

Abstract
How RANKL evokes [Ca2+]i oscillations and leads to osteoclast differentiation is unclear. We identified a new signaling protein, RGS12, and found that RGS12 is essential for [Ca2+]i oscillations and osteoclast differentiation induced by RANKL. RGS12 may play a critical role in the RANKL-evoked PLCγ– calcium channels–[Ca2+]i oscillation–NFAT2 pathway.
Introduction
RANKL-induced [Ca2+]i oscillations play a switch-on role in NFAT2 expression and osteoclast differentiation. However, RANKL evokes [Ca2+]i oscillations and leads to osteoclast differentiation by an unknown mechanism. In this study, we identified a new RANKL-induced signaling protein, regulator of G signaling protein 12 (RGS12), and investigated its effect on osteoclast differentiation in vitro.
Materials and Methods
We used a genome-wide screening approach to identify genes that are specifically or prominently expressed in osteoclasts. To study the role of the RGS12 in osteoclast differentiation, we used vector and lentivirus-based RNAi gene silencing technology to silence the RGS12 gene in the monocyte progenitor cell lines and primary bone marrow–derived monocytes (BMMs). The interaction between RGS12 and N-type calcium channels was elucidated using co-immunoprecipitation and immunoblotting.
Results
We found that RGS12 was prominently expressed in osteoclast-like cells (OLCs) induced by RANKL. This result was further confirmed at both the mRNA and protein level in human osteoclasts and mouse OLCs. Silence of RGS12 expression using vector and lentivirus based RNA interference (RNAi) impaired phosphorylation of phospholipase C (PLC)γ and blocked [Ca2+]i oscillations, NFAT2 expression, and osteoclast differentiation in RANKL-induced RAW264.7 cells and BMMs. We further found that N-type calcium channels were expressed in OLCs after RANKL stimulation and that RGS12 directly interacted with the N-type calcium channels.
Conclusions
These results reveal that RGS12 is essential for the terminal differentiation of osteoclasts induced by RANKL. It is possible that RGS12 regulates osteoclast differentiation through a PLC γ–calcium channel–[Ca2+]i oscillation–NFAT2 pathway.
Keywords: regulator of G signaling protein 12, RANKL, calcium oscillations, osteoclast differentiation, calcium channels
Introduction
Osteoclasts are the principal, if not exclusive, bone Oresorbing cells, and their activity has a profound impact on skeletal health. Disorders of skeletal insufficiency, such as osteoporosis, are typically characterized by enhanced osteoclastic bone resorption relative to bone formation. A more complete understanding of the mechanisms by which osteoclasts differentiate from their precursors and degrade the skeleton is therefore critical to developing therapies for these often-debilitating diseases.
In the past several years, important progress has been made in the mechanisms of osteoclast differentiation and activation, particularly the role of RANKL.(1–6) RANKL occupying RANK on the surface of osteoclast precursors leads to a recruitment of TRAF family proteins such as TRAF6, which activates the NF-κB, c-fos, and Jun N-terminal kinase (JNK) pathways.(7,8) The essential role of these factors in bone homeostasis is fully underscored.(1,9,10) Recently, Takayanagi et al.(11) reported that NFAT2 plays an essential and sufficient role in osteoclastogenesis. They showed that RANKL induces and activates NFAT2 through calcium signaling. Both the transient initial release of Ca2+ from intracellular stores and the influx through specialized Ca2+ channels control the dephosphorylation of the cytoplasmic components (NFAT2 proteins) and lead to their nuclear localization, which is followed by the activation of osteoclast-specific genes. Despite the importance of the calcium signaling–NFAT pathway, it remained unclear how RANKL activates calcium signals leading to the induction of NFAT2. Most recently, Koga et al.(12) reported that immunoreceptor tyrosine-based activation motif (ITAM)-dependent co-stimulatory signals activated by multiple immunoreceptors are essential for the maintenance of bone homeostasis. In osteoclast precursor cells, FcRγ and DAP12 associate with multiple immunoreceptors and activate calcium signaling through phospholipase Cγ (PLCγ). RANKL-induced phosphorylation of PLCγ and [Ca2+]i oscillations were impaired in DAP12−/−FcRγ−/− cells, suggesting that both proteins are involved in the PLCγ pathway and phosphorylation of PLCγ is the critical component involved in the RANKL-induced [Ca2+]i oscillations–NFAT2 pathway. However, the question remains as to how RANKL evokes the NFAT2 signaling pathway. The molecular linkage between RANKL and [Ca2+]i oscillations is still unknown.
We hypothesized that related signaling proteins critical to osteoclast differentiation may be induced by RANKL and involved in the [Ca2+]i oscillation–NFAT2 pathway. To test the hypothesis, we used a genome-wide microarray (Affimetrix) to screen osteoclast-specific genes and identified a new RANKL-induced signaling protein, regulator of G signaling protein 12 (RGS12), which is prominently expressed in RANKL-induced osteoclast-like cells (OLCs). We further determined the effect of RGS12 on [Ca2+]i oscillations and osteoclast differentiation in vitro. Our results showed that RGS12 plays an essential role in RANKL-regulated osteoclast differentiation possibly through the PLCγ–calcium channel– [Ca2+]i oscillation–NFAT2 pathway.
Materials and Methods
Cells and cell culture
MOCP-5 and RAW264.7 are the monocyte progenitor cell lines. MOCP-5 was generated in our laboratory.(13) RAW264.7 was obtained from the American Type Culture Collection (ATCC, Bethesda, MD, USA). Human osteoclastoma tumors were obtained courtesy of Dr Andrew Rosenberg.(14,15) Stromal cells from tumors were obtained as described.(16)
GeneChip analysis
MOCP-5 cells were stimulated with RANKL (10 ng/ml) and macrophage-colonly stimulating factor (M-CSF; 20 ng/ml) for 96 h. Total RNA was extracted from these cells using Trizol reagent (Life Technologies) as described by the manufacturer. Total RNA (15 μg) was used for cDNA synthesis by reverse transcription followed by synthesis of biotinylated cRNA through in vitro transcription. After cRNA fragmentation, hybridization with mouse U74Av2 Gene-Chip (Affymetrix) displaying probes for 12,000 mouse genes/ESTs was performed according to the manufacturer's protocol. Chips were washed, stained with streptavaidinphycoerythrin (SA-PE), and analyzed using an Affymetrix GeneChip scanner and accompanying gene expression software.
RNA blotting analysis
Total RNA from mouse tissues, human osteoclast and stromal cells obtained from human osteoclastoma tumors,(14,15) and deferent cell lines were isolated using the method of Chomczynski and Sacchi.(17) A 1.2-kb mouse RGS12 cDNA fragment was used as a probe. Hybridization was performed as described.(18)
RT-PCR and sequencing analysis
Total RNA from RANKL-induced OLCs was isolated using Trizol reagent. One-step RT-PCR was performed using Access RT-PCR system (Promega).
Construction of RGS12 RNAi vectors
Oligonucleotides encoding shRNA directed against the RGS12 gene (accession no. NM_173402) were designed using Insert Design Tool for the Vectors (Ambion) and BLOCK-iT RNAi Designer (Invitrogen) and cloned into the SalI/XbaI site of pAVU6 + 27 RNAi expression vector (gift of Paul D Good, Michigan University, Ann Arbor, MI, USA) and pENTR/H1/TO vector (Invitrogen). The LR recombination reaction was completed between the pENTR/H1/TO-RGS12 vector and pLenti6/BLOCK-iT-DEST construct to generate pLenti6/BLOCK-iT expression constructs according to the manufacturer's instructions. By transfecting these vectors into RAW264.7 cell lines and bone marrow–derived monocytes (BMMs), we performed a quick screen analysis of shRNA expression with TRACP staining and selected five constructs (pAVU-R12a or pAVU-R12b; pAVU-R12c; pLenti-R12a; or pLenti-R12b) for further study. The oligonucleotides for these constructs are located at site 298–318 (a), 671–691 (b), and 2182–2200 bp (c), respectively, in the RGS12 gene locus.
Production and titer of lentivirus
Lentiviral production was completed according to the manufacturer's instructions. The 293FT producer cell line was co-transfected with the expression constructs. The viral supernatant was harvested after 48–72 h, and titers were determined by infecting Hela or 293T cells with serial dilutions of concentrated lentivirus. The viral supernatant was added to the primary mouse BMMs and mature osteoclasts. After 24 h, the virus-containing media was removed and replaced with fresh, complete medium. After incubating the cells at 37°C for 24–48 h, we started to assay the cells for silence of the RGS12 gene.
Target cell transduction
The RAW264.7 cells were plated on 35-mm dishes at a density of 2.5 × 105 cells/cm2. After 24 h, the cells were transfected with LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instructions. Each transfection contained 0.5 μg of the expression vector. The cells were cultured in growth media for 24 h and switched to a medium containing 400 μxg/ml G418 to select stable colonies.
In vitro mouse osteoclastogenesis
Mouse BMMs containing osteoclast precursors were obtained from female BalB/c mice as described.(19,20) BMMs and RGS12-silenced stable cells were seeded at 5 × 104 cells/well in a 24-well plate and cultured in α-MEM (GIBCO-BRL) with 10% FBS (GIBCO-BRL) containing M-CSF (20 ng/ml). After 24 h, RGS12-silenced stable cells were further cultured in the presence of 10 ng/ml soluble RANKL (Peprotech) and 20 ng/ml M-CSF to generate osteoclasts. BMMs were infected with pLenti-R12a or pLenti-R12b viral supernatant plus 8 μg/ml polybrene (Sigma) for 24 h. They were treated with the same concentration of RANKL/M-CSF. Ninety-six hours later, the cells were fixed for determination of differentiation. M-CSF and RANKL/M-CSF were used at these concentrations throughout the paper unless otherwise described.
TRACP+ staining
TRACP was used as a marker for osteoclasts. Preosteoclasts and OLCs derived from BMMs and RGS12-silenced stable cell lines were fixed and stained for TRACP activity using a commercial kit (Sigma Chemical). Multinucleated (more than three nuclei) TRACP+ cells (MNCs) appeared as dark purple cells and were counted by light microscopy. The size of a “random field of view” is 50 mm(2). Six fields were counted for each group. All data are expressed as mean ± SD.
Bone resorption assay
BMMs were plated on dentine slices and infected with pLenti-scrambled and pLenti-R12a lentiviruses followed by RANKL/M-CSF induction for 96 h. The resorption pits were determined by immunostaining with collagen I antibody and compared with control cells.(2)
Immuno cell staining
The cells were induced as indicated above, sequentially fixed with 100% methanol, and incubated with 5% BSA/PBS for 30 minutes with their respective antibodies, goat anti-RGS12 antibody (1 μg/ml; Santa Cruz Biotechnology), rabbit anti-Atp6i antibody, anti-cathepsin K antibody (both produced in our laboratory), mouse anti-NFAT2 (1 μg/ml; Santa Cruz Biotechnology), or anti-α1B calcium-channel antibody (Alomone Laboratories; ACC-002) in TBST for 60 minutes. Positive reactions were identified following reaction with anti-IgG-FITC or horseradish peroxidase (HRP; 1 (μg/ml; Santa Cruz Biotechnology) for 60 minutes. For cells with FITC-conjugated antibody, coverslips were washed three times in PBS, mounted on glass slides, and examined with microscopes. For cells with horseradish peroxidase–conjugated antibody, coverslips were further stained with VECTASTAIN Elite ABC kit (Vector Laboratories).
Ca2+ measurements
Ca2+ measurements were performed as described.(11) The cells were first incubated with M-CSF or RANKL/M-CSF for 24, 48, or 72 h and then with 5 μM fluo-4 AM, 5 μM Fura Red AM, and 0.05% pluronic F127 for 30 minutes. The cells were postincubated in DMEM media with 20 ng/ml M-CSF for 20 minutes and mounted on a confocal microscope (Leica). The images of cells were scanned and plotted with an interval of 5 s. To estimate intracellular Ca2+ concentration in single cells, the ratio of fluorescence intensity of the fluo-4 to Fura Red was calculated with an interval of 5 s.
Co-immunoprecipitation assay
RANKL-induced BMMs were washed with PBS and lysed in buffer containing 50 mM Tris-HCl (pH 8.0), 0.15 M NaCl, 1% Nonidet P-40, and phosphatase and protease inhibitors.(18) For immunoprecipitations, lysates were incubated for 2 h at 4°C with the anti-α1B calcium-channel antibody (ACC-002; Alomone Laboratories) or an anti-RGS12 antibody and protein G-Sepharose beads (Amersham Biosciences). The precipitates were washed three times with lysis buffer and resuspended in SDS sample buffer. The samples were separated by 4–15% SDSPAGE.
Western blot
The cells were incubated with RANKL/M-CSF as above for 60 minutes or 96 h. Cell lysates containing 15 μg protein were electrophoresed, transferred to blots, and probed by reaction with antibodies as described.(18) Immunoreactivity was detected by enhanced chemiluminescence (ECL; Amersham), and blots were visualized and quantified using Fluor-S MultiImager and MULTIANALYST software (Bio-Rad).
Acridine orange staining
Acid production was determined using acridine orange, according to the method of Baron et al.(21) The cells were observed under a fluorescence microscope with a 490-nm excitation filter and a 525-nm arrest filter.
Apoptosis assay
Apoptosis was measured by Hoechst 33258 staining of condensed chromatin.(22,23) pAVU-scrambled and pAVU-R12a transfected stable cells were, respectively, treated with RANKL for 48 and 96 h. Next, the cells were fixed with 2% glutaraldehyde solution (WAKO) for 10 minutes and stained with 0.2 mM Hoechst 33258 to visualize the localization of DNA. Cells were examined under a fluorescence microscope for determination of osteoclasts with chromatin condensation and/or nuclear fragmentation.
Statistical analysis
Where indicated, experimental data are reported as means ± SD of triplicate independent samples. Studies using multiple groups were analyzed using ANOVA followed by Tukey-Kramer multiple comparisons test to determine statistically significant differences between groups. Statistical significance for two groups was assessed using Student's t-test.
Results
RGS12 is prominently expressed in mouse OLCs and human osteoclasts
To identify genes selectively activated by RANKL, we performed a genome-wide screening of mRNA in mouse monocyte progenitor cell line (MOCP-5)(13) with or without RANKL stimulation, using an oligonucleotide array (Affymetrix GeneChip). As shown in Fig. 1A, RGS12 was highly induced by RANKL in OLCs. To evaluate whether RGS12 is prominently expressed in authentic osteoclasts, Northern blots were performed on human osteoclasts and various mouse tissues (Fig. 1B). The results showed that RGS12 was prominently expressed in human osteoclast. The expression of RGS12 was also observed in the brain, and to a far lesser extent, in the spleen, calvaria, and lungs of mice. The expression of RGS12 was undetectable in the liver, heart, kidneys, and skeletal muscles of mice and in human stromal cells (Fig. 1B), indicating that RGS12 was prominently and fairly selectively expressed in OLCs. These results were further confirmed by RT-PCR in RAW264.7 cells with the induction of different doses of RANKL. As shown in Fig. 1C, the expression of RGS12 is dose-dependent to RANKL. To analyze RGS12 expression in osteoclasts at the protein level, we examined the expression of RGS12 in OLCs derived from RANKL-induced BMMs. The result indicated that RGS12 is prominently expressed in mouse OLCs but is undetectable in BMMs without RANKL stimulation (Fig. 1D). Consistent with this result, Western blotting analysis revealed that the expression of RGS12 is undetectable in BMMs without RANKL stimulation and dominant at 2 h after RANKL/M-CSF induction. In addition, compared with the expression of RGS12, the prominent expression of NFAT2 begins at 24 h and reaches its peak at 72 h after RANKL induction (Fig. 1E). Our results show that RGS12 is prominently expressed in RANKL-induced mouse OLCs and human authentic os-teoclasts at both the mRNA and protein levels, and RGS12 plays its role upstream of NFAT2 during osteoclast differentiation in vitro.
Fig. 1.
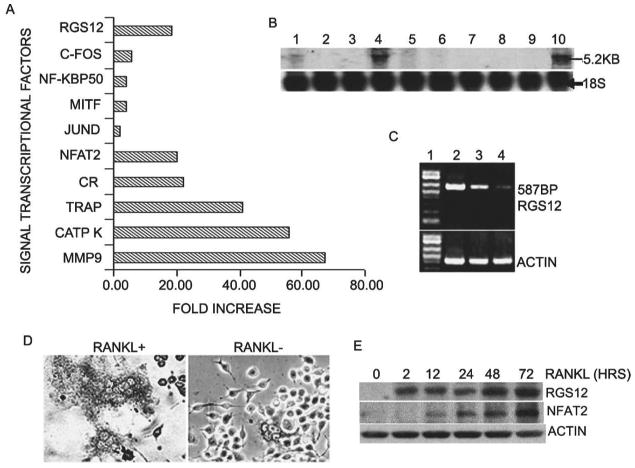
RGS12 is prominently expressed in osteoclasts and osteoclast precursors induced by RANKL/M-CSF. (A) Genome-wide screening of RANKL-inducible genes by GeneChip. MOCP-5 cells were stimulated for 96 h with and without RANKL (10 ng/ml) and M-CSF (20 ng/ml). mRNA of osteoclast marker genes (such as TRACP, calcitomin receptor [CR], cathepsin K [CATP K], matrix metalloprotease [MMP-9], and NFAT2) was strongly induced by RANKL in osteoclasts, confirming the validity of our screening protocol. Compared the other factors (c-fos, NF-kBp50, etc.), which are involved in osteoclast differentiation, RGS12 was significantly induced by RANKL. (B) RNA blotting analysis of RGS12 mRNA. Total RNA was extracted from mouse and human tissue. Lane 1, mSpleen; lane 2, mLiver; lane 3, mHeart; lane 4, mBrain; lane 5, mCalvaria; lane 6, mLung; lane 7, mKidney; lane 8, mMuscle; lane 9, human stromal cells; lane 10, human osteoclasts. (C) RT-PCR analysis of RGS12 mRNA in OLCs derived from RAW264.7 cells induced with 0, 5, and 10 ng/ml of RANKL, respectively, for 96 h. Lane 1, 1-kb plus marker; lane 2, RANKL 10 ng/ml; lane 3, RANKL 5 ng/ml; lane 4, no RANKL. (D) Immuno cell staining of RGS12 protein in BMM cells stimulated with RANKL (10 ng/ml)/M-CSF (20 ng/ml) for 96 h to produce OLCs. RGS12 is prominently expressed in OLCs. (E) Western blotting analysis for the expression of RGS12 and NFAT2 in RANKL-induced OLCs at the indicated time. The expression of RGS12 is undetectable in BMMs without RANKL stimulation and dominant at 2 h after RANKL/M-CSF induction. The prominent expression of NFAT2 begins at 24 h and reaches its peak at 72 h after RANKL induction.
RGS12 silencing blocks RANKL-induced osteoclast differentiation in osteoclast precursor cell line
To study the role of the RGS12 in osteoclast differentiation, we used vector-based RNAi gene silencing technology in RAW264.7 cells.(24) After stable transfection with RGS12 RNAi constructs, we performed a quick screening analysis of shRNA expression with TRACP staining and chose for passage the colonies carrying three constructs (pAVU-R12a, pAVU-R12b, and pAVU-R12c), which had inserts that were located at 298–318, 671–691, and 2182– 2200 bp (Fig. 2A). To assess silencing efficiency, we performed RT-PCR (Fig. 2B), Western blotting (Fig. 2C, lane 1–4), and immuno cell staining (Fig. 2D). As shown in Figs. 2A–2D, RGS12 mRNA transcription and protein expression were silenced in stably transfected RGS12-silenced cell lines after RANKL stimulation compared with control cells transfected with the pAVU-scrambled vector. Cells stably transfected with pAVU-scrambled vector, pAVU-R12a, pAVU-R12b, and pAVU-R12c, were induced with RANKL/M-CSF for 96 h. Approximately 80% of the cells differentiated into TRACP+ MNCs cells when transfected with the control vector (Fig. 3A). In contrast, only 3–4% of the cells were induced to differentiate into TRACP+ MNCs cells with pAVU-R12a, pAVU-R12b, or pAVU-R12c transfection (Fig. 3A). The number of TRACP+ MNCs in RGS12-silenced groups was 18.7 times lower than that of the control groups (Fig. 3B), indicating that RGS12 silencing almost completely blocks osteoclast differentiation from osteoclast precursor cells.
Fig. 2.
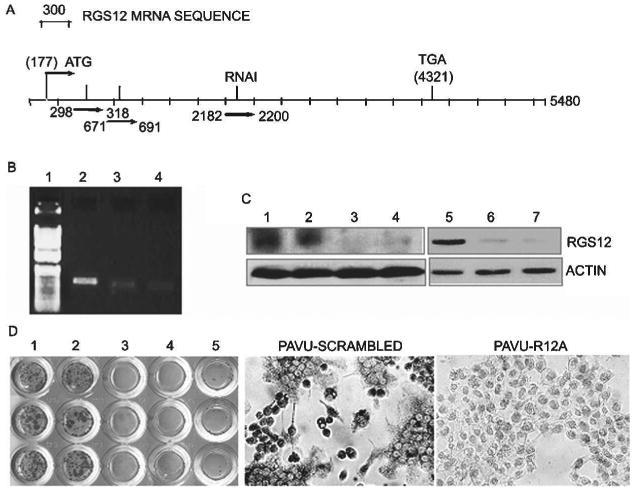
RGS12 RNAi blocks RGS12 expression. (A) Oligonucleotides for the constructs are located at sites 298–318, 671–691, and 2182–2200. (B) RT-PCR analysis of RGS12 transcription in RAW264.7 cells induced by RANKL (10 ng/ml)/M-CSF (20 ng/ml) for 96 h with or without RGS12 silencing. The RGS12 gene was strongly detected in control cells (lane 2) and weakly detected in RGS12-silenced cells (lanes 3 and 4). Lane 1, 1-kb plus marker. (C) Western blotting of RGS12 protein in RGS12-silenced RAW264.7 cells (lanes 1–4) and BMMs (lanes 5–7) stimulated with RANKL (10 ng/ml)/M-CSF (20 ng/ml) for 96 h. The signals were strong in control cells (lanes 1, 2, and 5) and weak in RGS12-silenced cells (lanes 3, 4, 6, and 7). Lane 1, mock; lane 2, pAVU6-scrambled shRNA; lane 3, pAVU-R12a; lane 4, pAVU-R12b; lane 5, pLenti-scrambled shRNA; lane 6, pLenti-R12a; lane 7, pLenti-R12b. (D) Immuno cell staining of RGS12 protein in control (1, mock; 2, pAVU-scrambled) or RGS12 RNAi expression vectors (3, pAVU-R12a; 4, pAVU-R12b; 5, pAVU-R12c) induced by RANKL. RGS12 RNAi blocks osteoclast differentiation.
Fig. 3.
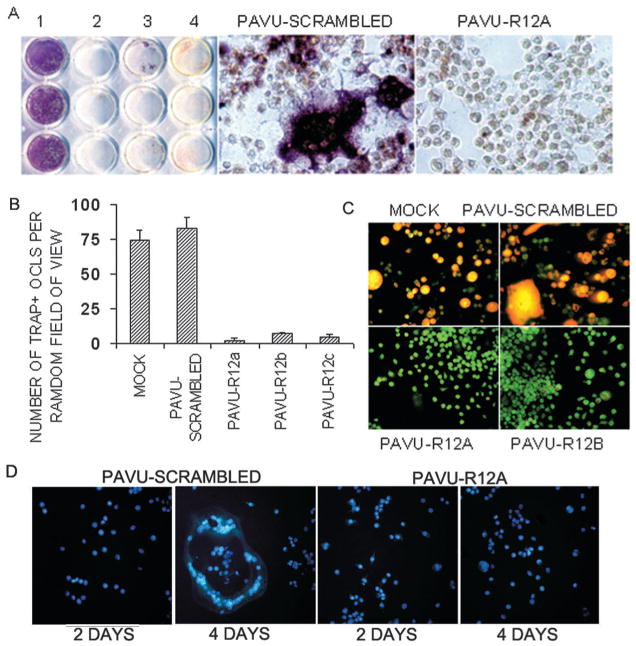
RGS12 silencing blocks RANKL-induced osteoclast differentiation in osteo-clast precursor cell lines. (A) TRACP+ MNCs were formed in RANKL-induced RAW264.7 cells transfected with pAVU-scrambled shRNA but not in the cells trans-fected with pAVU-R12a, b, or c. (B) Quantitative analysis of TRACP+ MNCs as in A. TRACP+ MNCs in the pAVU-scrambled group are >18.2 times (p < 0.05) that in the pAVU-R12a, b, and c groups. (C) Acridine orange staining. Strong orange fluorescence indicates extracellular acidification in the mock or pAVU-scrambled, but not in pAVU-R12a or pAVU-R12b. (D) Hoechst 33258 staining, apoptotic changes in the nuclei. There was no significant difference in survival between normal cells and silencing cells or multinucleated cells formed from RGS12-silenced cells.
To characterize the effects on osteoclast function, we studied whether extracellular acid compartments were formed beneath RGS12-silenced cells. Acridine orange staining, indicative of acid transport, showed strong orange fluorescence in control cells but not in the RGS12-silenced cells (Fig. 3C). This result shows that the RGS12-silenced cells induced by RANKL had lost their extracellular acidification function.
Because RANKL is a survival factor for osteoclasts, the question remains whether RGS12 is also required for cell survival. Accordingly, pAVU-scrambled and pAVU-R12a– transfected stable cells were, respectively, stimulated with RANK/M-CSF for 48 and 96 h as indicated. Using Hoechst 33258 staining, we detected characteristic apoptotic changes in the nuclei. There was no significant difference in survival between normal cells and silencing cells or multinucleated cells formed from RGS12-silenced cells as showed in Fig. 3D.
RGS12 silencing blocks osteoclast differentiation in authentic osteoclasts
To confirm our findings on authentic osteoclasts derived from primary bone marrow cells induced by RANKL, we used the same RNAi sequences at sites of 298–318 (a) and 671–691 bp (b) to generate recombinant lentiviruses carrying pLenti-scrambled shRNA, denoted as pLenti-scrambled, and RGS12 shRNA, denoted as pLenti-R12a and pLenti-R12b. The effect of silencing RGS12 expression was confirmed by Western blotting (Fig. 2C, lanes 6 and 7). Similar to the results with the osteoclast precursor cell line, as many as 80% of the cells differentiated into TRACP+ MNCs when infected with control viruses, but only 5% with pLenti-R12a or pLenti-R12b (Figs. 4A and 4B). To determine the effect of RGS12 RNAi on osteoclast gene expression, we also examined the expression of osteoclast marker genes, Cathepsin K and Atp6i, using immunofluorescent staining. As shown in Fig. 4C, RGS12 silencing blocked both Cathepsin K and Atp6i gene expression. To further characterize the bone-resorbing activity of TRACP+ MNCs, we performed a bone resorption assay. As shown in Fig. 4D, pLenti-scrambled–infected cells underwent differentiation into TRACP+ cells, which are multinucleated and have bone-resorbing activity; however, a few pLenti-R12a– infected cells differentiated into TRACP+ but have no bone-resorbing activity on dentine slices.
Fig. 4.
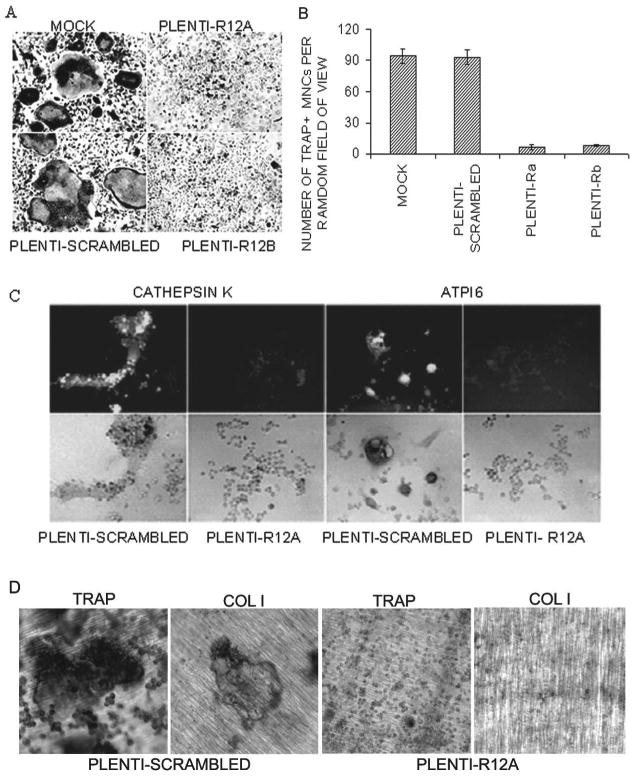
RGS12 silencing blocks RANKL-induced osteoclast differentiation in primary bone marrow cells. (A) Formation of TRACP+ MNCs in RANKL-induced BMMs infected with pLenti-scrambled and the absence of TRACP+ MNCs in the cells infected with pAVU-R12a or pAVU-R12b are shown. (B) Quantitative analysis of TRACP+ MNCs as in Fig. 3C, pLenti-R12a or b vs. RGS12-silenced groups (p < 0.05). (C) Immunostaining of cathepsin K and Atp6i in RGS12-silenced BMMs stimulated with RANKL/M-CSF. RGS12 silencing blocks the expression of cathepsin K and Atp6i as shown in pLenti-R12a groups. (D) Bone resorption assay. pLenti-scrambled infected cells underwent differentiation into TRACP+ cells, which are multinucleated and have bone-resorbing activity; however, a few pLenti-R12a infected cells differentiated into TRACP+, but have no bone-resorbing activity on dentine slices.
RGS12 silencing impairs osteoclast differentiation by blocking the RANKL-evokingPLCγ–[Ca2+]i oscillation–NFAT2 signaling pathway
The above observations suggest that RGS12 is essential for RANKL-induced osteoclast differentiation. Because other RGS proteins play an important role in the regulation of [Ca2+]i oscillations in acinar cells,(25) T lymphocytes,(26) neurons,(27) and cardiac myocytes,(28) we hypothesized that RGS12 may be a specific regulator of [Ca2+]i oscillations during osteoclast differentiation. To test this hypothesis, we visualized [Ca2+]i fluctuations using confocal microscopy in the cells with pAVU-scrambled or pAVU-R12a after RANKL induction. [Ca2+]i changes in cells co-loaded with fluo-4 and Fura Red were estimated as the ratio of fluorescence intensity of the fluo-4 to Fura Red, and the percent maximum ratio increase was plotted with an interval of 5 s. We found a sustained [Ca2+]i oscillation was initiated as late as 24 h after RANKL induction and was sustained thereafter, provided that RANKL was present (data not shown). As shown in Figs. 5A and 5B, in control (pAVU-scrambled) cells, sustained oscillations occurred at a frequency of ∼2-minute intervals. However, in the RGS12-silenced cells, [Ca2+]i oscillations were completely blocked, indicating that RGS12 is an essential regulator of this function.
Fig. 5.
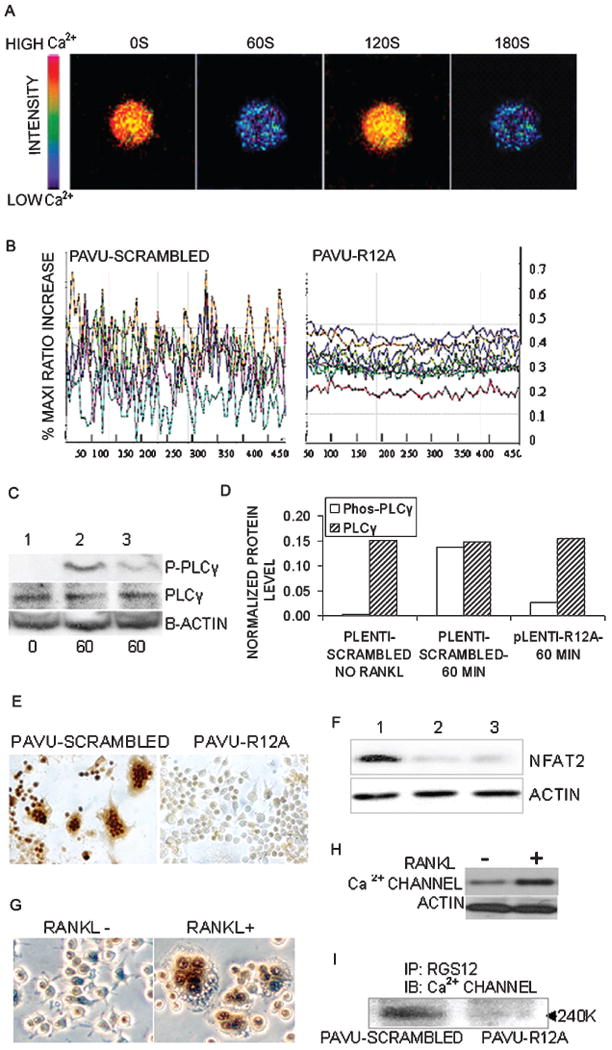
RGS12 silencing impairs osteoclast differentiation by blocking the PLCγ–[Ca2+]i oscillation–NFAT2 signaling pathway. (A and B) [Ca2+]i oscillations were observed in RANKL-induced BMMs or RAW264.7 cells ∼24–72 h after RANKL/M-CSF stimulation. [Ca2+]i oscillations was initiated as late as 24 h after RANKL/M-CSF induction and sustained thereafter with RANKL/M-CSF induction. [Ca2+]i changes were estimated as the ratio of fluorescence intensity of the fluo-4 to fura red plotted at 5-s intervals. (A) Example of successive pseudocolored Ca2+ images of normal cells treated with RANKL/M-CSF for 48 h. The cells show oscillations with a frequency of ∼2-minute intervals. (B) Ca2+ changes were traced in RGS12-silenced or control cells treated with RANKL/M-CSF for 72 h. Each color indicates a different cell in the same field. [Ca2+]i oscillations are blocked in RGS12-silenced cells (pAVU-R12a). (C) Western blotting. BMMs were infected with lentiviruses carrying pLenti-scrambled shRNA (lanes 1 and 2) or pLenti-R12a (lane 3) and stimulated with RANKL/M-CSF for 0 or 60 minutes. RGS12 silencing impaired phosphorylation of PLCγ1 (lane 3). (D) Quantification of PLCγ and Phosphor-PLCγ levels from immunblots, as in C. The protein levels of PLCγ and Phosphor-PLCγ were normalized to (β-actin. The levels of PLCγ were not significantly different between the RQS12-silenced and nonsilenced groups; however, the level of Phosphor-PLCγ in the silenced group (pLenti-R12a-60min) stimulated with RANKL/M-CSF was 5.1 times lower than that in the control (pLenti-scrambled-60min) at 60 minutes. (E) Immunoperoxidase cell staining revealed that the expression of NFAT2 was blocked in RGS12-silenced cells induced with RANKL/M-CSF for 96 h. (F) There were weak signals of NFAT2 protein detected in RGS12-silenced cells (lane 2, pAVU-R12a; lane 3, pAVU-R12b) compared with the control (lane 1, pAVU-scrambled). (G) Immuno cell staining revealed that the expression of N-type calcium channels is increased in RANKL-induced OLCs (pAVU-R12a). (H) Western blotting of α1B N-type calcium channel protein in OLCs. BMMs were treated with RANKL/M-CSF for 96 h. RANKL increased the expression of α1B N-type calcium channels. (I) Precipitation of RGS12 followed by Western blotting with an antibody against the α1B-subunit of the calcium channel. RGS12 directly interacts with the α1B-subunit of the calcium channel in RANKL-induced OLCs (pAVU-scrambled).
To gain further insight into the molecular mechanisms underlying RANKL-initiated [Ca2+]i oscillations, we analyzed the activation of PLCγ, which is a key participant in the PLCγ– [Ca2+]i oscillatioin–NFAT2 pathway.(12) BMMs were infected with pLenti-scrambled or pLenti-R12a viruses and induced with RANKL/M-CSF for 60 minutes. Activation of PLCγ was detected using anti-PLCγ and antiphospho-PLCγ antibodies. The results showed that RANKL-induced phosphorylation of PLCγ was impaired in RGS12-silenced cells (Figs. 5C and 5D).
To determine whether RGS12 silencing affects NFAT2 expression, we examined the expression of NFAT2 protein in RANKL/M-CSF-induced RGS12-silenced cells and control cells using immuno cell staining and Western blotting. As shown in Fig. 5E, the expression of NFAT2 was significantly blocked in RGS12-silenced cells, whereas it was highly expressed in control cells. The Western blotting result (Fig. 5F) showed that the expression of NFAT2 in RGS12-silenced cells was much lower than that in control cells, indicating that RGS12 acts upstream of NFAT2 in the RANKL-induced PLCγ– [Ca2+]i oscillation–NFAT2 pathway during osteoclast differentiation.
RGS12 interacts with the N-type calcium channel in OLCs
Recent evidence has shown that influx of extracellular Ca2+ through multiple channels, which include L- and N-type channels, is necessary for Ca2+ oscillations.(29–31) Previous studies of the involvement of RGS12 in modulating presynaptic GABAB-receptor signaling suggest that RGS12 is capable of direct interaction with the tyrosine-phosphorylated N-type calcium channel through its PTB domain. It modulates channel activity directly and accelerates G-subunit GTPase activity.(32–35)) Based on the literature and our data, we hypothesized that RGS12 may regulate the [Ca2+]i oscillation–NFAT2 pathway through N-type calcium channels. By examining the expression of N-type calcium channels in RANKL-induced RAW264.7 cells, we found that RANKL is able to increase the expression of N-type calcium channels in OLCs as shown in Figs. 5G and 5H. To further analyze whether RGS12 interacts with one of its binding partners, we examined the interaction of RGS12 with α1B N-type calcium channels by co-immunoprecipitation. As shown in Fig. 5I, RGS12 interacts directly with phosphorylated N-type calcium channels in OLCs, indicating that RGS12 regulates [Ca2+]i oscillations and osteoclast differentiation possibly through calcium channels.
Discussion
How RANKL evokes [Ca2+]i oscillations and leads to osteoclast differentiation is unclear. Several lines of evidence have shown that RGS proteins play an important part in the regulation of [Ca2+]i oscillations.(36–38) Luo et al. (25) reported that RGS proteins provide biochemical controls of agonist-evoked [Ca2+]i oscillations. [Ca2+]i oscillations could be evoked by G-protein–coupled receptors (GPCRs) and require the action of RGS proteins. Inhibition of endogenous RGS protein action disrupted agonist-evoked [Ca2+]i oscillations by a stepwise conversion to a sustained response. Dolmetsch et al.(26) reported that [Ca2+]i oscillations regulate gene expression and cell differentiation in T cells. Rapid oscillations stimulate all three transcriptional factors (NFAT, Oct/OAP, NF-κB), whereas infrequent oscillations activate only NF-κB. We have found that one of these proteins, RGS12, is prominently expressed in RANKL-induced osteoclast-like cells. Our further results provide the first evidence that RGS12 is involved in the RANKL-evoked signaling pathway as a critical regulator of [Ca2+]i oscillations and an essential factor in the terminal differentiation of osteoclasts induced by RANKL.
It is as yet unknown how RANKL induces the expression of RGS12 and how the RGS12 goes on to regulate the [Ca2+]i oscillations that finally lead to NFAT2 expression and activation and osteoclast differentiation from monocyte progenitor cells. It has been shown that RGS proteins share the conserved structure of ∼120 amino acids, called an RGS domain, which is responsible for their GAP activity.(39) RGS proteins act as GTPase-activating proteins (GAPs) specific to the Gα subunit of the G-protein and play a crucial role in shutting off G-protein–mediated cell responses in eukaryotes. Occupation of receptor with agonist initiates a cycle of inhibition of RGS protein action by binding to phosphoinositide (PIP3) and relief of the inhibition by binding to Ca2+-CaM to promote cyclical activation of PLC, inosital 1,4,5-trisphosphate (IP3) production, and Ca2+ release. RGS cyclically regulates [Ca2+]i signaling to lead to production of [Ca2+]i oscillations.(25) RGS12 is the largest member of the RGS protein family and has the potential to serve as a signal transduction “nexus” that modulates multiple signaling pathway components by virtue of its multidomain architecture. It contains the RGS domain, the Go Loco motif, a pair of Ras-binding domains (RBDs),(40) an N-terminal PDZ (PSD-95/Dlg/ZO-1) domain, and a phosphotyrosine-binding (PTB) domain.(32) Our results showed that silencing of RGS12 impaired phosphorylation of PLCγ, blocked [Ca2+]i oscillations, and inhibited NFAT2 expression and osteoclast differentiation, suggesting RGS12 may be involved in PLCγ activation and [Ca2+]i oscillations evoked by the GAP activity of its RGS domain.
Some studies(29–31) have shown that initiation and maintenance of [Ca2+]i oscillations need not only Ca2+ release from internal stores of endoplasmic reticulum (ER) but also influx of extracellular Ca2+ through multiple channels, which include L- and N-type channels. A large body of evidence(41–46) has suggested that agonist binding to receptor initiates activation of PLC and IP3 production, which promotes a brief spike of [Ca2+]i increase by depleting ER Ca2+ store; store depletion is sensed by an as yet uncharacterized signaling mechanism that triggers a plasma membrane calcium entry pathway through calcium channels. Schiff et al.(32) showed that, in chick dorsal root ganglion neurons, the PTB domain of RGS12 interacts directly with the N-type calcium channel, which is tyrosine phosphorylated in response to GABAB receptor stimulation.(33) They further found that RGS12 and the N-type calcium channel form a complex in a src-like kinase-dependent manner. In the presence of genistein, a tyrosine kinase inhibitor, the calcium channel did not co-precipitate with RGS12. Similarly, our data showed that the expression levels of RGS12 and tyrosine phosphorylated N-type calcium channels are increased in RANKL-induced OLCs and that RGS12 directly interacted with the tyrosine phosphorylated N-type calcium channels. In addition, we found that the expression of RGS12 is dominant at 2 h after RANKL induction, whereas [Ca2+]i oscillations appeared between 24 and 72 h after RANKL induction and that RGS12 silencing blocked [Ca2+]i oscillations and osteoclast differentiation. These results suggested that, before [Ca2+]i oscillations begin, a signaling protein is induced by RANKL to regulate [Ca2+]i oscillations (i.e., RGS12). RGS12 interacts with N-type calcium channels to regulate the transient changes in intracellular calcium, leading to the generation of [Ca2+]i oscillations and triggering osteoclast differentiation.
Based on these results and cited works, it is reasonable to propose that RGS12 may play dual functions in regulating [Ca2+]i oscillations and triggering osteoclast differentiation, such as (1) RGS12 protein acts through an interaction between the RGS domain of RGS12 and G-protein to promote cyclical activation of IP3 and Ca2+ release from ER; and (2) RGS12 interacts with tyrosine-phosphorylated N-type Ca2+ channel by binding its PTB domain and regulates extracellular Ca2+ influx. Whether either or both of these functions are in operation needs to be further determined in vitro by functional dissection of RGS12 domains and in vivo gene knockout, which are being studied in our laboratory.
Acknowledgments
We thank Lukas Cheney and Carrie Soltanoff for assistance with the manuscript. This work was supported by NIH Grant AR-44741 (Y-PL), AR-48133 (Y-PL), and ED016857 (SY).
Footnotes
The authors state that they have no conflicts of interest.
References
- 1.Alliston T, Derynck R. Medicine: Interfering with bone remodelling. Nature. 2002;416:686–687. doi: 10.1038/416686a. [DOI] [PubMed] [Google Scholar]
- 2.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447–451. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 3.Matsuo K, Owens JM, Tonko M, Elliott C, Chambers TJ, Wagner EF. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. 2000;24:184–187. doi: 10.1038/72855. [DOI] [PubMed] [Google Scholar]
- 4.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 5.Wagner EF, Karsenty G. Genetic control of skeletal development. Curr Opin Genet Dev. 2001;11:527–532. doi: 10.1016/s0959-437x(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 6.Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC. The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA. 2004;101:6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 8.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 10.Teitelbaum SL. RANKing c-Jun in osteoclast development 15. J Clin Invest. 2004;114:463–465. doi: 10.1172/JCI22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 12.Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- 13.Chen W, Li YP. Generation of mouse osteoclastogenic cell lines immortalized with SV40 large T antigen. J Bone Miner Res. 1998;13:1112–1123. doi: 10.1359/jbmr.1998.13.7.1112. [DOI] [PubMed] [Google Scholar]
- 14.Flanagan AM, Nui B, Tinkler SM, Horton MA, Williams DM, Chambers TJ. The multinucleate cells in giant cell granulomas of the jaw are osteoclasts. Cancer. 1988;62:1139–1145. doi: 10.1002/1097-0142(19880915)62:6<1139::aid-cncr2820620617>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Chambers TJ, Fuller K, Darby JA, Pringle JA, Horton MA. Monoclonal antibodies against osteoclasts inhibit bone resorption in vitro. Bone Miner. 1986;1:127–135. [PubMed] [Google Scholar]
- 16.Li YP, Alexander M, Wucherpfennig AL, Yelick P, Chen W, Stashenko P. Cloning and complete coding sequence of a novel human cathepsin expressed in giant cells of osteoclastomas. J Bone Miner Res. 1995;10:1197–1202. doi: 10.1002/jbmr.5650100809. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate- phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Yang S, Wei D, Wang D, Phimphilai M, Krebsbach PH, Franceschi RT. In vitro and in vivo synergistic interactions between the Runx2/Cbfa1 transcription factor and bone morphogenetic protein-2 in stimulating osteoblast differentiation. J Bone Miner Res. 2003;18:705–715. doi: 10.1359/jbmr.2003.18.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly KA, Tanaka S, Baron R, Gimble JM. Murine bone marrow stromally derived BMS2 adipocytes support differentiation and function of osteoclast-like cells in vitro. Endocrinology. 1998;139:2092–2101. doi: 10.1210/endo.139.4.5915. [DOI] [PubMed] [Google Scholar]
- 20.Kurland JI, Kincade PW, Moore MA. Regulation of Blymphocyte clonal proliferation by stimulatory and inhibitory macrophage-derived factors. J Exp Med. 1977;146:1420–1435. doi: 10.1084/jem.146.5.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baron R, Neff L, Louvard D, Courtoy PJ. Cell-mediated extracellular acidification and bone resorption: Evidence for a low pH in resorbing lacunae and localization of a 100-kD lysosomal membrane protein at the osteoclast ruffled border. J Cell Biol. 1985;101:2210–2222. doi: 10.1083/jcb.101.6.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kameda T, Miyazawa K, Mori Y, Yuasa T, Shiokawa M, Nakamaru Y, Mano H, Hakeda Y, Kameda A, Kumegawa M. Vitamin K2 inhibits osteoclastic bone resorption by inducing osteoclast apoptosis. Biochem Biophys Res Commun. 1996;220:515–519. doi: 10.1006/bbrc.1996.0436. [DOI] [PubMed] [Google Scholar]
- 23.Wu X, McKenna MA, Feng X, Nagy TR, McDonald JM. Osteoclast apoptosis: The role of Fas in vivo and in vitro. Endocrinology. 2003;144:5545–5555. doi: 10.1210/en.2003-0296. [DOI] [PubMed] [Google Scholar]
- 24.Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo X, Popov S, Bera AK, Wilkie TM, Muallem S. RGS proteins provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol Cell. 2001;7:651–660. doi: 10.1016/s1097-2765(01)00211-8. [DOI] [PubMed] [Google Scholar]
- 26.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 27.Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- 28.Olson EN, Williams RS. Calcineurin signaling and muscle remodeling. Cell. 2000;101:689–692. doi: 10.1016/s0092-8674(00)80880-6. [DOI] [PubMed] [Google Scholar]
- 29.Kohda M, Komori S, Unno T, Ohashi H. Carbacholinduced [Ca2+]i oscillations in single smooth muscle cells of guinea-pig ileum. J Physiol. 1996;492:315–328. doi: 10.1113/jphysiol.1996.sp021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am J Physiol. 1997;272:C966–C975. doi: 10.1152/ajpcell.1997.272.3.C966. [DOI] [PubMed] [Google Scholar]
- 31.Gao ZY, Chen M, Collins HW, Matschinsky FM, Lee VM, Wolf BA. Mechanisms of spontaneous cytosolic Ca2+ transients in differentiated human neuronal cells. Eur J Neu-rosci. 1998;10:2416–2425. doi: 10.1046/j.1460-9568.1998.00252.x. [DOI] [PubMed] [Google Scholar]
- 32.Schiff ML, Siderovski DP, Jordan JD, Brothers G, Snow B, De Vries L, Ortiz DF, Diverse-Pierluissi M. Tyrosine-kinasedependent recruitment of RGS12 to the N-type calcium channel. Nature. 2000;408:723–727. doi: 10.1038/35047093. [DOI] [PubMed] [Google Scholar]
- 33.Richman RW, Strock J, Hains MD, Cabanilla NJ, Lau KK, Siderovski DP, verse-Pierluissi M. RGS12 interacts with the SNARE-binding region of the Cav2.2 calcium channel. J Biol Chem. 2005;280:1521–1528. doi: 10.1074/jbc.M406607200. [DOI] [PubMed] [Google Scholar]
- 34.Richman RW, verse-Pierluissi MA. Mapping of RGS12-Cav2.2 channel interaction. Methods Enzymol. 2004;390:224–239. doi: 10.1016/S0076-6879(04)90015-8. [DOI] [PubMed] [Google Scholar]
- 35.Anantharam A, verse-Pierluissi MA. Biochemical approaches to study interaction of calcium channels with RGS12 in primary neuronal cultures. Methods Enzymol. 2002;345:60–70. doi: 10.1016/s0076-6879(02)45007-0. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Huang G, Luo X, Penninger JM, Muallem S. Role of regulator of G protein signaling 2 (RGS2) in Ca(2+) oscillations and adaptation of Ca(2+) signaling to reduce excitability of RGS2−/− cells. J Biol Chem. 2004;279:41642–41649. doi: 10.1074/jbc.M406450200. [DOI] [PubMed] [Google Scholar]
- 37.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 38.Ishii M, Inanobe A, Kurachi Y. PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltagedependent control of the G protein cycle in a cardiac K+ channel. Proc Natl Acad Sci USA. 2002;99:4325–4330. doi: 10.1073/pnas.072073399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishii M, Kurachi Y. Physiological actions of regulators of G-protein signaling (RGS) proteins. Life Sci. 2003;74:163–171. doi: 10.1016/j.lfs.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Ponting CP. Raf-like Ras/Rap-binding domains in R. J Mol Med. 1999;77:695–698. doi: 10.1007/s001099900054. [DOI] [PubMed] [Google Scholar]
- 41.Berridge MJ, Lipp P, Bootman MD. Signal transduction. The calcium entry pas de deux. Science. 2000;287:1604–1605. doi: 10.1126/science.287.5458.1604. [DOI] [PubMed] [Google Scholar]
- 42.Parekh AB, Terlau H, Stuhmer W. Depletion of InsP3 stores activates a Ca2+ and K+ current by means of a phosphatase and a diffusible messenger. Nature. 1993;364:814–818. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- 43.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 44.Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 45.Putney JW, Jr, Bird GS. The signal for capacitative calcium entry. Cell. 1993;75:199–201. doi: 10.1016/0092-8674(93)80061-i. [DOI] [PubMed] [Google Scholar]
- 46.Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]