

Abstract
Diallyl disulfide (DADS), a garlic organosulfur compound (OSC), has been researched as a cancer prevention agent; however, the role of DADS in the suppression of cancer initiation in non-neoplastic cells has not been elucidated. To evaluate DADS inhibition of early carcinogenic events, MCF-10A cells were pretreated (PreTx) with DADS followed by the ubiquitous carcinogen benzo(a)pyrene (BaP), or co-treated (CoTx) with DADS and BaP for up to 24 hours. The cells were evaluated for changes in cell viability/proliferation, cell cycle, induction of peroxide formation, and DNA damage. BaP induced a statistically significant increase in cell proliferation at 6 hours, which was attenuated with DADS CoTx. PreTx with 6 and 60 μM of DADS inhibited BaP-induced G2/M arrest by 68 and 78%, respectively. DADS, regardless of concentration or method, inhibited BaP-induced extracellular aqueous peroxide formation within 24 hours. DADS attenuated BaP-induced DNA single strand breaks at all time points through both DADS Pre- and CoTx, with significant inhibition for all treatments sustained after 6 hours. DADS was effective in inhibiting BaP-induced cell proliferation, cell cycle transitions, ROS, and DNA damage in a normal cell line, and thus may inhibit environmentally induced breast cancer initiation.
INTRODUCTION
Diallyl disulfide (DADS), CH2=CH-CH2-S-S-CH2CH=CH2, is an Allium sativum (garlic) organosulfide compound with potential as a natural chemopreventive agent. DADS is derived from allicin, a compound that is formed when the integrity of the garlic membrane is disrupted by cutting, chopping, or mashing [1]. The concentration of DADS in the average garlic bulb ranges from 530 to 610 micrograms per gram of garlic [2], and DADS is found primarily in steam distilled garlic oil preparations, but can also be detected in garlic powder and aged garlic extract [3, 4]. DADS has been shown to have antifungal [5], antibacterial [6], antiviral [7], antioxidant [8], antineoplastic [9, 10], and antimutagenic [11] properties. DADS’ antibacterial properties are theorized to contribute to its effectiveness in protection from Helicobacter pylori infections and gastric cancers [6]. It has been shown to inhibit colon [12, 13, 14], lung [14], renal [12], breast [15], and skin carcinogenesis [14, 16]. DADS inhibited 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary cancer in rats by significantly lowering both the ductal carcinoma incidence and multiplicity of total tumors [17]. DADS has demonstrated anticarcinogenic properties in numerous human cancer cell lines including HepG2 hepatoma [18], HL-60 leukemia [19], non-small cell lung cancer [20], SH-SY5Y neuroblastoma [21], MCF-7 breast cancer [22], as well as Caco-2 and HT-29 colon tumor cells [23, 24]. The role of DADS in preventing carcinogenesis in normal cells has not been fully elucidated so further research is needed to evaluate its capacity for inhibiting early stage cancer initiation in non-neoplastic cells.
Anti-neoplastic foods and compounds may prevent cancer initiation through the suppression of procarcinogen metabolism; inhibition of cell viability through apoptosis in damaged cells; induction of cell cycle arrest to enhance DNA repair mechanisms; pro-oxidant activity in precancerous cells, while selectively displaying antioxidant properties in normal cells; and the inhibition DNA adduct formation which can lead to DNA strand breaks, chromosomal aberrations, and DNA mutations. To evaluate these potential mechanisms of chemoprevention, analysis of normal cells during initial carcinogenic insults will help to unravel the mechanisms of cancer induction and cancer prevention by potentially chemopreventive compounds.
Benzo(a)pyrene (BaP) is a known ubiquitous environmental carcinogen that has been shown to transform cells from a normal to cancerous phenotype, in vitro [25]. BaP is a polycyclic aromatic hydrocarbon (PAH), a class of compounds produced during the incomplete combustion of organic material [26]. BaP has been found in emissions from coal, oil, incinerators and wood burning, asphalt applications, vehicle exhaust, tobacco smoke, and in foods cooked at high temperatures [27]. BaP is considered an occupational, food, and general carcinogen, and is listed as a Group I or known human carcinogen by the International Agency for Research on Cancer [28]. As a lipophilic compound, BaP can accumulate in fatty tissue in the body, specifically in ductal cells of the breast making it a potential candidate to induce breast carcinogenesis [29]. However, BaP requires metabolic activation to display its full carcinogenic potential. Its metabolites and intermediates are primarily responsible for the induction of DNA adducts and strand breaks, formation of mutations, induction of chromosomal aberrations and promotion of tumorgenesis [25, 29, 30].
The focus of this study is to evaluate DADS as an inhibitor of early environmentally-induced neoplastic transformation in a normal cell line. Cultures of a spontaneously immortalized normal human breast epithelial cell line, MCF10-A, were treated with BaP to induce early carcinogenic events in the initiation phase. To inhibit BaP-induced carcinogenic initiation, the cells were also treated with varying doses of DADS as a pretreatment (PreTx) or as a co-treatment (CoTx) with BaP. The cells were assessed for changes in cell viability, cell cycle, extracellular reactive oxygen species (ROS) formation, and DNA damage. The results indicate that DADS can suppress initial BaP-induced carcinogenic initiation events.
MATERIALS AND METHODS
Cell Line, Chemicals and Reagents
MCF-10A cells, a normal human breast epithelial cell line, were obtained from American Type Culture Collection (ATCC, Rockville, Maryland). Phenol red-free DMEM/F-12 media, horse serum, penicillin/streptomycin, antibiotic/antimycotic, epidermal growth factor, human insulin (Novolin R), trypsin-EDTA (10X), Hanks Balanced Salt Solution (HBSS), and Phosphate Buffered Saline (PBS) were all purchased from Invitrogen (Carlsbad, CA). Cholera toxin was obtained from Enzo Life Sciences (Plymouth Meeting, Pennsylvania). The CellTiter 96® AQueous One Solution Cell Proliferation Assay was obtained from Promega (Madison, Wisconsin). Benzo(a)pyrene (BaP), diallyl disulfide (DADS), PeroxiDetect™ Kit, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, Missouri).
Cell Culture
MCF-10A cells cultured in DMEM/F12 media supplemented with cholera toxin (100 ng/ml), epidermal growth factor (20 ng/ml), horse serum (5%), human insulin (10 μg/ml), hydrocortisone (0.5 μg/ml), and penicillin-streptomycin to were grown to 90–100% confluence in a 37°C, 5% CO2 humidified incubator. The supplemented media was changed every 2–3 days, and the cells were sub-cultured every 5–7 days.
Cell Treatments and Harvesting
MCF-10A cells were categorized into two groups, DADS PreTx and DADS CoTx. Cultures in the PreTx group were treated with 6, 60, or 600 μM of DADS for four hours, followed by 1μM of BaP for 3, 6, 12, or 24 hours. The CoTx group was treated with mixtures of 1 μM BaP and 6, 60, or 600 μM of DADS for 3, 6, 12, or 24 hours. Both the DADS and BaP were dissolved in DMSO, and for all experiments a cells only, 0.1% DMSO (vehicle), and 1 μM BaP (positive) controls were also utilized, unless otherwise stated. All treatments were prepared and conducted under low light conditions and incubated at 37°C, in a 5% CO2 humidified incubator. Cells were harvested by trypsinization and the cells suspended in PBS without Mg2+ or Ca2+ and frozen until further use.
MTS Assay for Cell Viability
A total of 20,000 untreated MCF-10A cells were placed into 88 wells of a flat bottom 96-well plate, covered with 100 μl of supplemented serum-free media, and allowed to adhere to the bottom of the wells overnight. The media was then aspirated from each well, followed by the application of 100 μl of supplemented serum-free media prepared with the treatments described above. Each treatment was conducted for eight replicates. Following incubation, the protocol of the CellTiter 96® Aqueous One Solution Reagent (MTS Assay) was followed to determine cell viability [31] using a PowerWave X-340 96-well plate reader (Bio-Tek Instruments, Inc, Winooski, Vermont). The cells only control was used to indicate 100% cell viability, in which all other treatments were compared.
Flow Cytometry
MCF-10A cells were treated as described above for 24 hours only and harvested at ~90% confluence from T-75 cell culture flasks. For cell cycle analysis, cells were fixed with 100% ethanol, and stained with propidium iodide in the presence of RNaseA. The cells were subjected to flow cytometry using the Becton-Dickson FACSCalibur Flow Cytometer (Becton Dickson (BD) Biosciences, Franklin Lakes, NJ), which evaluated 20,000 cells per sample. The samples were analyzed using ModFit software (Verity Software House, Topsham, ME) to separate and quantify the cells according to their current life cycle stage (G1, G2/M or S phase).
Aqueous Peroxide Detection
One hundred microliters of the cell treatments described above were prepared in serum-free media and applied to 10,000 MCF-10A cells in each well of a 96-well flat bottom plate, in triplicate. The extracellular media was analyzed by the protocol of the PeroxiDetect Kit for the determination of aqueous hydroperoxides [32] and analyzed spectrophotometrically at 560nm for nanomoles of hydrogen peroxide based on a standard curve (R2= 0.96).
Single Cell Gel Electrophoresis (Comet Assay)
Cells were treated and harvested as described above, with the addition of a 0.03% H2O2 positive control. The Comet assay was performed following the protocol of Aboyade-Cole [33], in triplicate using Komet 5.5 software (Andor Technology, PLC Belfast Northern Ireland), and Kinetic Imaging (Kinetic Imaging Ltd, Merseyside, United Kingdom).
Statistical Analysis
The data from all experiments were analyzed by one-way analysis of variance (ANOVA) followed by the Bonferroni’s Multiple Comparison Test using GraphPad Prism 5.0 software. The results are displayed as the average values +/− SEM to determine significant differences (P<0.05) between the treatment groups and the DMSO vehicle (*) and BaP only (#) controls, unless otherwise stated.
RESULTS
MTS Assay
To determine cell viability, the Cell Titer 96 AQueous One Solution Cell Proliferation Assay (MTS Assay) was utilized. In the PreTx study, neither BaP nor DADS PreTx induced a statistically significant increase in cell viability, suggesting that the treatments were not toxic to the cells (Figure 1). Although not significantly different, DADS pretreatment and the BaP alone control caused a slight induction of cell proliferation through 24 hours. In the CoTx samples, BaP alone induced a statistically significant increase in cell proliferation, when compared to the vehicle control, which was attenuated when given in combination with all three concentrations of DADS. This increase in cell proliferation was not significantly sustained though there was an increase in the cell viability observed at 12 and 24 hours, which was most effectively attenuated by the higher (60 and 600 μM) concentrations of DADS.
Figure 1.
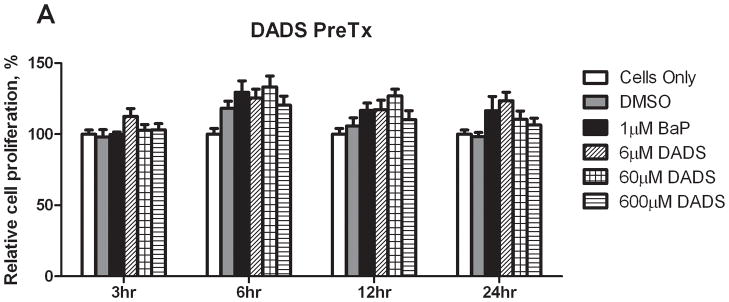
Relative cell proliferation of cells treated with BaP and DADS. MCF-10A cells were pretreated with DADS for four hours followed by the addition of 1 μM BaP (A) or treated concomitantly with 1 μM BaP and DADS (B). To determine cell viability, the MTS solution was applied to the cells for 2–3 hours and analyzed at an absorbance of 490nm. The absorbance of each treatment group was normalized relative to the cells only control for 100% cell viability. The graphs represent the average relative cell proliferation in quadruplicate for N=3, +/− the SEM (* and # indicate a P<0.05 significant difference from the DMSO control and the 1 μM BaP only control, respectively).
Cell Cycle Analysis
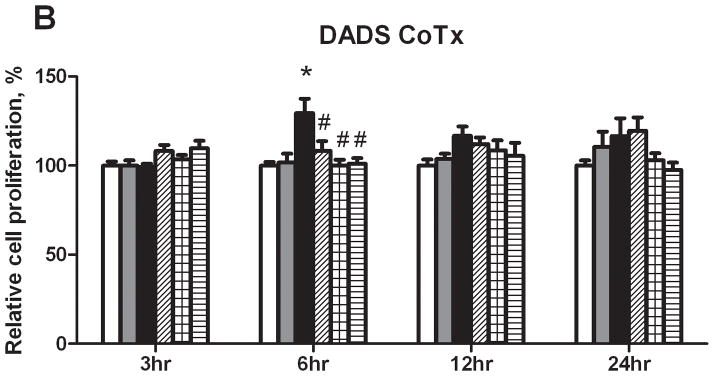
A cell cycle analysis was performed to determine the percentage of individual cells progressing through a complete cycle as the result of a particular exposure. Treatment with BaP induced cellular transition from G1 to the G2/M and S phases, and resulted in an average of 19 and 22% respective increase in cell phase count relative to the PreTx DMSO control. Similar results were seen in the CoTx group (Figure 2). Pretreatment with 6 μM and 60 μM of DADS inhibited BaP-induced G2/M arrest by 68 and 78%, respectively. The BaP-induced S-phase shift was inhibited by 60 μM DADS pretreatment, reducing the cell number in this phase by 105%. At 600 μM, DADS PreTx did not alter G2/M induction, but rather significantly shifted cells from the S phase, inducing G1 arrest. In the CoTx study, BaP alone, and 6, 60 and 600 μM DADS and BaP CoTx induced 87, 135, 184, and 175% respective increases in cells in the G2/M phase, as compared to the DMSO vehicle control. At 600 μM DADS was also effective in significantly reducing cells in S-phase by 64 and 66%, relative to the DMSO and BaP only controls, respectively, and shifted cells into G1 and G2/M phases.
Figure 2.

Results of cell cycle analysis of MCF-10A cells treated with BaP and DADS. MCF-10A cells were either PreTx with DADS for four hours, followed by treatment with 1 μM BaP for 24 hours (A) or CoTx with DADS and BaP for 24 hours (B). The cells were fixed in ethanol, stained with propidium iodide, and analyzed by flow cytometry. The values represent the average percent of cells in each phase, G1, G2/M, and S +/− SEM.
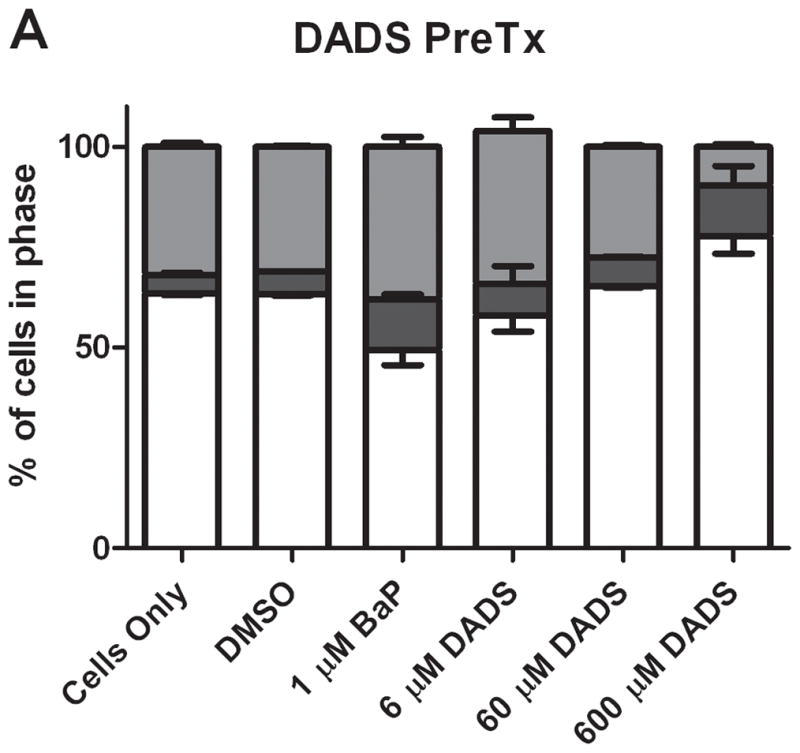
In comparing the ratio of cells in G2/M phase versus G1 phase, the results from the different types of treatments are vastly different. In both groups, BaP caused a noticeable increase in the ratio of cells in G2/M versus G1. DADS (60 μM) pretreatment effectively attenuated this increase; however, CoTx at 6 and 60 μM caused a statistically significant increase in the average ratio, all relative to the DMSO only control (Figure 3). DADS CoTx was ineffective in preventing BaP-induced G2/M phase transitions, and may have enhanced the BaP effect.
Figure 3.
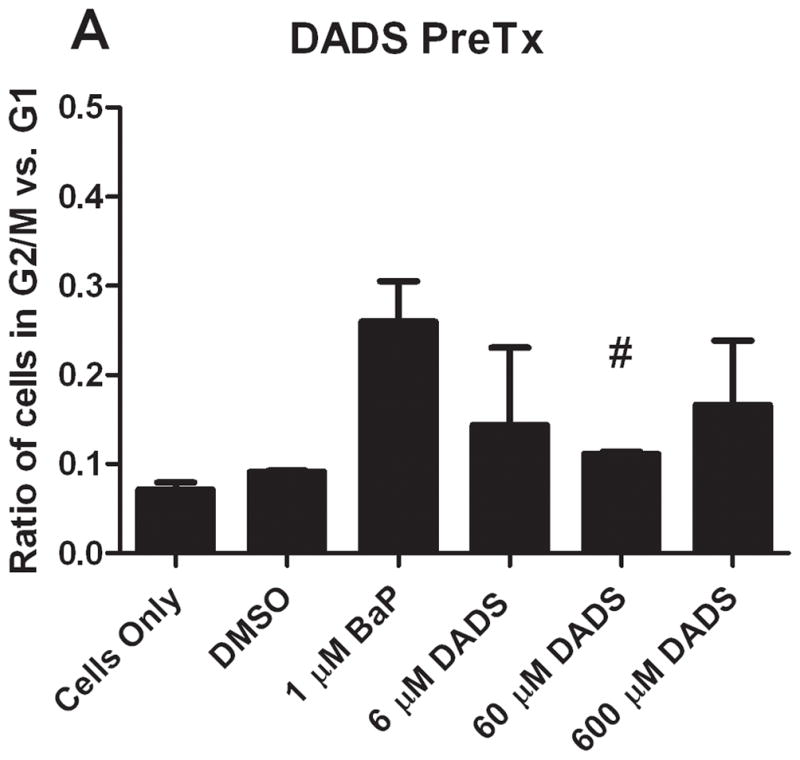
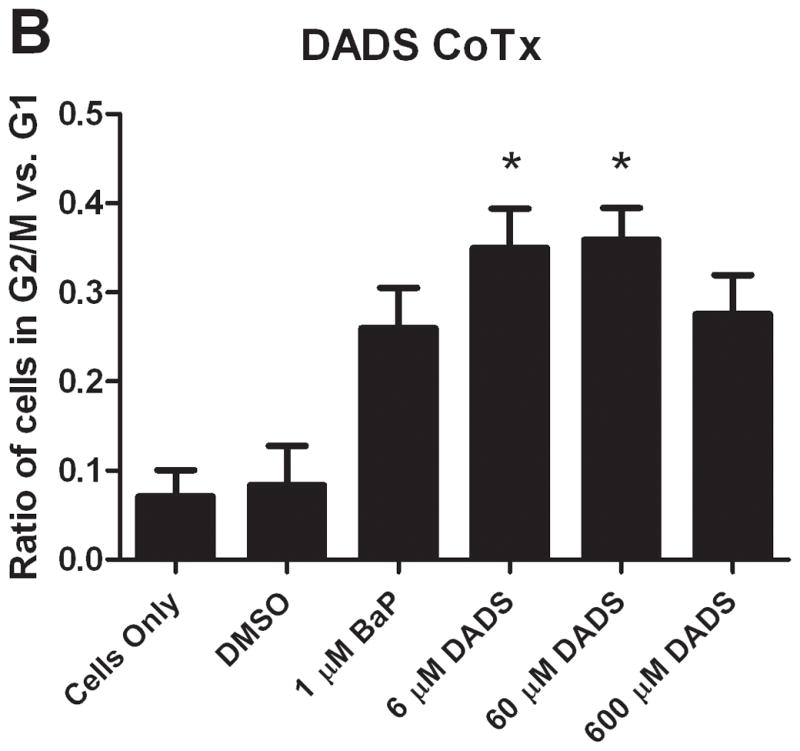
The ratio of cells in G2/M and G1 phases at 24 hours. The bars indicate the average percentage of cells in G2/M divided by the average percentage of cells in G1 for DADS PreTx (A) and DADS CoTx (B), +/− SEM (* indicates a P<0.05 significant difference from the DMSO control, and # indicates a P<0.05 significant difference from the 1 μM BaP only control).
Reactive Oxygen Species Detection
The levels of aqueous peroxides resulting from PreTx and CoTx with DADS and BaP are compared in Figure 4. BaP was shown to significantly increase AQP at all time points evaluated. PreTx with 6 and 60 μM of DADS attenuated BaP-induced AQP at all time points, but were most effective at 24 hours, with 105 and 99% inhibition, respectively. The 600 μM PreTx was effective in reducing BaP-induced AQP at 12 and 24 hours, with 40 and 89% respective attenuation of ROS formation. CoTx studies illustrated the effectiveness of DADS at all time points and concentrations in the sustainable inhibition of BaP-induced AQP to levels not significantly different than the DMSO and cells only controls by 6 hours. The cells only control was not shown for graphical purposes; however, the values for the DMSO vehicle control and cells only control did not vary significantly (P>0.05) at any of the time points evaluated..
Figure 4.

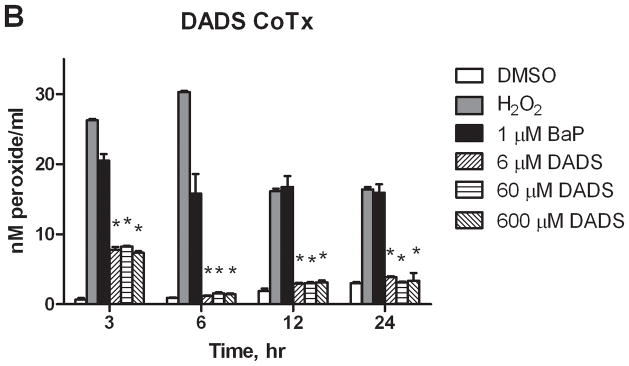
Inhibition of BaP-induced aqueous peroxide formation by DADS. MCF-10A cells were exposed to a pretreatment of DADS followed by 1 μM BaP (A), or with a concomitant combination of 1 μM BaP and DADS (B). The results deem the average aqueous peroxides, +/−SEM, as detected by the PeroxiDetect Kit in triplicate for N=3. (* indicates a P<0.01 significant difference from the DMSO control).
DNA Damage
In this study, BaP was shown to induce significant DNA strand breaks at 3, 6, 12, and 24 hours by 5.4, 2.5, 4.0, and 4.0 times that of the DMSO only control, respectively (Figure 5). DADS attenuated BaP-induced DNA damage at all time points through both Pre- and CoTx, with significant inhibition for all treatments sustained after 6 hours. DADS CoTx at 60 and 600 μM further produced a sustained protective effect on the cells by reducing DNA damage to levels below that of the DMSO vehicle control. The cells only control was not shown for graphical purposes, however, the values for the DMSO vehicle control and cells only control did not vary significantly.
Figure 5.
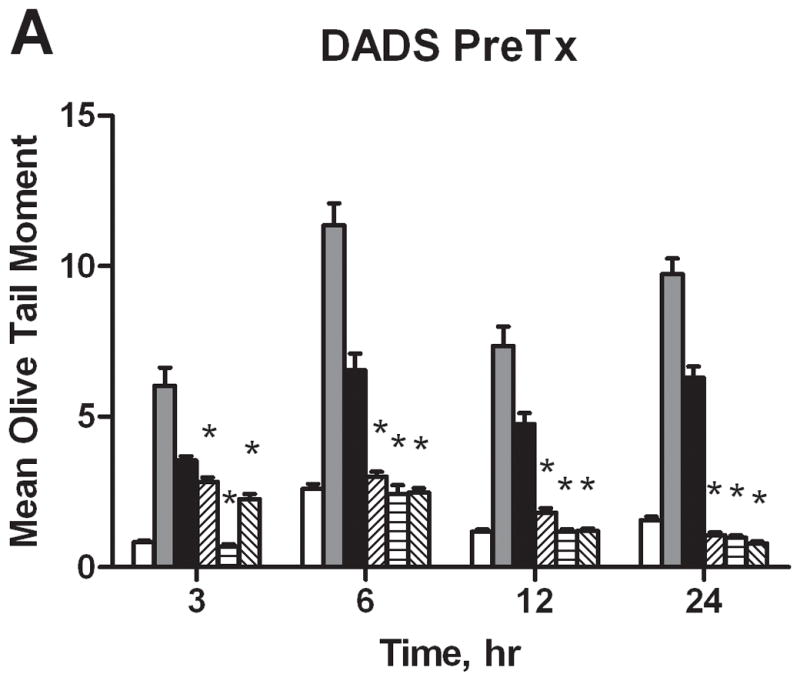
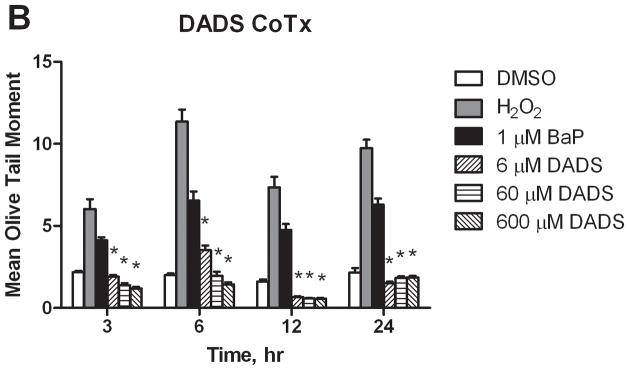
DNA strand breaks resulting from BaP and DADS treatment of MCF-10A cells. DADS Inhibits BaP-induced DNA damage as demonstrated by Comet assay. MCF-10A cells were treated MCF-10A cells were exposed to a PreTx of DADS followed by 1 μM BaP (A), or with a CoTx of 1 μM BaP and DADS (B). The results demonstrate the mean olive tail moment (OTM), as an indicator of DNA damage, +/− SEM for 150 cells for N=3. (* indicates a P<0.05 significant difference from the DMSO control).
DISCUSSION
Garlic organosulfide compounds (OSCs), including DADS, have been previously shown to inhibit benzo(a)pyrene induced carcinogenesis [34–36]. As a potential chemopreventive compound, DADS needs to be evaluated for its potential to prevent environmentally-induced carcinogenesis. In this study, DADS cotreatment attenuated BaP-induced cell proliferation at all concentrations evaluated, and 60 and 600 μM of DADS returned cell counts to that of the controls at six hours. BaP-induced cell cycle transitions at 24 hours were most effectively attenuated by 60 μM DADS pretreatment, whereas DADS concurrent treatment induced G1 and G2/M cell cycle transitions. DADS CoTx was more effective than DADS PreTx in attenuating BaP-induced peroxide formation, however, by 24 hours, all concentrations of DADS, regardless of application method, significantly inhibited BaP-induced peroxidation. Additionally, all concentrations of DADS evaluated suppressed BaP-induced genotoxicity, observed as DNA strand breaks at 12 and 24 hours, and at 24 hours DADS may have played a protective role on the MCF-10A cells, as detected DNA damage was less than that of the controls.
One suggested mechanism of suppression of BaP-induced carcinogenesis is the inhibition of metabolic enzymes responsible for the biotransformation of the parent compound to its ultimate carcinogenic metabolite benzo(a)pyrene-7,8-diol-9,10-epoxide (BPDE) [34]. BaP metabolism requires the enzymatic activation of several phase I and phase II enzymes including cyp1A1, cyp1A2, cyp1B1, and epoxide hydrolase [37]. DADS was shown to inhibit this metabolism in HepG2 hepatoma cells, in which 100–1000 μM DADS attenuated BaP-induced CYP1A2 protein expression, reducing 7,8-diol, 9,10-diol, and 4,5-diol metabolites [18]. BaP, its metabolites and intermediates have been shown to induce oxidative stress and DNA adducts [38–41], thus inhibition of BaP metabolism is important in suppressing its carcinogenic potential.
Human mammary epithelial cells (HMECs) metabolize BaP in vitro, preferentially biotransforming the parent compound to BPDE [42]. Indirect BaP-induced DNA insults and carcinogenesis in HMECs may be mediated by the ROS and free radicals produced as BaP intermediates [43]. To inhibit BaP-induced early carcinogenesis, MCF-10A cells were treated with BaP and DADS, a potential garlic chemopreventive compound for 24 hours and evaluated for changes in cell viability, cell cycle arrest, the production of ROS, and DNA damage expressed as DNA strand breaks. According to Belloir and colleagues, pretreatment with chemopreventive agents promotes modulation of drug metabolism enzymes, whereas co-treatment reveals the ability of the compounds to scavenge direct-acting compounds [36], thus we evaluated DADS as a chemopreventive compound through both pretreatment and concomitant treatment with the environmental carcinogen.
Changes in cell viability can demonstrate a compound’s toxicity or proliferative capacity. Studies vary as to the extent of proliferation observed with BaP in various normal and neoplastic breast cell lines. In normal HMECs, and several neoplastic breast cell lines (MCF-7, HCC 1806, T47-D, and MDA MB 231), BaP did not significantly alter cell viability [44–47]. However Siriwardhana and Wang demonstrated that MCF-10A cells treated for 10–20 rounds of BaP for resulted in an increase in cell proliferation [48]. Tannheimer and colleagues concluded that BaP induced growth factor signaling pathways and cell proliferation in MCF-10A cells, in the absence of additional growth factors [49].
In this study, 1 μM, BaP induced an increase in cell proliferation, which was found to be statistically significant at six hours, relative to the DMSO control in the CoTx study. The potential for BaP to induce growth factor signaling may explain the increase in cell population in BaP treated cells [49]. To inhibit cell proliferation, cells were pre- or concurrently treated with DADS. At six hours, 60 and 600 μM DADS CoTx were the most effective attenuators of BaP-induced cell proliferation. The 6 μM DADS concentration in both the PreTx and CoTx groups maintained a slight increase in cell viability compared to the DMSO control, and the average cell count surpassed that of BaP only in the 24 hour PreTx group. DADS has been shown to induce a protective mechanism inhibiting aflatoxin B1 and hydrogen peroxide induced cellular toxicity [50, 51], and may exhibit a similar mechanism in BaP-induced carcinogenesis.
Cell cycle arrest is a protective response to cellular stress, which occurs through signal transduction pathway or checkpoints [52]. The checkpoints activated in the G1/S phase inhibit the replication of damaged DNA and in the G2/M phase to inhibit the segregation of damaged chromosomes during mitosis [4]. Compounds that induce cell cycle arrest of damaged or cancer cells may inhibit cell proliferation, induce apoptosis, and prevent disease progression.
BaP has been previously shown to alter cell cycle progression in human breast cells [47, 53], though gaps still exist in our understanding of the role of BaP-induced cell cycle changes in normal breast cells. Here, we found that BaP induced a cell cycle transition of MCF-10A cells from the G1 to the S and G2/M phases. Induction of S phase accumulation was shown to result from DNA adducts stalling the replication forks at the sites of DNA damage [54], and resulting in an increase in S-phase accumulation [47]. The inhibition of the S-phase accumulation in MCF-10A cells was most prevalent in DADS pretreatment at 6 and 60 μM. At 600 μM, DADS-induced G2/M and G1 phase transitions, which was most apparent in the PreTx study. The cells were therefore, highly arrested, primarily in G1-phase, potentially allowing for DNA repair. DADS CoTx was not effective in the inhibition of BaP-induced phase shifts, and caused an increase in G2/M cells, relative to both the vehicle control and BaP only. Comparison of the G1 and G2/M arrest ratios, it is apparent that 60 μM DADS PreTx is the most effective inhibitor of BaP-induced cell cycle changes, where as BaP and DADS CoTx are more likely to increase cells in G2/M. It is likely that at 6 and 60 μM DADS, PreTx induced a protective effect on the cells that inhibited changes in the cell cycle, though stalling cells in G2/M as seen in the CoTx may have induced DNA repair mechanism to prevent the transfer of damaged chromosomes during mitosis [4]. Further research on DNA repair enzymes is needed to elucidate this potential mechanism.
DADS has been shown to be an effective inducer of G2/M cell cycle arrest at G2/M and growth inhibition in several cancer cell lines including SW480 colon cancer cells [55], human gastric cancer MGC803 cells [56], and PC3 prostate cancer cells [57, 58]. DADS induced G2/M phase accumulation in HCT-15 human colon tumor cells by inhibiting Cdk1 kinase activity and increased cyclin B1 protein expression [9, 59]; and in COLO 205 colon cancer cells by also increasing cyclin B1, without altering CDK1 protein expression [60]. In the human liver J5 tumor cell line, DADS arrested cells in G2/M, as a likely result of increased cyclin B1 and suppressed Cdk7 kinase expression [61]. In MGC803 cells, DADS induced G2/M arrest was attributed to activation of the p38 MAP kinase pathway and decreased Cdc25C protein expression [56]. Our research demonstrates that DADS has the potential of inhibiting BaP-induced S phase accumulation. Future studies will evaluate cell cycle genes and protein expression.
Active oxygens that lead to lipid peroxidation can impact carcinogenesis by causing chromosomal damage and rearrangements and by modulating epigenetic mechanisms that can alter cell growth and differentiation [43]. This is beneficial in damaged and cancer cells, in which excessive ROS mediates apoptosis. DADS is reduced by GSH to thiols which can generate ROS in the presence of transition metal ions under favorable biological conditions [4, 62]. DADS was shown to increase intracellular hydrogen peroxide and other ROS in human leukemia HL-60 cells and SH-SY5Y neuroblastoma cells resulting in apoptosis [21, 63]. DADS has also been shown to induce apoptosis in colon cancer cells through promotion of caspase-3, -8, and -9 [60]. Nagaraj and collegues determined that DADS is toxic to cancer cells by mediating ROS-induced caspase-dependent apoptosis through Bax-triggered mitochondria-mediated signaling pathways [64]. Tumor cells possess poor antioxidant defense systems relative to normal cells that can lead to ROS induced apoptosis [21]; thus non-neoplastic cells need to be evaluated for ROS induced DNA damage that can lead cancer initiation and cellular transformation
BaP has been previously shown to induce the formation of ROS, potentially as a result of its metabolism to quinone intermediates that act as free radicals [40]. The quantification of free radicals can be estimated by the level of peroxides in a sample. Aqueous peroxides represent the extracellular ROS with the potential to induce the cellular injury that can lead to lipid peroxidation, DNA damage, and potentially cancer. In this study, BaP significantly induced extracellular peroxides from 3 to 24 hours. All concentrations of DADS were effective in attenuating BaP-induced aqueous peroxides by 24 hours, regardless of method, however, only the CoTx was effective at all concentrations by 3 hours. In contrast, ROS was induced by DADS at 3 hours with PreTx, but lead to the attenuation of BaP-induced ROS at 24 hours. The DADS used in this study did not induce apoptosis, as seen in cancer cells, nor were the elevated peroxide levels maintained in the cells. Sallmyr et al. [65] suggests that endogenous chemicals that increase ROS initiate a cycle of genomic instability leading to DNA strand breaks and altered repair, resulting in genomic changes that can induce carcinogenesis. Thus the increases in BaP-induced ROS in this study were further evaluated for potential induced DNA damage.
An increase in oxidative-induced base lesions and DNA adducts have been found in breast cancer tumors as compared to normal tissues [66, 67]. High levels of DNA adducts can lead to double strand breaks in DNA and with an increase in S-phase accumulation that can promote the initiation of carcinogenesis [47]. DNA strand breaks, as demonstrated by the Comet assay, are markers for induced DNA damage and genotoxicity [36]. In the Comet Assay, under alkaline conditions, alkali labile sites and single-strand DNA damage form a comet-like structure when electrophoresed. The olive tail moment (OTM) integrates the quantity of DNA damage with the size of the strands and is defined as the product of the tail length and the fraction of total DNA in the tail relative to that in the head. The comet assay is a useful assay to demonstrate DNA damage as a biomarker for carcinogenesis and its inhibition through chemoprevention [68].
BaP was shown to induce a statistically significant level of DNA strand breaks in MCF-10A cells at 3, 6, 12, and 24 hours. DADS was effective at all concentrations for both treatment methods in the attenuation of BaP-induced DNA strand breaks by 6 hours. CoTx with 60 and 600 μM DADS at 3 and 6 hours, and all three concentrations at 12 and 24 hours further prevented normally occurring strand breakage as seen in the cells only control. Therefore, we have demonstrated that DADS inhibits BaP-induced DNA strand breaks, inhibiting the promotion of carcinogenesis. These results are mirrored in the literature in which DADS inhibited aflatoxin B1-induced DNA damage in primary rat hepatocytes [50]. DADS was also shown to be more effective than other garlic OSC (diallyl sulfide, allicin, S-allyl cysteine, and allyl mercaptan) at inhibiting BaP, hydrogen peroxide, N-nitrosopiperidine, N-nitrosodibutlamine and N-nitrosamine-induced DNA strand breaks and hydrogen peroxide induced genotoxicity, in human HepG2 hepatoma cells [36, 69, 70].
CONCLUSION
BaP was shown to induce significant DNA damage in MCF-10A cells, without inducing concurrent cellular toxicity, thereby demonstrating its carcinogenic potential in a normal cell line. In comparing DADS PreTx and CoTx inhibition of BaP-induced early carcinogenesis potential in MCF-10A cells, CoTx overall, was more effective in attenuating BaP-induced changes in the cells. CoTx inhibited BaP-induced cell proliferation at six hours, inhibited peroxide formation, and inhibited DNA strand breaks, at all evaluated concentrations and time points. CoTx also induced G2/M and G1 cell cycle arrest, potentially protecting the cells from potentiating BaP-induced damage. PreTx was also shown to be effective in reducing BaP-induced cellular damage and alterations by inhibiting BaP-induced G2/M transition, and maintaining the cell cycle pattern of the controls, inhibiting aqueous peroxides at all time points at 6 and 60 μM DADS, and by 12 and 24 hours at 600 μM, and by inhibiting DNA strand breaks by six hours for all concentrations.
For both the PreTx and CoTx methods, 60 μM DADS appears to have the most protective effective against BaP-induced carcinogenesis for this study. This is significant in that 60 μM is a potential physiological dose of DADS that assuming a one-compartment physiological model can be obtained through dietary consumption, as estimated in 12–14 garlic cloves or 6–7 tablespoons of minced garlic. However, actual pharmacokinetic studies are needed using human and animal models to determine the actual daily consumption to attenuate breast cancer. This research is novel in its evaluation of DADS in the inhibition of BaP-induced carcinogenesis in a normal cell model, and other cell types should be evaluated to determine its DADS effectiveness in preventing the induction of other cancers.
We have demonstrated, for the first time that DADS can inhibit carcinogenesis initiation in a normal breast epithelial cell line. This research shows that a physiological dietary concentration of DADS, as found in garlic cloves and minced garlic, can potentially inhibit benzo(a)pyrene induced early breast carcinogenesis. This research further demonstrates the effectiveness of garlic as a potential breast cancer attenuator.
Acknowledgments
This research is a continuation of the legacy and mission of the late Dr. Ronald D. Thomas; we love you and we miss you. We would like to acknowledge the FAMU College of Pharmacy and Pharmaceutical Sciences Faculty and Staff, especially Drs. Karam Soliman, Barak Abonyo, Carl Goodman, and Tracy Womble for their assistance, and use of their supplies and equipment. Additionally, we would like to thank the Florida Education Fund’s McKnight Doctoral Fellowship Program for honoring Dr. Yasmeen Nkrumah-Elie as a McKnight Fellow. This study was funded in part by NIH RCMI Grant # 5G12RR003020-25.
Contributor Information
Yasmeen M. Nkrumah-Elie, Email: yasmeen1.barnesnkrum@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, R110, Tallahassee, Florida 32307
Jayne S. Reuben, Email: JReuben@ghs.org, Department of Biomedical Sciences, University of South Carolina School of Medicine-Greenville, 701 Grove Road, HAS Building, MIPH, Greenville, SC 29605
Alicia M. Hudson, Email: atucker78@aol.com, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, R110, Tallahassee, Florida 32307
Equar Taka, Email: equar.taka@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307.
Ramesh Badisa, Email: ramesh.badisa@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307.
Tiffany Ardley, Email: tiffany.ardley@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307.
Bridg’ette Israel, Email: Bridgette.israel@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307.
Sakeenah Y. Sadrud-Din, Email: Ssadrud690@aol.com, College of Health Professions, South University – Montgomery, AL, 5355 Vaughn Road, Montgomery, Alabama 36116-1120
Ebenezer T. Oriaku, Email: ebenezer.oriaku@famu.edu, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307
Selina F. Darling-Reed, Email: selina.darling@famu.edu, sdarlingreed@gmail.com, College of Pharmacy and Pharmaceutical Sciences, Florida Agricultural and Mechanical University, 1415 S. Dr. Martin Luther King Jr., Blvd, Room 300, Tallahassee, Florida 32307, 1-850-412-5078 (office) 1-850-561-2786 (lab), 1-850-599-3347 (fax)
References
- 1.Block E. The chemistry of garlic and onions. Sci Am. 1985;252:94–99. doi: 10.1038/scientificamerican0385-114. [DOI] [PubMed] [Google Scholar]
- 2.Minami T, Boku T, Inada K, Morita M, Okazaki Y. Odor components of human breath after the ingestion of grated raw garlic. J Food Sci. 1989;54:763–765. [Google Scholar]
- 3.Corzo-Martinez M, Corzo N, Villamiel M. Biological properties of onions and garlic. Trends in Food Sci Technol. 2007;18:609–625. [Google Scholar]
- 4.Iciek M, Kwiecien I, Wlodek L. Biological properties of garlic and garlic-derived organosulfur compounds. Environ Mol Mutagen. 2009;50:247–265. doi: 10.1002/em.20474. [DOI] [PubMed] [Google Scholar]
- 5.Tansey MR, Appleton JA. Inhibition of fungal growth by garlic extract. Mycologia. 1975;67:409–413. [PubMed] [Google Scholar]
- 6.You WC, Zhang L, Gail MH, Ma JL, Chang YS, et al. Helicobater pylori infection, garlic intake and precancerous lesions in a Chinese population at low risk of gastric cancer. Int J Epidemiol. 1998;27:941–944. doi: 10.1093/ije/27.6.941. [DOI] [PubMed] [Google Scholar]
- 7.Weber ND, Anderson DO, North JA, Murray BK, Lawson LD, et al. In vitro virucidal activity of Allium sativum (garlic) extract and compounds. Planta Med. 1992;58:417–423. doi: 10.1055/s-2006-961504. [DOI] [PubMed] [Google Scholar]
- 8.Chung LY. The antioxidant properties of garlic compound. allyl cysteine, alliin, allicin, and allyl disulfide. J Med Food. 2006;9:205–213. doi: 10.1089/jmf.2006.9.205. [DOI] [PubMed] [Google Scholar]
- 9.Knowles LM, Milner JA. Depresses p34cdc2 kinase activity and G2/M phase arrest induced by diallyl disulfide in HCT-15 cells. Nutr Cancer. 1998;30:169–174. doi: 10.1080/01635589809514659. [DOI] [PubMed] [Google Scholar]
- 10.Milner JA. A Historic Perspective on Garlic and Cancer. J Nutr. 2001;131:1027S–1031S. doi: 10.1093/jn/131.3.1027S. [DOI] [PubMed] [Google Scholar]
- 11.Guyonnet D, Belloir C, Suschetet M, Siess H, Le Bon A-M. Antimutagenic activity of organosulfur compounds from Allium is associated with phase II enzyme induction. Muta Res. 2001;495:135–145. doi: 10.1016/s1383-5718(01)00205-4. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi S, Hakoi K, Yada H, Hirose M, Ito N, et al. Enchancing effects of diallyl sulfide on hepatocarcinogenesis and inhibitory actions of the related diallyl disulfide on colon and renal carcinogenesis in rats. Carcinogenesis. 1992;13:1513–1518. doi: 10.1093/carcin/13.9.1513. [DOI] [PubMed] [Google Scholar]
- 13.Sundaram SG, Milner JA. Diallyl disulfide suppresses the growth of human colon tumor cell xenografts in athymic nude mice. J Nutr. 1996;126:1355–1361. doi: 10.1093/jn/126.5.1355. [DOI] [PubMed] [Google Scholar]
- 14.Sundaram SG, Milner JA. Diallyl disulfide inhibits the proliferation of human tumor cells in culture. Biochim Biophys Acta. 1996;1315:15–20. doi: 10.1016/0925-4439(95)00088-7. [DOI] [PubMed] [Google Scholar]
- 15.Suzui N, Sugie S, Rahman KM, Ohnishi M, Yoshimi N, et al. Inhibitory effects of diallyl disulfide or aspirin on 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine-induced mammary carcinogenesis in rats. Jpn J Cancer Res. 1997;88:705–711. doi: 10.1111/j.1349-7006.1997.tb00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dwivedi C, Rohlfs S, Jarvis D, Engineer FN. Chemoprevention of chemically induced skin tumor development by diallyl sulfide and diallyl disulfide. Pharm Res. 1992;9:1668–1670. doi: 10.1023/a:1015845315500. [DOI] [PubMed] [Google Scholar]
- 17.Mori H, Sugie S, Rahman W, Suzui N. Chemoprevention of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-induced mammary carcinogenesis in rats. Cancer Lett. 1999;143:195–198. doi: 10.1016/s0304-3835(99)00124-x. [DOI] [PubMed] [Google Scholar]
- 18.Chun HS, Kim HJ, Choi EH. Modulation of cytochrome P4501-mediated bioactivation of benzo[a]pyrene by volatile allyl sulfides in human hepatoma cells. Biosci Biotechnol Biochem. 2001;65:2205–2212. doi: 10.1271/bbb.65.2205. [DOI] [PubMed] [Google Scholar]
- 19.Lin JG, Chen GW, Su CC, Hung CF, Yang CC, et al. Effects of garlic components diallyl sulfide and diallyl disulfide in arylamine N-acetyltransferase activity and 2-aminofluorene-DNA adducts in human promyelocytic leukemia cells. Am J Chin Med. 2002;30:315–325. doi: 10.1142/S0192415X02000338. [DOI] [PubMed] [Google Scholar]
- 20.Hong YS, Ham YA, Choi JH, Kim J. Effects of allyl sulfur compounds and garlic extract on the expression of Bcl-2, Bax, and p53 in non small cell lung cancer cell lines. Exp Mol Med. 2000;32:27–34. doi: 10.1038/emm.2000.22. [DOI] [PubMed] [Google Scholar]
- 21.Filomeni G, Aquilano K, Rotilio G, Ciriolo M. Reactive oxygen species-dependent c-Jun NH2-terminal kinase/c-Jun signaling cascade mediates neuroblastoma cell death induced by diallyl disulfide. Cancer Res. 2003;63:5940–5949. [PubMed] [Google Scholar]
- 22.Li G, Qiao CH, Lin RI, Pinto JT, Osbourne MP, et al. Antiproliferative effects of garlic constituents on cultured human breast cancer cells. Oncol Rep. 1995;2:787–791. doi: 10.3892/or.2.5.787. [DOI] [PubMed] [Google Scholar]
- 23.Drusene N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, et al. Diallyl disulfide (DADS) increases Histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 24.Robert V, Mouille B, Mayeur C, Michaud M, Blachier F. Effects of the garlic compound diallyl disulfide on the metabolism, adherence, and cell cycle of HT-29 colon carcinoma cells: evidence of sensitive and resistant sub-populations. Carcinogenesis. 2001;22:1155–1161. doi: 10.1093/carcin/22.8.1155. [DOI] [PubMed] [Google Scholar]
- 25.Caruso JA, Reiners JJ, Emond J, Schultz T, Tainsky MA, et al. Genetic alteration of chromosome 8 is a common feature of human mammary epithelial cell lines transformed in vitro with benzo[a]pyrene. Muta Res. 2001;473:85–99. doi: 10.1016/s0027-5107(00)00140-8. [DOI] [PubMed] [Google Scholar]
- 26.Edwards NT. Polycyclic aromatic hydrocarbons (PAHs) in the environment: a review. J Environ Qual. 1983;12:427–441. [Google Scholar]
- 27.Luttrell W, Thomas C. Toxic tips: Benzo[a]pyrene. Chem Health and Saf. 2007;14:21–22. [Google Scholar]
- 28.IARC Monogr Eval Carcinog Risks Hum. Vol. 92. World Health Organization, IARC Press; Lyon France: 2010. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures; p. 773. [PMC free article] [PubMed] [Google Scholar]
- 29.Lightfoot TJ, Coxhead JM, Cupid BC, Nicholson S, Garner RC. Analysis of DNA adducts by accelerator mass spectrometry in human breast tissue after administration of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and benzo[a]pyrene. Muta Res. 2000;472:119–127. doi: 10.1016/s1383-5718(00)00134-0. [DOI] [PubMed] [Google Scholar]
- 30.Hanelt S, Helbig R, Hartmann A, Lang M, Seidel A, et al. A comparative investigation of DNA adducts, DNA strand breaks and gene mutations induced by benzo[a]pyrene and (±)-anti-benzo[a]pyrene-7,8-diol 9,10-epoxide in cultured human cells. Muta Res. 1997;390:179–188. doi: 10.1016/s0165-1218(97)00019-0. [DOI] [PubMed] [Google Scholar]
- 31.CellTiter 96 AQueous One Soultion Cell Proliferation Assay: Instructions for Use of Products G3580, G3581, and G3582 2005. Promega; Madison, WI: 2005. Technical Bulletin; pp. 1–2. [Google Scholar]
- 32.PeroxiDetect Kit: For the Determination of Aqueous and Lipid Hydroperoxides. Sigma-Aldrich; St. Louis, MO: 2005. Technical Bulletin. [Google Scholar]
- 33.Aboyade-Cole A, Darling-Reed S, Oriaku E, McCaskill M, Thomas R. Diallyl sulfide inhibits PhiP-induced cell death via the inhibition of DNA strand breaks in normal breast epithelial cells. Oncol Rep. 2008;20:319–323. [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava SK, Hu X, Xia H, Zaren HA, Chatterjee ML, et al. Mechanism of differential efficacy of garlic organosulfides in preventing benzo(a)pyrene-induced cancer in mice. Cancer Lett. 1997;118:61–67. doi: 10.1016/s0304-3835(97)00237-1. [DOI] [PubMed] [Google Scholar]
- 35.Guyonnet D, Belloir C, Suschetet M, Siess MH, Le Bon A-M. Liver subcellular fraction from rats treated by organosulfur compounds from allium modulate mutagen activation. Muta Res. 2000;466:17–26. doi: 10.1016/s1383-5718(99)00234-x. [DOI] [PubMed] [Google Scholar]
- 36.Belloir C, Singh V, Daurat C, Siess MH, Le Bon AM. Protective effects of garlic sulfur compounds against DNA damage induced by direct- and indirect-acting genotoxic agents in HepG2 cells. Food Chem Toxicol. 2006;44:827–834. doi: 10.1016/j.fct.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Patri M, Padmini A, Babu PP. Polycyclic aromatic hydrocarbons in air and their neurotoxic potency in association with oxidative stress: A brief perspective. Annals of Neurosciences. 2009:16. doi: 10.521/ans.0972.7531.2009.160109. [DOI] [Google Scholar]
- 38.Sagredo C, Ovrebo S, Haugen A, Fujii-Kuriyama Y, Baera R, et al. Quantitative anlysis of benzo[a]pyrene biotransformation and adduct formation in Ahr knockout mice. Toxicol Lett. 2006;167:173–182. doi: 10.1016/j.toxlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 39.Perlow RA, Kolbanovskii A, Hingerty BE, Geacintov NE, Broyde S, et al. DNA adducts from a tumorigenic metabolite of benzo[a]pyrene block human RNA polymerase II elongation in a sequence- and stereochemistry-dependent manner. J Mol Biol. 2002;321:29–47. doi: 10.1016/s0022-2836(02)00593-4. [DOI] [PubMed] [Google Scholar]
- 40.Kim HS, Kwack SJ, Lee BM. Lipid peroxidation, antioxidant enzymes and benzo[a]pyrene-quinones in the blood of rats treated with benzo[a]pyrene. Chem Biol Interact. 2000;127:139–150. doi: 10.1016/s0009-2797(00)00177-0. [DOI] [PubMed] [Google Scholar]
- 41.Spink DC, Wu SJ, Spink BC, Hussain MM, Vakharia DD, et al. Induction of CYP1A1 and CYP1B1 by benzo(k)fluoranthene and benzo(a)pyrene in T-47D human breast cancer cells: Roles of PAH interactions and PAH metabolites. Toxicol Appl Pharmacol. 2008;226:213–224. doi: 10.1016/j.taap.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stampfer MR, Bartley JC. Human Mammary Epithelial Cells in Culture: Differentiation and Transformation. In: Dickson R, Lippman M, editors. Breast Cancer: Cellular and Molecular Biology. Kluwer Academic Publishers; Boston, MA: 1988. pp. 1–24. [DOI] [PubMed] [Google Scholar]
- 43.Leadon SA, Stampfer MR, Bartley J. Production of oxidative DNA damage during the metabolic activiation of benzo[a]pyrene in human mammary epithelial cells correlates with cell killing. Proc Natl Acad Sci USA, Cell Biol. 1988;85:4365–4368. doi: 10.1073/pnas.85.12.4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keshava C, Whipkey D, Weston A. Transcriptional signatures of environmentally relevant exposures in normal human mammary epithelial cells: benzo[a]pyrene. Cancer Lett. 2005;221:201–211. doi: 10.1016/j.canlet.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 45.Sigounas G, Hairr JW, Cooke CD, Owen JR, Asch AS, et al. Role of benzo[a]pyrene in generation of clustered DNA damage in human breast tissue. Free Radic Biol Med. 2010;49:77–87. doi: 10.1016/j.freeradbiomed.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 46.Tampio M, Markkanen P, Puttonen KA, Hagelberg E, Heikkinen H, et al. Induction of PUMA-α and down-regulation of PUMA-β expression is associated with benzo(a)pyrene-induced apoptosis in MCF-7 cells. Toxicol Lett. 2009;188:214–222. doi: 10.1016/j.toxlet.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 47.Sadikovic B, Rodenhiser DI. Benzopyrene exposure disrupts DNA methylation and growth dynamics in breast cancer cells. Toxicol Appl Pharmacol. 2006;216:458–468. doi: 10.1016/j.taap.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 48.Siriwardhana N, Wang H-CR. Precancerous carcinogenesis of human breast epithelial cells by chronic exposure to benzo[a]pyrene. Mol Carcinog. 2008;47:338–348. doi: 10.1002/mc.20392. [DOI] [PubMed] [Google Scholar]
- 49.Tannheimer SL, Lauer FT, Lane J, Burchiel SW. Factors influencing elevation of intracellular Ca2+ in the MCF-10A human mammary epithelial cell line by carcinogenic polycyclic aromatic hydrocarbons. Mol Carcinog. 1999;25:48–54. doi: 10.1002/(sici)1098-2744(199905)25:1<48::aid-mc6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 50.Sheen L-Y, Wu C-C, Li C-K, Tsai S-J. Effect of diallyl sulfide and diallyl disulfide, the active principles of garlic, on the aflatoxin B1-induced DNA damage in primary rat hepatocytes. Toxicol Lett. 2001;122:45–52. doi: 10.1016/s0378-4274(01)00347-2. [DOI] [PubMed] [Google Scholar]
- 51.Kim J-G, Koh S-H, Lee YJ, Lee K-Y, Kim Y, et al. Differential effects of diallyl disulfide on neuronal cells depend on its concentration. Toxicology. 2005;211:86–96. doi: 10.1016/j.tox.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 52.Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 53.Vanparys C, Maras M, Lenjou M, Robbens J, Van Bockstaele D, et al. Flow cytometric cell cycle analysis allows for rapid screening of estrogenicity in MCF-7 breast cancer cells. Toxicol In Vitro. 2006;20:1238–1248. doi: 10.1016/j.tiv.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Bi X, Slater DM, Ohmori H, Vaziri C. DNA polymerase K is specifically required for the recovery from benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. JBiol Chem. 2005;280:22343–22355. doi: 10.1074/jbc.M501562200. [DOI] [PubMed] [Google Scholar]
- 55.Xiao D, Pinto JT, Gundersen GG, Weinstein IB. Effects of a series of organosulfur compounds on mitotic arrest and induction of apoptosis in colon cancer cells. Mol Cancer Ther. 2005;4:1388–1398. doi: 10.1158/1535-7163.MCT-05-0152. [DOI] [PubMed] [Google Scholar]
- 56.Yuan JP, Wang GH, Ling H, Su Q, Yang YH, et al. Diallyl disulfide-induced G2/M arrest of human gastric cancer MGC803 cells involves activation of p38 MAP kinase-pathways. World J Gastroenterol. 2004;10:2731–2734. doi: 10.3748/wjg.v10.i18.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gunadharini DN, Arunkumar A, Krishnamoorthy G, Muthuvel R, Vijaybabu, et al. Antiproliferative effect of diallyl disulfide (DADS) on prostate cancer cell line LNCaP. Cell Biochem Funct. 2006;24:407–412. doi: 10.1002/cbf.1262. [DOI] [PubMed] [Google Scholar]
- 58.Arunkumar A, Vijayababu MR, Srinivasan N, Aruldhas MM, Arunakaran J. Garlic compound, diallyl disulfide induces cell cycle arrest in prostate cancer cell line PC-3. Mol Cell Biochem. 2006;288:107–113. doi: 10.1007/s11010-006-9126-6. [DOI] [PubMed] [Google Scholar]
- 59.Knowles LM, Milner JA. Diallyl disulfide inhibits p34cdc2 kinase activity through changes in complex formation and phosphorylation. Carcinogenesis. 2000;21:1129–1134. [PubMed] [Google Scholar]
- 60.Yang J-S, Chen G-W, Hsia T-C, HO H-C, Ho C-C, et al. Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through induction of reactive oxygen species, endoplasmic reticulum stress, caspases casade and mitochondrial-dependent pathways. Food Chem Toxicol. 2009;47:171–179. doi: 10.1016/j.fct.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 61.Wu CC, Chung JG, Tsai SY, Yang JH, Sheen LY. Differential affects of allyl sulfides from garlic essential oil on cell cycle regulation in human liver tumor cells. Food Chem Toxicol. 2004;42:1937–1947. doi: 10.1016/j.fct.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Munday R, Munday JS, Munday CM. Comparative effects of mono-, di-, tri-, and tetrasulfides derived from plants of the Allium family: Redox cycling in vitro and hemolytic activity and phase 2 enzyme induction in vivo. Free Radical Biol Med. 2003;34:1200–1211. doi: 10.1016/s0891-5849(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 63.Kwon KB, Yoo SJ, Ryu DG, Yang JY, Rho HW, et al. Induction of apoptosis by diallyl disulfide through activation of caspase-3 in human leukemia HL-60 cells. Biochem Pharmacol. 2002;63:41–47. doi: 10.1016/s0006-2952(01)00860-7. [DOI] [PubMed] [Google Scholar]
- 64.Nagaraj NS, Anilakumar KR, Singh OV. Diallyl sulfide causes caspase-dependent apoptosis in human cancer cells through a Bax-triggered mitochondrial pathway. J Nutr Biochem. 2010;21:405–412. doi: 10.1016/j.jnutbio.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 65.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs), and error-prone repair. Cancer Lett. 2008;270:1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 66.Blasiak J, Arabski M, Krupa R, Wozniak K, Rykala J, et al. Basal, oxidative, and alkylative DNA damage, DNA repair efficacy and mutagen sensitivity in breast cancer. Muta Res. 2004;554:139–148. doi: 10.1016/j.mrfmmm.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 67.Smith TR, Miller MS, Lohman KK, Case LD, Hu JL. DNA damage and breast cancer risk. Carcinogenesis. 2003;24:883–889. doi: 10.1093/carcin/bgg037. [DOI] [PubMed] [Google Scholar]
- 68.Wasson GR, McKelvey-Martin VJ, Downes CS. The use of the comet assay in the study of human nutrition and cancer. Mutagenesis. 2008;23:153–162. doi: 10.1093/mutage/gen003. [DOI] [PubMed] [Google Scholar]
- 69.Garcia A, Haza AI, Arranz N, Delgado ME, Rafter J, et al. Organosulfur compounds alone or in combination with vitamin C protect towards N-nitrosopiperidine and N-nitrosodibutylamine-induced oxidative DNA damage in HepG2 cells. Chem Biol Interact. 2008;173:9–18. doi: 10.1016/j.cbi.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 70.Arranz N, Haza AI, Garcia A, Moller L, Rafter J, et al. Protective effects of organosulfur compounds towards N-nitrosamine-induced DNA damage in the single-cell gel electrophoresis (SCGE)/HepG2 assay. Food Chem Toxicol. 2007;45:1662–1669. doi: 10.1016/j.fct.2007.02.032. [DOI] [PubMed] [Google Scholar]