

Abstract
Smoking is the only modifiable risk factor associated with development, expansion, and rupture of abdominal aortic aneurysm (AAA), a leading cause of death in the human population. However, the causative link between cigarette smoke and AAA remains to be established. Here we report that, like angiotensin II (AngII), acute infusion of nicotine, a major component of cigarette smoke, resulted in significantly increased elastin degradation, enlargement of the aorta, aberrant expression of matrix metalloproteinase 2 (MMP2), and an increased incidence of AAA in ApoE knockout (ApoE−/−) and deficient in both ApoE and AMP-activated kinase (AMPK) α1 subunit (ApoE−/−/AMPKα1−/−) mice. Importantly, genetic deletion of AMPKα2 (ApoE−/−/AMPKα2−/−) markedly reduced the increase of maximal aortic diameter and incidence of AAA caused by nicotine or AngII in vivo. Transplantation of bone marrow cells from ApoE−/− mice into ApoE−/−/AMPKα2−/− mice or vice versa did not alter nicotine/AngII-induced AAA formation. Mechanistically, we found that both nicotine and AngII activated AMPK in cultured vascular smooth muscle cells (VSMC) and increased the nuclear co-localization of AMPKα2 and AP-2α, a key transcriptional factor essential for MMP2 expression. Biochemical and biological analysis revealed that AMPKα2 directly phosphorylated AP-2α at serine 219 and this phosphorylation increased AP-2α-dependent MMP2 gene expression in VSMC. We conclude that nicotine increases the incidence of AAA in vivo through activation of AMPKα2 and AP-2α. Moreover, our results provide the first demonstration of a causative link between nicotine and AAA in vivo.
INTRODUCTION
Abdominal aortic aneurysm (AAA), characterized by permanent, localized dilatation (ballooning) of the abdominal aorta exceeding the normal diameter by more than 50%, is the most common form of aortic aneurysm. AAA is found in up to 8% of men aged >65 years, but usually remain asymptomatic until they rupture. Rupture of an AAA and its associated catastrophic physiological insult carries an overall mortality rate in excess of 80%; ruptured AAAs are the 13th leading cause of death in the United States1. The exact cause of AAA is unknown. Among all identified risk factors, smoking, which increases the risk of AAA 7.6-fold, is the only modifiable factor associated with the development, expansion, and rupture of AAA. To date, however, no causative link has been proven between smoking and AAA formation.
Pathologically, AAAs are characterized by dilatation of all layers of the arterial wall as a result of loss of elastin, smooth muscle cell apoptosis, and compensatory collagen deposition2–4. Inflammation and matrix degradation in the vasculature are crucial for AAA formation. Reactive oxygen species (ROS) and reactive nitrogen species are reported to be a common link between inflammation, matrix degradation, and AAA pathogenesis. For example, angiotensin II (AngII) increases NAD(P)H oxidase-derived ROS5, activates matrix metalloproteinase (MMPs) protein and mRNA expressions6, and induces AAA formation in animal experiments via inflammation7. Although increased ROS in AAA is well documented, the molecular mechanisms by which ROS induce AAA remain poorly characterized.
AMPK is a serine/threonine kinase consisting of α, β, and γ subunits. The α subunit (isoforms α1 and α2) contains the catalytic activity, while the β and γ regulatory subunits maintain the stability of the heterotrimer complex. As indicated by its name, AMPK is activated by increased intracellular AMP; thus, AMPK is considered to be an energy sensor in the regulation of whole body energy homeostasis and dysfunctional AMPK has been reported in multiple diseases, including obesity, diabetes, and cardiovascular dysfunction. Recently, Birnbaum MJ’s group8,9 and ours10,11 have found that AMPK can be activated by oxidants or hypoxia including hydrogen peroxide and peroxynitrite, which activate AMPK via LKB1-dependent or -independent pathways11,12. Accordingly, oxidant-mediated AMPK activation in vascular smooth muscle cells (VSMCs) inhibits cell proliferation and restenosis induced by AngII or wire injury12,13. However, whether AMPK is involved in AAA formation remains unknown. The aim of the present study was to determine the roles and underlying molecular mechanisms of AMPK in smoking-enhanced AAA formation. We report that nicotine, a major component of cigarette smoke, activates AMPK in VSMCs and that AMPKα2 phosphorylates AP-2α at serine 219 resulting in aberrant expression of MMP2 and consequent AAA formation.
RESULTS
Deletion of AMPKα2 suppresses nicotine-induced AAA formation in vivo
Nicotine is a major component of cigarette smoke and is rapidly absorbed into the bloodstream, with blood concentrations ranging from 40–100 ng/ml after each cigarette14. In regular smokers, nicotine is present in the blood at 90–100 nM over the first 6 to 8 h of the day15. Although there is overwhelming epidemiological evidence linking smoking and AAA16, there is no evidence that smoking or nicotine causes AAA formation in humans or in animals. We firstly investigated whether nicotine infusion (1, 5 mg/kg/day, 6 weeks) increased the incidence of AAA in ApoE−/−, ApoE−/−/AMPKα1−/− or ApoE−/−/AMPKα2−/− mice. None of the animals died during this time, and nicotine infusion did not affect hemodynamic parameters (heart rate, systolic blood pressure, and diastolic blood pressures) or metabolic indexes (serum cholesterol, triglyceride, and glucose levels) in mice (Supplementary Tab. 1,2, online).
All ApoE−/−, ApoE−/−/AMPKα1−/− or ApoE−/−/AMPKα2−/− mice did not exhibit AAA when infused with vehicle (Fig. 1a–d). However, both low-dose and high-dose of nicotine infusions for 6 weeks significantly increased the incidence of AAA, the maximal aortic diameter, and total aortic weight in ApoE−/− and ApoE−/−/AMPKα1−/− mice (Fig. 1a–d and Supplementary Fig. 1a–c, Online). Compared with mice infused with vehicle, nicotine markedly increased the size of the aortic lumen and the wall thickness of ApoE−/− and ApoE−/−/AMPKα1−/− mice, but not ApoE−/−/AMPKα2−/− mice (Fig. 1e). As expected, the elastic lamina was typically disrupted and degraded in nicotine-infused ApoE−/− and ApoE−/−/AMPKα1−/− mice.
Figure 1.

AMPKα2 deficiency prevents nicotine-induced AAA formation in ApoE−/− mice. ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice were infused with nicotine (1 mg/kg/day) for 6 weeks using an osmotic pump. N=10–15 in each group. (a) Representative photographs showing macroscopic features of aneurysms induced by nicotine. The arrow indicates a typical AAA. (b) The incidence of nicotine-induced AAA, (c) Maximal abdominal aortic diameter, and (d) Total aortic weights in ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice after nicotine infusion. (e) Representative staining with HE, elastin, and α-actin in suprarenal aortas of ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. (f) Grade of elastin degradation in aorta wall. *P<0.05 vs. vehicle-infused ApoE−/− mice. #P<0.05 vs. vehicle-infused ApoE−/−/AMPKα1−/− mice. $P<0.05 vs. nicotine-infused ApoE−/− mice.
Deletion of AMPKα2 markedly reduced the incidence of nicotine-induced AAA, compared to nicotine-infused ApoE−/− mice (AAA incidence: 20% for ApoE−/− vs. 0% for ApoE−/−/AMPKα2−/−, P<0.05, Fig. 1b). The maximal aortic diameter and total aortic weight in nicotine-infused ApoE−/−/AMPKα2−/− mice was also significantly smaller than that in nicotine-infused ApoE−/− mice (Fig. 1c,d), and nicotine infusion failed to cause significant elastic lamina degradation and aortic expansion in ApoE−/−/AMPKα2−/− mice (Fig. 1e,f). Positive staining for α-actin indicated that VSMCs were the major cellular component in the area of the AAA (Fig. 1e). Overall, these results suggest that deletion of AMPKα2 protects against nicotine-enhanced AAA formation in ApoE−/− mice in vivo.
AMPKα2 deficiency attenuates AngII-induced AAA formation in vivo
AAA induced by AngII infusion is a well-characterized mouse model17,18. To further validate the role of AMPKα2 in AAA, we examined the effects of 4-week AngII infusion in ApoE−/−, ApoE−/−/AMPKα1−/−, and ApoE−/−/AMPKα2−/− mice. As shown in Online Supplementary Tab. 3,4, AngII infusion had no effects on heart rate, serum cholesterol, triglyceride, and glucose levels. AngII infusion markedly increased both systolic and diastolic blood pressure in ApoE−/−, ApoE−/−/AMPKα1−/−, and ApoE−/−/AMPKα2−/− mice. In saline-infused mice, there was no difference in the gross morphology of aortas among control ApoE−/−, ApoE−/−/AMPKα1−/−, and ApoE−/−/AMPKα2−/− mice (Supplementary Fig. 2a, Online).
Consistent with previous reports2,7,19, the incidence of AAA induced by AngII infusion was 85% and 80% in ApoE−/− mice and ApoE−/−/AMPKα1−/− mice (Supplementary Fig. 2a,b). Concomitantly, both the maximal abdominal aortic diameter (Supplementary Fig. 2c) and total aortic weight (Supplementary Fig. 2d) were markedly increased in AngII-treated ApoE−/− mice and ApoE−/−/AMPKα1−/− mice, similar to Daugherty’s reports which AngII induced AAA formation as well as increased maximal abdominal aortic diameter in ApoE−/− or LDLr−/− mice mice20–22. Histological analysis showed that ApoE−/− and ApoE−/−/AMPKα1−/− mice infused with AngII exhibited the increased size of the aortic lumen and wall thickness (Supplementary Fig. 2e) and the elastic lamina was frequently disrupted and degraded (Supplementary Fig 2f).
In contrast, only 17% of the AngII-infused ApoE−/−/AMPKα2−/− mice developed AAA (Supplementary Fig. 2b). The maximal abdominal aortic diameter (Supplementary Fig. 2c) and total aortic weight (Supplementary Fig. 2d) were markedly reduced in AngII-treated ApoE−/−/AMPKα2−/− mice, compared to AngII-treated ApoE−/− mice or ApoE−/−/AMPKα1−/− mice. The aortas of AngII-infused ApoE−/−/AMPKα2−/− mice showed no changes in aortic wall thickness/aortic expansion or elastic lamina degradation (Supplementary Fig. 2e,f). These results suggest that deletion of AMPKα2 reduces AngII-induced AAA formation in vivo.
AMPKα2 deficiency abolishes nicotine- or AngII-induced MMP2 upregulation in vivo
MMPs are important in the initiation and progression of AAA23 because they degrade the extracellular matrix (ECM), thus decreasing the resistance of the vessel wall to blood flow24. In particular, VSMC-derived MMP2 and macrophage-derived MMP9 are key enzymes in AAA formation25,26. Thus, we next determined the relative contributions of MMP2 and MMP9 in nicotine-enhanced AAA formation. The expression of MMP2 was significantly increased in nicotine-infused ApoE−/− and ApoE−/−/AMPKα1−/− mice (Fig. 2a). The expression of MMP9 protein was also increased by nicotine, but to a lesser extent than that of MMP2. In contrast, ApoE−/−/AMPKα2−/− mice displayed reduced MMP2 protein expression after nicotine infusion.
Figure 2.

Nicotine infusion results in oxidative stress and enhances MMP2/9 expressions via AMPKα2 in vivo. ApoE−/, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice were infused with nicotine (1 mg/kg/day) for 6 weeks. N is 10–15 in each group. (a) Representative stainings of MMP2, MMP9, MDA, and 3-NT in suprarenal aortas in nicotine-infused mice. (b, c and d) MMPs activity, protein expressions, and mRNA levels assayed by Zymagraphy, western blot and RT-PCR in mice aortas. (e) ROS productions detected by DHE/HPLC in mice aortas. P<0.05 vs. control ApoE−/− mice. *P<0.05 vs. vehicle-infused ApoE−/− mice. #P<0.05 vs. vehicle-infused ApoE−/−/AMPKα1−/− mice. $P<0.05 vs. nicotine-infused ApoE−/− mice.
We next assayed MMP activity, mRNA and protein levels in isolated aortas from ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. Both MMP2 and MMP9 activities were detectable, although MMP2 appeared to be the major MMP in vehicle- or nicotine-treated ApoE−/− and ApoE−/−/AMPKα2−/− mice (Fig. 2b). Nicotine infusion increased the activity of MMP2 markedly in ApoE−/− and ApoE−/−/AMPKα1−/− mice and to a lesser extent in ApoE−/−/AMPKα2−/− mice, but appeared to have minimal effects on MMP9 in ApoE−/−, ApoE−/−/AMPKα1−/− or ApoE−/−/AMPKα2−/− mice. In addition, nicotine infusion significantly increased MMP2 protein and mRNA levels in ApoE−/−, ApoE−/−/AMPKα1−/− or ApoE−/−/AMPKα2−/− mice (Fig. 2c,d and Supplementary Fig. 3a, Online). Similarly, AngII infusion markedly increased the protein and mRNA expressions, and activity (Supplementary Fig. 4a,b, Online) of both MMP2 and MMP9 in ApoE−/− and ApoE−/−/AMPKα1−/− mice but not in ApoE−/−/AMPKα2−/− mice.
Vascular AMPKα2-deficiency is essential for AAA Inhibition
Both VSMC-derived MMP2 and macrophage-derived MMP-9 use elastin as their substrate. Because AMPKα1, rather than α2, is the predominant isoform in macrophages and deletion of AMPKα1 did not alter Nicotine/AngII-induced AAA formation, we hypothesized that AMPKα2 in VSMCs plays a major role in AAA formation. To test this, we transplanted ApoE−/−/AMPKα2−/− bone marrow (BM) cells into irradiated ApoE−/− mice or ApoE−/−/AMPKα2+/+ BM cells into irradiated ApoE−/−/AMPKα2−/− mice. After 42 days of engraftment, flow cytometric analysis indicated that >90% of circulating cells in recipient mice were donor-derived (Data not shown). We then treated the mice with nicotine (1 mg/kg/day, 42 days). As expected, formation of AAA was observed in ApoE−/− mice transplanted with either AMPKα2−/− or AMPKα2+/+ BM cells, but not in ApoE−/−/AMPKα2−/− mice transplanted with either AMPKα2−/− or AMPKα2+/+ BM cells (Fig. 3a). The incidence of AAA was identical in ApoE−/− mice transplanted with AMPKα2−/− or AMPKα2+/+ BM cells (appropriately 20%). Importantly, no AAA (AAA incidence: 0%) was seen in ApoE−/−/AMPKα2−/− mice transplanted with either AMPKα2−/− or AMPKα2+/+ BM cells (Fig. 3a) and there was no difference in the maximum abdominal aortic diameter (Fig. 3b) and total aortic weight (Fig. 3c). Similar to nicotine, AngII infusion caused AAA formation, increased AAA incidence (Fig. 3d), maximum abdominal aortic diameter (Fig. 3e), and total aortic weight (Fig. 3f) in ApoE−/− mice transplanted with either AMPKα2−/− or AMPKα2+/+ BM cells, but not in ApoE−/−/AMPKα2−/− mice transplanted with either AMPKα2−/− or AMPKα2+/+ BM cells (Fig. 3d–f). Taken together, these data suggest that loss of AMPKα2 expression in vascular cells, rather than BM-derived cells, is crucial for the development of AAA.
Figure 3.

Bone marrow (BM) reconstitution shows a key role for vascular-specific AMPKα2 deficiency in AAA formation. After irradiation, ApoE−/− and ApoE−/−/AMPKα2−/− mice were immediately injected with BM-derived cells from ApoE−/− and ApoE−/−/AMPKα2−/− mice. (a–c) 6 weeks later, mice were infused with nicotine (1 mg/kg/day) for 6 weeks. (a) The incidence of AAA, (b) Maximal abdominal aortic diameters, and (c) Total aortic weights in nicotine-infused mice. N=10–15 in each group. *P<0.05 vs. nicotine-infused ApoE−/− plus ApoE−/−AMPKα2+/+ BM-derived cells. NS indicates no significant difference. (d–f) 1 week later, mice were infused with AngII (1.44 mg/kg/day) for 4 weeks by using an osmotic pump. (d) The incidence of AAA, (e) Maximal abdominal aortic diameters, and (f) Total aortic weights in AngII-infused mice. N=10–15 in each group. *P<0.05 vs. AngII-infused ApoE−/− plus ApoE−/−AMPKα2+/+ BM-derived cells. NS indicates no significant difference.
MMP2 is crucial for nicotine/AngII-induced elastin degradation
In order to further support the key role of VSMC-derived MMP2 in AAA formation, we generated acute MMP2-deficient mice by in vivo siRNA delivery. As depicted in Online Supplementary Fig. 5a, MMP2 siRNA significantly silenced MMP2 protein expression in mice aortas and both AngII and nicotine enhanced Pro-MMP2 protein levels in control siRNA-transfected mice. The content of collagen IV, a direct substrate of MMP2, was reduced by AngII or nicotine in control siRNA mice but not in MMP2 siRNA mice (Supplementary Fig. 5a,b). Accordingly, MMP2 siRNA but not control siRNA blocked AngII or nicotine-induced elastin degradation (Supplementary Fig. 5b,c), indicating MMP2, rather than MMP9, mediates AngII/nicotine-induced elastin degradation.
Infusion with nicotine or AngII increases ROS in vivo
Increased ROS and the associated vascular inflammation are considered as key mediators of AngII-induced AAA formation7. We first measured the levels of malondialdehyde (MDA) and 3-nitrotyrosine (3-NT), two markers of oxidative stress27, in ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. As shown in Fig. 2a and Supplementary Fig. 4a, infusion with nicotine or AngII significantly increased the levels of MDA and 3-NT in ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. Serum levels of cytokines (IL-6, TNFα, IFNγ, and CypA) were also elevated in AngII/nicotine-infused mice (Supplementary Tab. 5 and 6, Online). The increased ROS levels were further confirmed by assaying superoxide with dihydroethidium (DHE) by HPLC. As depicted in Fig. 2e and Supplementary Fig. 4c, either nicotine or AngII increased DHE fluorescence in ApoE−/−, ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. These data suggest that both AngII and nicotine trigger oxidative stress in vivo and ROS may be the upstream activator of AMPK.
Activation of AMPK by nicotine or AngII increases the expression of pro-MMP2 in VSMCs
Since VSMCs are the major cell type in AngII- and nicotine-triggered AAA in mice and AMPK expression in VSMCs appears to be essential in MMP2 expression, it seemed highly likely that nicotine and AngII would activate AMPK and that this activation would modulate MMP2 expression and activity in VSMCs. Indeed, AMPK was activated by exposure of cells to low concentrations (0.01–0.1 μM) of nicotine that are equivalent to the blood concentrations in habitual smokers (Supplementary Fig. 6a,b, Online). The degree of AMPK activation was similar to that caused by the potent AMPK activators 5-amino-4-imidazole carboxamide riboside (AICAR, 2 mM) or peroxynitrite (50 μM). Nicotine also increased phosphorylation of AMPK Thr172 in a time-dependent manner (Supplementary Fig. 6c). Likewise, AngII also activated AMPK and its substrate ACC in VSMCs in a time-dependent manner (Supplementary Fig. 6d,e), consistent with previously published data12. Treatment with AngII or nicotine did not alter the total level of AMPKα, suggesting that changes in AMPK Thr172 phosphorylation were not due to altered expression of the protein.
To further establish the role of AMPKα2 in MMP2 expression in the vascular wall, we tested the effects of nicotine or AngII on the protein and mRNA expressions and activity of MMP2 in cultured human VSMCs. As indicated in Supplementary Fig. 3c,d and Supplementary Fig. 7a,b, exposure of VSMCs to AngII or nicotine markedly increased MMP2 mRNA and protein expressions in whole cell lysates and the activity of MMP2 in cultured medium. Compound C, a known AMPK inhibitor, abolished the effects of AngII or nicotine on MMP2, indicating that AMPK mediates MMP2 expression induced by AngII or nicotine.
AMPKα2, but not AMPKα1, mediates upregulation of MMP2 by AngII in VSMCs
The observation that deletion of AMPKα2, but not AMPKα1, provided protection from Nicotine-induced AAA formation suggests that only AMPKα2 regulates MMP2 expression (Fig. 1a–c). To further investigate AMPK isoform-specific MMP2 expression, we silenced AMPKα1 or AMPKα2 in VSMC by transfection with AMPKα1-specfic or AMPKα2-specific siRNAs. As depicted in Fig. 4a,c and Supplementary Fig. 3e,g, transfection with AMPKα2 siRNA, but not AMPKα1 siRNA, abolished nicotine, metformin, AngII, or AICAR-enhanced MMP2 protein and mRNA expressions and activity in cultured medium of human VSMCs.
Figure 4.
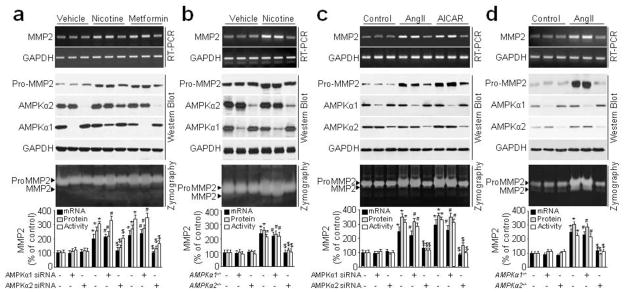
Inhibition of AMPKα2, but not AMPKα1, abolished nicotine/AngII-induced pro-MMP2 mRNA, protein expression, and MMP2 activity in VSMCs. (a) Cultured human VSMCs were transfected with control siRNA, AMPKα1 siRNA, or AMPKα2 siRNA for 48 h and then incubated with nicotine (0.1 μM) or metformin (2 mM) for 24 h. pro-MMP2 protein and MMP2 mRNA level in cell lysates, and MMP2 activity in culture medium were detected by Western blot, RT-PCR, and zymography, respectively. N=3. *P<0.05 vs. control siRNA. #P<0.05 vs. AMPKα1 siRNA. $P<0.05 vs. control siRNA plus nicotine or metformin. (b) Cultured mouse VSMCs (WT, AMPKα1−/−, and AMPKα2−/−) from the descending aorta were incubated with nicotine (0.1 μM) for 24 h. pro-MMP2 protein and mRNA levels in cell lysates and MMP2 activity in culture medium were assayed. N=3. *P<0.05 vs. WT. #P<0.05 vs. AMPKα1−/−. $P<0.05 vs. WT plus nicotine. (c) Cultured human VSMCs were transfected with control siRNA, AMPKα1 siRNA, or AMPKα2 siRNA for 48 h and then incubated with AngII (1 μM) or AICAR (2 mM) for 24 h. pro-MMP2 protein and MMP2 mRNA level in cell lysates, and MMP2 activity in culture medium were detected by Western blot, RT-PCR, and zymography, respectively. N=3. *P<0.05 vs. control siRNA. #P<0.05 vs. AMPKα1 siRNA. $P<0.05 vs. control siRNA plus AngII or AICAR. (d) Cultured mouse VSMCs (WT, AMPKα1−/−, and AMPKα2−/−) from the descending aorta were incubated with AngII(1 μM) for 24 h. pro-MMP2 protein and mRNA levels in cell lysates and MMP2 activity in culture medium were assayed. N=3. *P<0.05 vs. WT. #P<0.05 vs. AMPKα1−/−. $P<0.05 vs. WT plus AngII.
We next performed these experiments in VSMCs isolated from WT, AMPKα1−/−, or AMPKα2−/− mice. As expected, AngII up-regulated MMP2 levels in AMPKα2−/− VSMCs, but not in WT and AMPKα1−/− cells (Fig. 4b,d and Supplementary Fig. 3f,h). These data are in agreement with those obtained with AMPKα2-specific siRNA and ApoE−/−/AMPKα2−/− mice. Overall, our results support the notion that AMPKα2, but not AMPKα1, is required for MMP2 expression in VSMCs.
Upregulation of MMP2 by nicotine or AngII is AP-2α-dependent
Activator protein 2α (AP-2α) is a transcription factor that is required for MMP2 expression in endothelial cells28. AP-2α−/− mice die after birth due to abnormal vascular development, suggesting an important role of AP-2α in regulating vascular angiogenesis29,30. Currently, there is no evidence that AP-2α can regulate MMP2 gene expression in VSMCs. We hypothesized that nicotine- or AngII-induced MMP2 upregulation occurs through AP-2α-dependent gene transcription and expression of MMP2. Indeed, transfection of VSMCs with AP-2α-specific or AMPKα2-specific siRNAs, but not control siRNA, inhibited nicotine-induced pro-MMP2 protein expression and MMP2 activity (Fig. 5a), implying that the upregulation of MMP2 by nicotine in VSMCs requires AP-2α. Similarly transfection of VSMCs with AP-2α siRNA, but not control siRNA, also abolished AngII-induced MMP2 upregulation (Fig. 5b).
Figure 5.
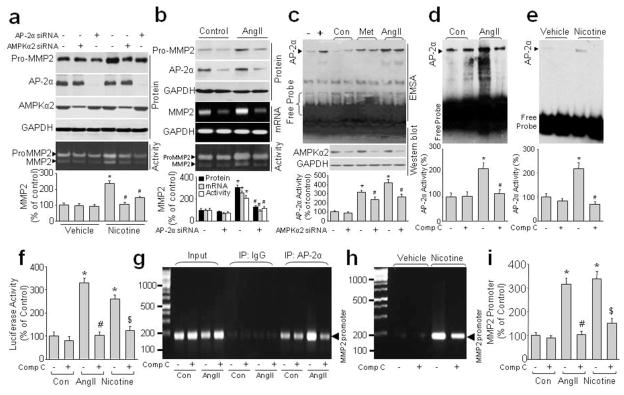
AP-2α mediates nicotine/AngII-induced AMPKα2-dependent MMP2 protein expression in VSMCs. (a) Cultured human VSMCs were transfected with control siRNA, AMPKα2 siRNA, or AP-2α siRNA for 48 h and then incubated with nicotine (0.1 μM) for 24 h. MMP2 activity in culture medium and pro-MMP2 protein expression in cell lysates were detected by Zymography and western blot, respectively. Representative results from three independent experiments are shown. *P<0.05 vs. Vehicle, #P<0.05 vs. Nicotine alone. (b) Cultured human VSMCs were transfected with control siRNA and AP-2α siRNA for 48 h and then incubated with AngII (1 μM) for 24 h. pro-MMP2 and AP-2α protein expression in cell lysates, mRNA level in cells, and MMP2 activity in culture medium were detected by western blot, RT-PCR, and zymography, respectively. N=3. *P<0.05 vs. control siRNA, #P<0.05 vs. control siRNA plus AngII. (c) Cultured human VSMCs were transfected with control siRNA or AMPKα2 siRNA for 48 h and then incubated with AngII (1 μM) or metformin (2 mM) for 2 h. Nuclear extract was subjected to EMSA to measure AP-2α DNA affinity. Negative control (−) consists of biotin-labeled DNA probe without extract. Positive control (+) consists of biotin-labeled DNA probe with nuclear extract provided by the kit. N=3. *P<0.05 vs. control siRNA, #P<0.05 vs. control siRNA plus metformin or AngII. (d–h) Cultured human VSMCs were pre-incubated with or without compound C (10 μM) for 30 min and then treated with nicotine (0.1 μM) or AngII (1 μM) for 2 h. AP-2α activity was detected by EMSA (d, e) or luciferase reporter gene (f) and binding of AP-2α to the MMP2 gene promoter were detected by ChIP (g, h), respectively. (d, e) N=3, *P<0.05 vs. control or vehicle, #P<0.05 vs. AngII or Nicotine. (f) N is 3. *P<0.05 vs. control, #P<0.05 vs. AngII alone, $P<0.05 vs. Nicotine alone. For ChIP experiments, the complex of chromatin/protein was pulled down by AP-2α primary antibody and the MMP2 promoter was amplified by PCR. Positive control, 10% of the total chromatin in the absence of immunoprecipitation (lane 1–4, input). Negative control, chromatin immunoprecipitated with IgG and amplified with MMP2 promoter primers (IgG, lane 5–8). The expected PCR product is 200 bp. Representative results from three independent experiments are shown. (i) Quantifications of ChIP. N is 3. *P<0.05 vs. control, #P<0.05 vs. AngII alone, $P<0.05 vs. Nicotine alone.
Nicotine and AngII increase AP-2α-binding to the MMP2 promoter
Since AP-2α is required for nicotine- and AngII-induced MMP2 gene transcription, we reasoned that AngII and nicotine might activate AP-2α to increase its binding affinity for the MMP2 gene promoter. As shown in Fig. 5c–f, an increased DNA shift and activity of AP-2α detected by EMSA and luciferase reporter gene was observed in AngII- or nicotine-stimulated VSMC, suggesting that AngII and nicotine increased AP-2α activity and the affinity to DNA. Inhibition of AMPKα2 with either siRNA or compound C significantly suppressed the AngII- or nicotine-enhanced AP-2α activity and the binding of AP-2α to DNA. Conversely, metformin, a well-characterized AMPK activator, increased AP-2α/DNA binding affinity (Fig. 5c). Finally, binding was inhibited by AMPKα2 siRNA (Fig. 5c). Overall, these data suggest that AMPK mediates nicotine/AngII-induced activation of AP-2α and its binding to DNA.
We next investigated whether AP-2α specially binds to the MMP2 gene promoter in VSMCs using ChIP analysis. In human VSMCs, the MMP2 gene promoter was positively amplified in samples from AngII-treated cells following immunoprecipitation with AP-2α primary antibody but not with control IgG (Fig. 5g), suggesting that the positive amplification of the MMP2 gene promoter is specific to AP-2α. Further, compound C attenuated the AngII-induced binding of AP-2α to the MMP2 gene promoter, implying a requirement for AMPK activation in this binding. Similar effects were observed in nicotine-treated VSMCs (Fig. 5g–i).
Nicotine and AngII promote the co-localization and association of AMPKα2 and AP2α in VSMCs
To characterize the mechanisms by which AMPK participates in the activation of AP-2α by AngII or nicotine and AP-2α is located in the nucleus, we first determined the subcellular location of AMPKα. As depicted in Fig. 6a, the nicotine/AngII-induced nuclear translocation of AMPKα2, but not AMPKα1, was observed by western blot analysis of separated cytoplasmic and nuclear fractions. Treatment of cells with compound C inhibited the nicotine/AngII-promoted AMPKα2 nuclear translocation.
Figure 6.

Nicotine/AngII promotes the binding of AMPKα2 to AP-2α and AMPKα2-dependent AP-2α serine 219 phosphorylation. (a, b) Cultured human VSMCs were incubated with nicotine (0.1 μM) or AngII (1 μM) for 2 h in the presence of compound C (10 μM). (a) Cell fractions were subjected to western blot analysis to determine the subcellular localization of AMPKα1/2 and AP-2α. LDH, cytoplasmic marker; H2AX, nuclear marker. N=3. (b) AMPKα1, AMPKα2, or AP-2α proteins in total cell lysates were pulled down by the appropriate primary antibody and subjected to western blot analysis to detect the binding of AP-2α to AMPKα1/α2. The blot is representative of three independent blots. (c) GST-AMPKα2 and GST-AP-2α protein were co-incubated in reaction buffer for 30′. Mixture was subjected to run native gel or pulled down by primary antibody followed by western blot. 3 independent experiments were performed. (d) Serine phosphorylation of AP-2α was assayed by IP with primary AP-2α antibody followed by western blotting with anti-phospho-serine antibody. The experiment was repeated three times. (e) Cultured HEK293 cells were transfected with wild type AMPKα2 (WT-AMPKα2) or dominant negative (DN-AMPKα2) for 24 h. WT-AMPKα2 and DN-AMPKα2 protein were purified by IP with primary AMPKα2 antibody. AP-2α protein and IP-derived AMPKα2 protein were co-incubated in kinase buffer in the presence of P32-labeled ATP and phosphorylation of AP-2α was visualized by radioautography. N=3. (f) Peptides (SAMS, AP-2α 176-185/214-233/228-237/247-226/315-324) were co-incubated with AMPKα2 protein in reaction buffer in the presence of P32-ATP. Phosphorylation of AP-2α and SAMS peptides was visualized by radioautography. N=3. (g) Peptides (SAMS, SAMS S6A, AP-2α 214–223, AP-2α 214–233 S219A) were co-incubated with AMPKα2 protein in reaction buffer in the presence of P32-ATP. Phosphorylation of AP-2α and SAMS peptides was visualized by radioautography. N=3. (h) HA-tagged WT and site-directed mutants of full-length AP-2α were transfected into HEK293 cells for 24 h and then treated with AICAR (2 mM) for 2 h. Serine phosphorylation of AP-2α was determined by pull-down with anti-HA and western blot analysis with anti-phosphor-serine antibody. N=3.
The interaction of AMPKα2 and AP-2α after nicotine/AngII treatment was confirmed by immunoprecipitation experiments. As indicated in Fig. 6b, binding of AMPKα2/AP-2α was evident in the nuclear cell fraction pulled down by either AP-2α or AMPKα2. However, no binding was detected between AMPKα1 and AP-2α.
The nuclear import and co-localization of AMPKα2 and AP2α were further verified by immunofluorescence experiments (Supplementary Fig. 8, online). The protein-protein interaction of AMPKα2 and AP2α was also further confirmed by in vitro co-incubation and pull-down experiments (Fig. 6c). Together, these data suggest that AMPKα2 directly affects AP-2α through protein/protein interaction.
AMPK increases phosphorylation of AP-2α at serine 219
Based on our findings that AngII triggers nuclear import of AMPKα2 and its co-localization with AP-2α, we proposed a mechanism by which AMPK might directly regulate AP-2α function. Post-translational modifications of AP-2α, including serine phosphorylation, are known to regulate its transcriptional activity31. As AMPK is a Thr/Ser protein kinase, we reasoned that AMPK might directly phosphorylate AP-2α serine(s). To test this, we first measured total serine phosphorylation of AP-2α in VSMCs after AngII/nicotine treatment. As depicted in Fig. 6d, exposure of VSMC to AngII or nicotine increased the serine phosphorylation of AP-2α. Conversely, inhibition of AMPK by compound C dramatically inhibited AngII- or nicotine-induced AP-2α serine phosphorylation, implying that activation of AMPK may increase AP-2α serine phosphorylation.
Direct phosphorylation of AP-2α by AMPKα2 was demonstrated by measuring in vitro AMPK kinase activity (Fig. 6e). Wild type AMPKα2 (WT-AMPKα2), but not dominant negative AMPKα2 (DN-AMPKα2), significantly phosphorylated AP-2α recombinant protein as detected by P32-ATP radio-autography, indicating that AP-2α is a substrate of direct phosphorylation by AMPKα2.
Next, we determined which serine residue(s) in AP-2α protein are phosphorylated by AMPKα2 by comparison with an optimal AMPK substrate motif32. Analysis of AP-2α (Supplementary Fig 9a–c, Online) revealed potential AMPK target motifs at serines 181, 219, 233, 252, and 320 in the regulatory and helix-span-helix domains, that are similar to previously identified AMPK substrates (the SAMS peptide, eNOS, ACC, ULK1, H2B, and TSC2)33–35. When we compared in vitro phosphorylation of peptides corresponding to residues 176–185, 214–223, 228–237, 247–256, or 315–324 of AP-2α with that of SAMS peptide, a canonical AMPK target sequence, only peptide 214–223 of AP-2α was phosphorylated by AMPKα2 protein (Fig. 6f). Furthermore, mutation of serine to alanine at Ser6 of SAMS and Ser219 of AP-2α peptide 214–233 abolished AMPKα2-induced phosphorylation (Fig. 6g).
To further confirm S219 of AP-2α as the phosphorylation target of AMPKα2, we generated five site-directed mutant constructs of AP-2α (Serine to Alanine at S181A, S219A, S233A, S252A, and S320A). In HEK293 cells transfected with WT AP-2α, treatment with AICAR, which phosphorylates AMPK at Thr172, dramatically increased AP-2α serine phosphorylation (Fig. 6h). Among the AP-2α mutant constructs, AICAR-induced AP-2α serine phosphorylation was abolished only in cells transfected with AP-2α S219A, confirming that serine 219 is the target of AMPK-induced AP-2α serine phosphorylation.
Mutation of AP-2α Serine 219 to alanine abolishes AngII- or nicotine-induced upregulation of MMP2 in VSMCs
Since AMPKα2 directly phosphorylates AP-2α serine 219 in cells, we next tested whether serine 219 phosphorylation of AP-2α determines AP-2α activity and function. As shown in Online Supplementary Fig. 10a–c, transfection of human VSMCs with the AP-2α S219A mutant dramatically abrogated AngII/nicotine-enhanced upregulation of MMP2 (mRNA level, protein expression, and activity), AP-2α-DNA binding affinity, and binding of AP-2α to the MMP2 gene promoter. These results indicate that serine 219 phosphorylation of AP-2α by AMPKα2 promotes its binding to the MMP2 promoter resulting in aberrant MMP2 expression in VSMCs.
Increased AMPK α2 activity, AMPK Thr172 phosphorylation, and ACC Ser 172 phosphorylation in human AAAs
To establish human relevance, we further examined AMPKα2 activity, AMPK Thr172 phosphorylation, and Ser79 phosphorylation of acetyl CoA carboxynase (ACC), a well characterized downstream enzyme of AMPK. As shown in Supplemental Figure 11, human AAA samples exhibited higher AMPKα2 activity, AMPK Thr172 phosphorylation, and ACC Ser79 phosphoylation in aortic tissues from AAA than those from non-AAA control patients.
Cigarette smoke increases AMPK α2 activity, AMPK Thr172 phosphorylation, and ACC Ser 172 phosphorylation in humans
Finally, to address if cigarette smokers altered AMPK activity, we assayed AMPKα2 activity, p-AMPK, and p-ACC in the plasma of both active smokers and non-smokers. In acute smokers, AMPKα2 activity is higher than in non-smokers (Supplemental Figure 11). Taken together, our data suggest that smoke-activated AMPK is important in the initiation and progression of AAA.
DISCUSSION
In the present study, we provide the first evidence that nicotine increases AAA in ApoE−/− mice in vivo through activation of AMPKα2 (Supplementary Fig. 12). Mechanistically, we found that nicotine- or AngII-derived ROS trigger nuclear translocation of AMPKα2, which binds and phosphorylates AP-2α at serine 219 resulting in aberrant expression of MMP2 and consequent ECM degradation. This novel mechanism not only provides a causative link between nicotine and AAA, but also explains how pro-inflammatory factors such as ROS promote AAA formation.
One of our most important findings is that activation of AMPKα2 by nicotine and AngII triggers AAA formation in ApoE−/− mice. This conclusion is supported by several observations. First, gene deletion of AMPKα2 suppressed AngII/nicotine-induced MMP2 protein expression in cells or MMP2 activity in culture medium. Inhibition of AMPK with the pharmacological inhibitor compound C reproduced this effect. Second, activation of AMPK by AICAR produced similar effects to those induced by AngII or nicotine. Third, the formation of AAA following infusion with AngII or nicotine was reduced in ApoE−/−/AMPKα2−/− mice compared with ApoE−/− mice. We thus demonstrated that ablating AMPK activity by deleting the AMPK gene has a marked protective effect against aortic aneurismal disease. In fact, a clinical investigation has indicated that diabetes is negatively associated with AAA formation36, and there is evidence that hyperglycemia suppresses AMPK activity37,38 whereas insulin treatment promotes the enlargement of aortic diameter39,40 through the effect of lowering blood glucose. These findings suggest an important role for AMPK in the pathogenesis of AAA.
Another important finding is that AP-2α is a direct substrate for phosphorylation by AMPKα2 at serine 219. AP-2α is also phosphorylated at Ser233 by protein kinase A (PKA)31 and at both Ser252 and Ser320 by protein kinase D (PKD)41, based on the phosphorylation motifs (RRXS/T and LXRXXS/T) in its basic transactivation domain. By comparing the amino acid sequence of AP-2α with known substrates of AMPK, we identified serine 219 as a novel target of AMPKα2. Phosphorylation of S219 by AMPKα2 was demonstrated by the following evidence: (1) AP-2α is phosphorylated only in the presence of AMPKα2 in a cell-free system; (2) AMPKα2 specifically phosphorylated an AP-2α peptide containing S219; (3) Mutation of serine 219 to alanine abolished AMPK-induced AP-2α serine phosphorylation, increased DNA binding affinity, and MMP2 levels. In fact, AP-2α is very important for mouse embryonic development and mice lacking AP-2 die at or before birth as a result of severe defects29. Combined with our observations, we think defects of cardiovascular development by loss of MMP2 in AP-2a deletion mice may be one of factors to lethal. Our data also indicate that AP-2a may have a broader role in regulating genes critical to normal embryogenesis.
Our studies are in line with earlier reports8,34,35 that oxidants-mediated AMPK activation by concentrations of nicotine relevant to smokers in adipocytes as well as AngII in VSMC. In addition, our study has extended these observations by showing that AMPK activation might be detrimental in the initiation and progression of AAA, which contradicts the idea that AMPK activation is beneficial in cardiovascular diseases. For example, AMPKα2 deletion is reported to cause accelerated atherosclerosis36 and pharmacological activation of AMPK has been shown to ablate wire injury-induced vascular remodeling by inhibiting VSMC proliferation8. Although atherosclerosis, restenosis, and AAA are associated with vascular inflammation and have many of the same predisposing risk factors, there is an important difference between the pathways mediating atherogenesis and AAA formation. For example, early atherosclerosis is primarily characterized by pro-inflammatory factor-mediated foam cell accumulation in the intima37. In contrast, AAA involves degradation of the tunica media in which enhanced activation of the proteolytic system, such as MMPs, plays a crucial role38. In addition, reduced VSMC proliferation has also been found within fragmented media in human AAA biopsy specimens. Thus, VSMC proliferation, which leads to acceleration of atherosclerosis and restenoic vascular remodeling, might be paradoxically beneficial for AAA40. Therefore the inhibitory effects of AMPK activation on VSMC proliferation might accentuate the development of AAA. Furthermore, in a recent review paper, Golledge et al41 concluded that a plurality of studies indicate an independent effect of medications on aneurysms and atherosclerosis in animal models, further suggesting distinct pathogenetic mechanisms.
Another interesting issue of this study is the different effects of AngII and nicotine in AAA formation. From our observations, although both AngII and nicotine have a common pathway (ROS/AMPK/AP-2α/MMP2) in VSMC to induced AAA in ApoE−/− mice, the incidence and size of AAA induced by AngII is significantly larger than nicotine (Fig. 1a–c and Supplementary Fig. 2a–c). One might expect that the lower incidence of AAA in nicotine infused mice would correlate with lower ROS generation (Fig. 2e and Supplementary Fig. 4c). In addition, it is reported that AngII has other pathways to induce AAA, such as TGF-beta and other growth factors, which can be transcriptionally activated by MMP223,42. Whether or not these pathways contribute to nicotine-enhanced AAA warrants further investigation.
Smoking increases the risk of AAA 7.6-fold. Moreover, smoking is the only modifiable factor associated with the development, expansion, and rupture of AAA. The results presented in this work have for the first time established a causal effect of nicotine, a major component of cigarettes, on the formation, progression, and rupture of AAA in vivo. This finding is highly important, not only because it has established a causative link between cigarette smoke and AAA, but also because it has further identified nicotine as an active agent in smoke-triggered AAA. The finding that nicotine increases AAA formation might be clinically important. One common approach to the cessation of smoking is nicotine supplementation, such as with gum or patches. The rapid absorption of nicotine can increase plasma nicotine concentrations to as high as 5 × 10−7M within 15 min43, a concentration that, based on our findings, might increase AAA formation. Thus, we suggest that nicotine supplementation might be cautiously reconsidered and that the duration of nicotine supplementation should be minimized in populations with advanced age.
Methods
Animals
Male wild-type (C57BL6) and ApoE−/− mice (12–16 weeks of age; 20–25 g body weight) were obtained from Jackson Laboratories (Bar Harbor, ME). AMPKα1−/− and AMPKα2−/− mice were generated as previously described44. Western blot indicates over 90% of AMPKα1 or α2 is knockdown. AMPKα1−/− and AMPKα2−/− mice previously backcrossed to a C57BL6 background were crossed with ApoE−/− mice of C57BL6 background to generate ApoE−/−/AMPKα1−/− and ApoE−/−/AMPKα2−/− mice. ApoE−/−/AMPKα+/+ mice served as controls. Mice were housed in temperature-controlled cages with a 12-h light-dark cycle and given free access to water and food. The animal protocol was reviewed and approved by the University of Oklahoma Institute Animal Care and Use Committee.
Statistical analyses
Quantitative results are expressed as mean ± SEM. Comparisons of parameters among more than two groups were made by one-way analysis of variance, and comparisons of different parameters between each group were made by a post hoc analysis using the Bonferroni test. Chi-Square test was applied to comparisons of AAA incidence and survival rate. Statistical significance was evaluated with GraphPad Prism4. A value of P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (HL079584, HL074399, HL080499, HL089920, HL096032, HL105157 and HL110448) and research awards from the American Diabetes Association and Warren Endowed Chair in Diabetes Research, University of Oklahoma Health Sciences Center. Dr. M.H. Zou is a recipient of the National Established Investigator Award of the American Heart Association.
Footnotes
Author contributions
S.W. designed and conducted the experiments, analyzed data, and wrote the manuscript. C.Z. and J.Z. did the clinical experiments. B.L. partially performed the pathological experiments. M.Z. and L.X. did bone morrow transplantation. H.Z. and J.L. generated series mutants. B.V. provided the AMPK-deleted mice. M.-H.Z. conceived the project, designed the experiments, and wrote the manuscript.
Additional methods. Detailed methodology is described in the Supplementary Methods available online.
References
- 1.Vorp DA, Vande Geest JP. Biomechanical determinants of abdominal aortic aneurysm rupture. Arterioscler Thromb Vasc Biol. 2005;25:1558–1566. doi: 10.1161/01.ATV.0000174129.77391.55. [DOI] [PubMed] [Google Scholar]
- 2.Satoh K, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah PK. Inflammation, metalloproteinases, and increased proteolysis: an emerging pathophysiological paradigm in aortic aneurysm. Circulation. 1997;96:2115–2117. doi: 10.1161/01.cir.96.7.2115. [DOI] [PubMed] [Google Scholar]
- 4.Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421. [DOI] [PubMed] [Google Scholar]
- 5.Rajagopalan S, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browatzki M, et al. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res. 2005;42:415–423. doi: 10.1159/000087451. [DOI] [PubMed] [Google Scholar]
- 7.Thomas M, et al. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation. 2006;114:404–413. doi: 10.1161/CIRCULATIONAHA.105.607168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 9.Choi SL, et al. The regulation of AMP-activated protein kinase by H(2)O(2) Biochem Biophys Res Commun. 2001;287:92–97. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 10.Han Y, Wang Q, Song P, Zhu Y, Zou MH. Redox regulation of the AMP-activated protein kinase. PLoS One. 2010;5:e15420. doi: 10.1371/journal.pone.0015420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, et al. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagata D, et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004;110:444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 13.Song P, et al. AMPKalpha2 Deletion Exacerbates Neointima Formation by Upregulating Skp2 in Vascular Smooth Muscle Cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- 15.Benowitz NL. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Prog Cardiovasc Dis. 2003;46:91–111. doi: 10.1016/s0033-0620(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 16.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180. [DOI] [PubMed] [Google Scholar]
- 17.Lu H, Rateri DL, Cassis LA, Daugherty A. The role of the renin-angiotensin system in aortic aneurysmal diseases. Curr Hypertens Rep. 2008;10:99–106. doi: 10.1007/s11906-008-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 19.Wang YX, et al. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation. 2005;111:2219–2226. doi: 10.1161/01.CIR.0000163544.17221.BE. [DOI] [PubMed] [Google Scholar]
- 20.Daugherty A, et al. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gavrila D, et al. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1671–1677. doi: 10.1161/01.ATV.0000172631.50972.0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rateri DL, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor−/− mice. Circ Res. 2011;108:574–581. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearce WH, Shively VP. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann N Y Acad Sci. 2006;1085:117–132. doi: 10.1196/annals.1383.025. [DOI] [PubMed] [Google Scholar]
- 24.Aziz F, Kuivaniemi H. Role of matrix metalloproteinase inhibitors in preventing abdominal aortic aneurysm. Ann Vasc Surg. 2007;21:392–401. doi: 10.1016/j.avsg.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longo GM, et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pyo R, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, et al. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106:1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park MJ, et al. Nerve growth factor induces endothelial cell invasion and cord formation by promoting matrix metalloproteinase-2 expression through the phosphatidylinositol 3-kinase/Akt signaling pathway and AP-2 transcription factor. J Biol Chem. 2007;282:30485–30496. doi: 10.1074/jbc.M701081200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, et al. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 1996;381:238–241. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- 30.Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- 31.Garcia MA, Campillos M, Marina A, Valdivieso F, Vazquez J. Transcription factor AP-2 activity is modulated by protein kinase A-mediated phosphorylation. FEBS Lett. 1999;444:27–31. doi: 10.1016/s0014-5793(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 32.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bungard D, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golledge J, et al. Reduced expansion rate of abdominal aortic aneurysms in patients with diabetes may be related to aberrant monocyte-matrix interactions. Eur Heart J. 2008;29:665–672. doi: 10.1093/eurheartj/ehm557. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, et al. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension. 2008;52:484–490. doi: 10.1161/HYPERTENSIONAHA.108.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi HC, et al. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283:20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Miyama N, et al. Hyperglycemia limits experimental aortic aneurysm progression. J Vasc Surg. 2010;52:975–983. doi: 10.1016/j.jvs.2010.05.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dua MM, et al. Hyperglycemia modulates plasminogen activator inhibitor-1 expression and aortic diameter in experimental aortic aneurysm disease. Surgery. 2010;148:429–435. doi: 10.1016/j.surg.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 42.McMahon S, Laprise MH, Dubois CM. Alternative pathway for the role of furin in tumor cell invasion process. Enhanced MMP-2 levels through bioactive TGFbeta. Exp Cell Res. 2003;291:326–339. doi: 10.1016/s0014-4827(03)00407-5. [DOI] [PubMed] [Google Scholar]
- 43.Tundulawessa Y, Yongchaiyud P, Chutrthong W, Tundulawessa K. The bioequivalent and effect of nicotine formulation gum on smoking cessation. J Med Assoc Thai. 2010;93:574–579. [PubMed] [Google Scholar]
- 44.Viollet B, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.