froment MT, Verdier L, Debouzy JC, Brasme B, Gillon E, Schopfer LM, Lockridge O, Masson P. (2008) Binding and hydrolysis of soman by human serum albumin. Chem Res Toxicol 21:421–431 [DOI] [PubMed] [Google Scholar]
- Li B, Ricordel I, Schopfer LM, Baud F, Mégarbane B, Nachon F, Masson P and Lockridge O (2010) Detection of adduct on tyrosine 411 of albumin in humans poisoned by dichlorvos. Toxicol Sci 116:23–31. [DOI] [PMC free article] [PubMed]
- Li B, Schopfer LM, Hinrichs SH, Masson P, Lockridge O. (2007) Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay for organophosphorus toxicants bound to human albumin at Tyr411. Anal Biochem 361:263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockridge O, Schopfer LM. (2010) Review of tyrosine and lysine as new motifs for organophosphate binding to proteins that have no active site serine. Chem Biol Interact 187:344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M, Lanier M, Cashman J. (2010) Solid-phase synthesis of phosphonylated peptides. Synlett 2010:1951–1954 [Google Scholar]
- Marrs TC, Maynard RL, and Sidell FR, editors (2007) Chemical warfare agents: Toxicology and treatment, 2nd ed, John Wiley & Sons, Chichester, West Sussex, England. [Google Scholar]
- Marsillach J, Richter RJ, Kim JH, Stevens RC, MacCoss MJ, Tomazela D, Suzuki SM, Schopfer LM, Lockridge O, Furlong CE. (2011) Biomarkers of organophosphorus (OP) exposures in humans. Neurotoxicology 32:656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson P, Lockridge O. (2010) Butyrylcholinesterase for protection from organophosphorus poisons: Catalytic complexities and hysteretic behavior. Arch Biochem Biophys 494:107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray JS, Coleman DR, 4th, Wang W, Campbell ML. (2001) The synthesis of phosphopeptides. Biopolymers 60:3–31 [DOI] [PubMed] [Google Scholar]
- Peeples ES, Schopfer LM, Duysen EG, Spaulding R, Voelker T, Thompson CM and Lockridge O (2005) Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry. Toxicol Sci 83:303–312. [DOI] [PubMed]
- Read RW, Riches JR, Stevens JA, Stubbs SJ, Black RM. (2010) Biomarkers of organophosphorus nerve agent exposure: Comparison of phosphylated butyrylcholinesterase and phosphylated albumin after oxime therapy. Arch Toxicol 84:25–36 [DOI] [PubMed] [Google Scholar]
- Schopfer LM, Champion MM, Tamblyn N, Thompson CM, Lockridge O. (2005) Characteristic mass spectral fragments of the organophosphorus agent FP-biotin and FP-biotinylated peptides from trypsin and bovine albumin (Tyr410). Anal Biochem 345:122–132 [DOI] [PubMed] [Google Scholar]
- Sogorb MA, Vilanova E. (2002) Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol Lett 128:215–228 [DOI] [PubMed] [Google Scholar]
- Thompson CM, Prins JM, George KM. (2010) Mass spectrometric analyses of organophosphate insecticide oxon protein adducts. Environ Health Perspect 118:11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieseler S, Schopfer L, Lockridge O. (2006) Markers of organophosphate exposure in human serum. J Mol Neurosci 30:93–94 [DOI] [PubMed] [Google Scholar]
- Worek F, Koller M, Thiermann H, Szinicz L. (2005) Diagnostic aspects of organophosphate poisoning. Toxicology 214:182–189 [DOI] [PubMed] [Google Scholar]
- Zheng X, Okolotowicz K, Wang B, Macdonald M, Cashman JR, Zhang J. (2010) Direct detection of the hydrolysis of nerve agent model compounds using a fluorescent probe. Chem Biol Interact 187:330–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.