

Abstract
Purpose
Screening of mutations in the paired box 3 (PAX3) gene in three generations of a Turkish family with Waardenburg syndrome type 1 (WS1).
Methods
WS1 was diagnosed in a 13-month-old girl according to the WS Consortium criteria. Detailed family history of the proband revealed eight affected members in three generations. Routine clinical and audiological examination and ophthalmologic evaluation were performed on eight affected and five healthy members of the study family. Dystopia canthorum was detected in all affected patients; however, a brilliant blue iris was present in five patients who also had mild retinal hypopigmentation. Genomic DNA was extracted from the peripheral blood of affected and unaffected individuals in the family as well as 50 unrelated healthy volunteers. All coding exons and adjacent intronic regions of PAX3 were sequenced directly.
Results
A novel missense heterozygous c.788T>G mutation was identified in eight patients. This nucleotide alteration was not found in unaffected members of the study family or in the 50 unrelated control subjects. The mutation causes V263G amino-acid substitution in the homeodomain of the PAX3 protein, which represents the 45th residue of helix 3.
Conclusions
We identified a novel missense c.788T>G mutation in PAX3 in a family with Waardenburg syndrome with intrafamilial phenotypic heterogeneity.
Introduction
Waardenburg syndrome (WS), which equally affects both sexes and all races, is an inherited disorder characterized by varying degrees of sensorineural deafness and pigmentary abnormalities affecting the skin, hair, and eye with an incidence of 1 in 40,000 [1,2]. Five major and five minor diagnostic criteria for WS were proposed by the Waardenburg Consortium [3]. Two major or one major and two minor criteria must be found in an individual to diagnose WS [3,4]. WS is classified into four major types depending on the presence or absence of dystopia canthorum and additional symptoms [1,5-8].
WS shows genetic heterogeneity. WS1 and WS3 are caused by mutations in the paired box 3 (PAX3) gene [9-12]. WS2 is due to mutations in the microphthalmia-associated transcription factor (MITF) gene [13-17] and the encoding snail homolog 2 (SNAI2) gene [18]. However, the molecular etiology of most patients with WS2 is still unknown [19]. WS4 is associated with mutations in the endothelin receptor type B (EDNRB) gene [17,20,21], the endothelin-3 (EDN3) gene [22-25], and the SRY (sex determining region Y)-box 10 (SOX10) gene [6,7,19,24]. Recently, deletions in SOX10 were also identified in patients with WS2 and WS4 [26].
PAX3, located on the long arm of chromosome 2 (2q35), includes ten exons [27]. This gene is a member of the mammalian PAX gene family that encodes for DNA-binding transcription factors and plays a role in maintaining stem cell pluripotency, cell-lineage specification, proliferation, migration, apoptosis, and inhibition of terminal differentiation [28,29]. PAX3 is expressed in neural crest cells including the spinal ganglia, the craniofacial mesectoderm, and the limb mesenchyme during embryogenesis and plays an important role in the migration and differentiation of melanocytes, which originate from the embryonic neural crest [17,28].
More than 70 pathogenic PAX3 mutations including missense, nonsense, and frameshifts mutations, small insertions or deletions, and splice alterations have been reported in patients with WS1 [17]. Most PAX3 mutations are located in exon 2; no mutations have been described in exons 9 and 10. Approximately 50% of the described mutations of PAX3 are missense; the remaining are truncating variations [17]. Partial or total gene deletions have been reported in 10% of patients without identified point mutations [30-35]. The literature that studied patients with WS1 caused by mutations in PAX3 was reviewed in 2009 by Pingault et al., and no relationship was found between the severity of disease and the type of mutation [17]. In this report, clinical features and intrafamilial heterogeneity of eight affected individuals in a Turkish family with WS1 with a novel mutation are presented.
Methods
Family description
Eight affected including the proband, and 5 unaffected members of the study family, as well as 50 healthy volunteers were included in the present study. Informed consent conforming to the tenets of the Declaration of Helsinki, blood samples, and clinical evaluations were obtained from each study participant, under protocols approved by Dr. Behçet Uz Children's Hospital ethics committee (approval number and date: B-10-4-ISM-4-35-65-72; 29.03.2012/25). Informed consent conforming to the tenets of the Declaration of Helsinki, blood samples, and clinical evaluations were obtained from each study participant, under protocols approved by the Behçet Uz Children's Hospital ethics committee (approval number and date: B-10–4-ISM-4–35–65–72; 29.03.2012/25). The female patient was referred to us for genetic evaluation because of dysmorphic facial features at the age of 13 months. The proband and her family were evaluated at the Medical Genetics Clinic, Dr. Behçet Uz Children's Hospital, Izmir, Turkey. The proband (Patient IV:6) was diagnosed with WS1 according to the WS Consortium criteria [3]. Her family history revealed eight affected members in three generations (Figure 1). Routine clinical examination and detailed audiological and ophthalmologic evaluation were performed on eight affected and five healthy members of the study family. Two major or one major and two minor criteria must be found in an individual to diagnose WS. In four patients (IV:3, IV:4, IV:5, and IV:6), premature graying of the hair was not evaluated because of the patients’ youth.
Figure 1.
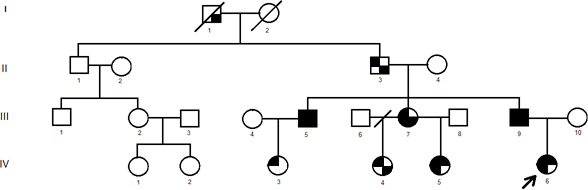
The pedigree of the family is shown. The squares indicate men, and the circles indicate women. Filled quadrants indicate phenotype associated with WS1. Upper left represents dystopia canthorum. Lower left represents brilliant blue iris. Upper right represents hearing loss. Lower right represents synophyris.
Hearing loss was assessed with pure tone audiometry or, in children, with brainstem-evoked response audiometry (BERA). Degree of hearing loss was computed by using a four-frequency average called the pure-tone average. The average of the hearing threshold levels (decibels) taken at 500 Hz, 1000 Hz, 2000 Hz, and 4000 Hz was calculated for each patient to find the pure-tone average level. Afterwards, hearing loss was graded according to the classification defined by J.G. Clark in 1981 [36]. Dystopia canthorum, which is characterized by an increase in the distance between the inner angles of the eyelids with normal distances between the pupils and the outer canthus, was defined for each affected member of the family with a W index that exceeds 1.95 [1].
Mutation analysis
Peripheral blood samples which were collected in EDTA tubes and stored fresh (-20 °C) were obtained from participating members of the family and 50 unrelated healthy volunteers. Genetic analyses were performed in Duzen Laboratory Groups, Genetics Division, Ankara, Turkey. Genomic DNA was extracted from leukocytes using the QIAamp DNA mini kit (Qiagen, 51304, Dusseldorf, Germany), according to manufacturer's instructions. Genomic fragments including coding regions and adjacent intronic regions of PAX3 were amplified with PCR, using nine primer pairs described previously [7] (Table 1). The amplicons from individual exons were purified and analyzed with cycle sequencing with ABI BigDye Terminator Cycle Sequencing Kit v3.1 (ABI Applied Biosystems, Foster City, CA) on an automatic DNA sequencer (ABI 3130 Genetic Analyzer, Applied Biosystems). Sequencing results from patients and the consensus sequences from the NCBI Human Genome Database (NCBI Reference Sequence: NG_011632.1) were imported into the ABI SeqScape program and aligned to identify variations. Each found mutation was confirmed with bidirectional sequencing. The mutation was named following the nomenclature recommended by the Human Genomic Variation Society.
Table 1. Primers for amplifying and sequencing PAX3 genomic fragments [7].
| Exon | Forward primers | Reverse primers | Product size (bp) |
|---|---|---|---|
| 1 |
5′ GATGGGAAGAGAAAGTGGTC 3′ |
5′ TGCAGAAAGGAAATCGAGTA 3′ |
788 |
| 2 |
5′ CCGATGTCGAGCAGTTTCAG 3′ |
5′ CGCACCTTCACAAACCTCAG 3′ |
503 |
| 3 |
5′ TGGGATGTGTTCTGGTCTG 3′ |
5′ TCCCAATAGCTGAGATCGA 3′ |
420 |
| 4 |
5′ CTGGAGAAGGATGAGGATGT 3′ |
5′ CGTCAGATCACCAATGTCAG 3′ |
383 |
| 5 |
5′ TACGGATTGGTTAGACTTGT 3′ |
5′ AACAATATGCATCCCTAGTAA 3′ |
508 |
| 6 |
5′ CAACACAGAAGGCAGAGA 3′ |
5′ ATAGGTACGTTCAGGACAA 3′ |
445 |
| 7 |
5′ TGTGCAGAGATAGGTGTGAC 3′ |
5′ TTTGATGAAGCCAGTAGGA 3′ |
586 |
| 8 |
5′ TCTCCTGGACAGCTCTTTAA 3′ |
5′ GGCATGTGTGGCTTAATCT 3′ |
480 |
| 9&10 | 5′ GGTCAGCTCCAGGATCATAT 3′ | 5′ GCAAATGGAATGTTCTAGCT 3′ | 580 |
Results
Phenotype analysis
Eight affected patients (five women [III:7, IV:3, IV:4, IV:5, and IV:6] and three men [II:3, III:5, and III:9]) and five nonaffected members (three women [III:2, IV:1, and IV:2] and two men [II:1 and III:1]) of three generations in the study family were enrolled in the present study (Figure 1). Case I:1 was not evaluated in this study as he died before enrollment. The diagnostic criteria for WS proposed by the Waardenburg Consortium and the clinical features of the study patients are shown in Table 2.
Table 2. Diagnostic criteria of WS, and clinical evaluation of affected family members. Two major, or one major and two minor criteria have to be found in an individual to be diagnosed as WS.
| DIAGNOSTIC CRITERIA | FEATURES | STUDY PARTICIPANTS |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| II-3 | III-5 | III-7 | III-9 | IV-3 | IV-4 | IV-5 | IV-6 | ||
| MAJOR
CRITERIA |
Sensorineural hearing loss |
- |
+ |
- |
+ |
- |
- |
+ |
+ |
| Iris pigmentary abnormality (heterochromia iridis, or segmentary heterochromia of the iris, or characteristic brilliant blue iris) |
- |
+ |
+ |
+ |
- |
- |
+ |
+ |
|
| Hair hypopigmentation (white forelock, white hairs at other sites on the body) |
- |
- |
- |
- |
- |
- |
- |
- |
|
| Dystopia canthorum |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
| First-degree relative previously diagnosed with Waardenburg syndrome |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
|
| MINOR CRITERIA | Skin hypopigmentation |
- |
- |
- |
+ |
- |
- |
- |
- |
| Synophrys |
+ |
+ |
+ |
+ |
- |
+ |
- |
- |
|
| Broad nasal root |
- |
- |
- |
- |
+ |
- |
+ |
+ |
|
| Hypoplasia alae nasi |
+ |
- |
+ |
- |
- |
- |
- |
+ |
|
| Premature graying of the hair (before the age of 30 years) | + | + | + | + | N/A | N/A | N/A | N/A | |
N/A: Not applicable
Dystopia canthorum was detected in all affected patients (Figure 2). A brilliant blue iris was present in five patients who also had mild retinal hypopigmentation (III:5, III:7, III:9, IV:5, and IV:6; Figure 2 and Figure 3). Orthoptic assessments were within normal limits, and nystagmus was not present in affected patients or nonaffected members of the family; however, astigmatic refractive errors were found frequently in all affected members. Synophrys was presented in five out of eight patients (II:3, III:5, III:7, III:9, and IV:4), and this finding was more prominent in two (II:3 and III:9; Figure 2). Clinical features of all unaffected family members (II:1, III:1, III:2, IV:1, and IV:2) were revealed as normal.
Figure 2.
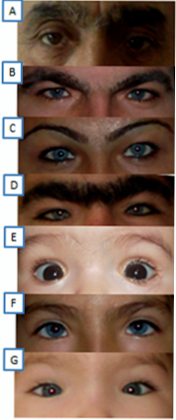
Photographs of eyes from patients with Waardenburg syndrome type 1. A: Dystopia canthorum (W index: 2.03) and synophyris were present in a 68-year old man (II:3). B: Dystopia canthorum (W index: 2.08), brilliant blue iris, and synophyris were present in a 32-year old man (III:5). C: Dystopia canthorum (W index: 1.96), brilliant blue iris, and synophyris were present in a 30-year old woman (III:7). D: Dystopia canthorum (W index: 2.35), brilliant blue iris, and synophyris were present in a 26-year old man (III:9). E: Dystopia canthorum (W index: 2.49) and broad nasal root were present in a 5-month old girl (IV:3). F: Dystopia canthorum (W index: 2.19), brilliant blue iris, and broad nasal root were present in a 10-year old girl (IV:5). G: Dystopia canthorum (W index: 2.39), brilliant blue iris, and broad nasal root. W index: 2.39.
Figure 3.
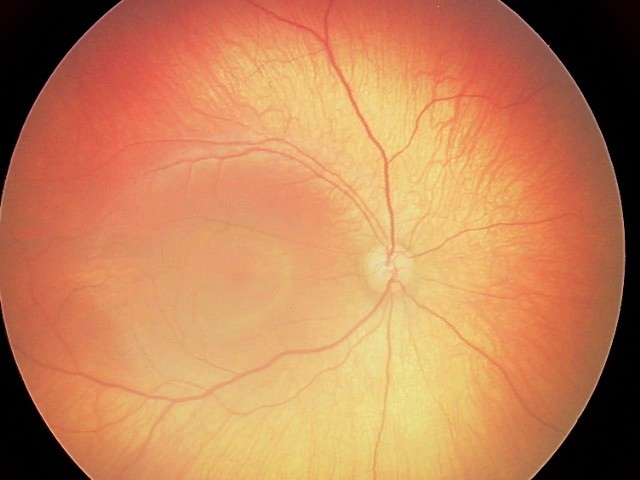
Retinal hypopigmentation was seen on the fundus photograph of the proband.
Four patients had a sensorineural hearing impairment. Three (III:5, IV:5, and IV:6) were profoundly and one (III:9) was moderately deaf. Out of the three profoundly deaf patients, two (IV:5 and IV:6) had received a cochlear implant. None had a white forelock, whereas premature graying of the hair (before the age of 30 years) was present in patients II:3, III:5, III:7, and III:9. Due to the patients’ youth, premature graying of the hair was not evaluated in four patients (IV:4 was 15 years old, IV:5 was 10 years old; IV:3 and IV:6 were younger than 2 years old). Hypopigmented patches of the skin were present in only one patient (III:9; Table 2). No patients had limb defects.
Mutation analysis
After direct sequencing of PAX3, a heterozygous change T>G in exon 5, at position 788 of the translation start site was detected in all affected patients (Figure 4A). This position belongs to helix 3 of the homeodomain in the PAX3 protein, and converts the 45th amino acid in this domain from valine to glycine. This mutation was not found in the other unaffected relatives (II:1, III:2, IV:1, and IV:2) in the healthy controls (Figure 4B). Case III:1 declined the molecular genetic analysis.
Figure 4.
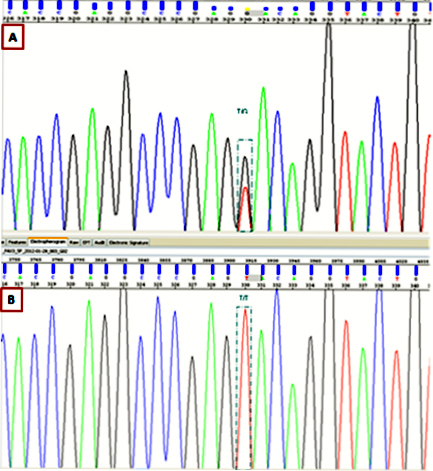
Sequence chromatography. A: The heterozygous change, c.788T>G, was identified in affected family members. B: Unaffected members and 50 healthy controls are wild-type at this position.
Discussion
A careful clinical evaluation is necessary to differentiate various types of WS. Dystopia canthorum is a commonly seen feature of WS with an incidence of 41.2% to 99% [1,2]. However, dystopia canthorum is also the most penetrant (%99) and the most distinguishing feature of WS1 [1,2,6,37,38]. All affected patients in the present study had dystopia canthorum.
In 2006, the frequencies of heterochromia iridis and synophrys were reported as 25% and 45% of WS1 patients, respectively [6]. However, incidence of synophrys in patients with WS1 was reported as 85% in a review published in 2009 [17]. Although a brilliant blue iris was present in five out of eight patients, heterochromia iridis was not detected in any members of the study family. However, synophrys, a minor criterion, was also present in five out of eight patients.
WS accounts for approximately 2% to 5% of congenital sensorineural deafness [30,39]. Congenital sensorineural deafness is a feature in approximately 25% to 75% of patients with WS1 [17,40,41]. Deafness related to WS can be moderate to severe, unilateral, or bilateral but most commonly non-progressive [40,42]. In our study, three patients had total bilateral severe sensorineural deafness, and one patient had moderate bilateral deafness. Hearing tests were normal in the other four affected patients and five unaffected members of the family. However, Wang et al. reported a lack of deafness among their Chinese patients with WS1, and indicated a possible ethnic specific variation in clinical expression of the syndrome [43]. The incidence of hearing loss among Turkish patients with WS1 was published as 75% by Oysu et al. in 2000 [40]. Hearing loss was present in 50% of our study patients with WS1, which is in accordance with the literature [17,30,39-42].
Mutations of PAX3 on chromosome 2q37 have been reported in 33% to 80% of patients with WS1 in familial and sporadic cases [3,7,13,44]. PAX3 is a transcription factor that plays a major role in embryogenesis [45]. By 2009, about 70 mutations of PAX3 related to WS had been introduced [17]. The PAX3 protein is a member of the family of paired domain proteins that bind DNA and regulate gene expression [7]. PAX3 encodes a paired domain and a homeodomain [6]. Missense mutations are almost exclusively located within the two DNA binding domains. One, the homeodomain, includes three α-helices. Most PAX3 mutations are located in the paired domain or in helix 3 of the homeodomain [6]. Helix 3 of the homeodomain makes sequence-specific DNA contacts and several phosphate contacts in the major groove [6,7]. Mutations that affect helix 3 of the homeodomain in the PAX3 protein may lead to a decrease in DNA binding affinity and/or specificity [7]. We identified the novel c.788T>G mutation in PAX3 leading to V→G substitution on the 45th position of helix 3.
The phenotypic variability among the eight affected patients with the same mutation is well matched with inheritance properties of the disease. PAX3 mutations are inherited dominantly with variable expressivity [43]. The disease is known as fully penetrant when at least one of its signs is detected, but the penetrance for each one is not complete. The fact that there is no obvious association between different types of mutations and the severity of disease could be due to the role of gene dosage as the pathophysiology of the syndrome. It is hypothesized that stochastic events were not solely responsible for its expression, so that genetic factors and/or the environmental background can modify the phenotype [17]. A few mutations of PAX3 have been tested for their functional consequences. The functional experiments mostly included DNA-binding activity and transactivation capabilities. Although missense mutations are thought to abolish PAX3 ability to bind and activate its transcriptional targets, further functional studies are necessary to evaluate the precise molecular mechanism caused by the c.788T>G mutation.
In conclusion, we identified a novel missense mutation in PAX3 that is associated with the occurrence of WS1. The new mutation, like all other defined mutations, lead to phenotypic variability within the same family, which is one of the most important features of the disease.
Acknowledgments
We certify that any affiliations with or involvement in any organization or entity with direct financial interest in the subject matter or materials discussed in the manuscript are disclosed in the paper. Financial support of the present study was provided by the Project Support Committee of Dr.Behçet Uz Children's Hospital. Neither this manuscript nor one with substantially similar content under our authorship has been published or is being considered for publication elsewhere.
References
- 1.Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–65. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi JH, Moon SK, Lee KH, Lew HM, Chang YH. Three cases of Waardenburg syndrome type 2 in a Korean family. Korean J Ophthalmol. 2004;18:185–9. doi: 10.3341/kjo.2004.18.2.185. [DOI] [PubMed] [Google Scholar]
- 3.Farrer LA, Kenneth M. Grundfast, Amos J, Arnos KS, Asher JH Jr, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB, Greenberg J, Hoth C, Marazita M, Milunsky A, Morell R, Nance W, Newton V, Ramesar R, San Agustin TB, Skare J, Stevens CA, Wagner RG, Wilcox ER, Winship I, Read AP. Waardenburg syndrome (WS) type I is caused by defects at multiple 2: first report of the WS consortium. Am J Hum Genet. 1992;50:902–13. [PMC free article] [PubMed] [Google Scholar]
- 4.Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. 1999;38:656–63. doi: 10.1046/j.1365-4362.1999.00750.x. [DOI] [PubMed] [Google Scholar]
- 5.Krishtul A, Galadari I. Waardenburg syndrome: Case report. Int J Dermatol. 2003;42:651–2. doi: 10.1046/j.1365-4362.2003.01949_3.x. [DOI] [PubMed] [Google Scholar]
- 6.Qin W, Shu A, Qian X, Gao J, Xing Q, Zhang J, Zheng Y, Li X, Li S, Feng G, He L. A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome. Mol Vis. 2006;12:1001–8. [PubMed] [Google Scholar]
- 7.Yang SZ, Cao JY, Zhang RN, Liu LX, Liu X, Zhang X, Kang DY, Li M, Han DY, Yuan HJ, Yang WY. Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients. Chin Med J (Engl) 2007;120:46–9. [PubMed] [Google Scholar]
- 8.Newton VE. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol. 2002;61:201–8. doi: 10.1159/000066810. [DOI] [PubMed] [Google Scholar]
- 9.Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am J Hum Genet. 1993;52:455–62. [PMC free article] [PubMed] [Google Scholar]
- 10.Baldwin CT, Hoth CF, Amos JA, da-Silva EO, Milunsky A. An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature. 1992;355:637–8. doi: 10.1038/355637a0. [DOI] [PubMed] [Google Scholar]
- 11.Tassabehji M, Read AP, Newton VE, Harris R, Balling R, Gruss P, Strachan T. Waardenburg’s syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature. 1992;355:635–6. doi: 10.1038/355635a0. [DOI] [PubMed] [Google Scholar]
- 12.Wollnik B, Tukel T, Uyguner O, Ghanbari A, Kayserili H, Emiroglu M, Yuksel-Apak M. Homozygous and heterozygous inheritance of PAX3 mutations causes different types of Waardenburg syndrome. Am J Med Genet A. 2003;122A:42–5. doi: 10.1002/ajmg.a.20260. [DOI] [PubMed] [Google Scholar]
- 13.Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–5. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- 14.Hughes AE, Newton VE, Liu XZ, Read AP. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat Genet. 1994;7:509–12. doi: 10.1038/ng0894-509. [DOI] [PubMed] [Google Scholar]
- 15.Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55:95–100. doi: 10.1002/ajmg.1320550123. [DOI] [PubMed] [Google Scholar]
- 16.Tassabehji M, Newton VE, Liu XZ, Brady A, Donnai D, Krajewska Walasek M, Murday V, Norman A, Obersztyn E, Reardon W. The mutational spectrum in Waardenburg syndrome. Hum Mol Genet. 1995;4:2131–7. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- 17.Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 18.Sánchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet. 2002;11:3231–6. doi: 10.1093/hmg/11.25.3231. [DOI] [PubMed] [Google Scholar]
- 19.Gad A, Laurino M, Maravilla KR, Matsushita M, Raskind WH. Sensorineural deafness, distinctive facial features, and abnormal cranial bones: a new variant of Waardenburg syndrome? Am J Med Genet A. 2008;146A:1880–5. doi: 10.1002/ajmg.a.32402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puffenberger EG, Hosoda K, Washington SS, Nakao K, deWit D, Yanagisawa M, Chakravart A. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell. 1994;79:1257–66. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 21.Syrris P, Carter ND, Patton MA. Novel nonsense mutation of the endothelin-B receptor gene in a family with Waardenburg-Hirschsprung disease. Am J Med Genet. 1999;87:69–71. [PubMed] [Google Scholar]
- 22.Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, Martelli H, Bidaud C, Munnich A, Lyonnet S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet. 1996;12:442–4. doi: 10.1038/ng0496-442. [DOI] [PubMed] [Google Scholar]
- 23.Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–85. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 24.Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A, Herbarth B. Hermans- Borgmeyer I, Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I, Romeo G, Smith JC, Read AP, Wegner M, Goossens M. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet. 1998;18:171–3. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 25.Pingault V, Bondurand N, Lemort N, Sancandi M, Ceccherini I, Hugot JP, Jouk PS, Goossens M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: is there a dosage effect of EDN3/EDNRB gene mutations on neurocristopathy phenotypes? J Med Genet. 2001;38:205–9. doi: 10.1136/jmg.38.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet. 2007;81:1169–85. doi: 10.1086/522090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogan KJ, Underhill DA, Gros P. An alternative splicing event in the Pax-3 paired domain identifies the linker region as a key determinant of paired domain DNA-binding activity. Mol Cell Biol. 1996;16:6677–86. doi: 10.1128/mcb.16.12.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008;21:627–45. doi: 10.1111/j.1755-148X.2008.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Fang WH, Krupinski J, Kumar S, Slevin M, Kumar P. Pax genes in embryogenesis and oncogenesis. J Cell Mol Med. 2008;12:2281–94. doi: 10.1111/j.1582-4934.2008.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MPLA in Waardenburg Syndrome. Genet Test. 2007;11:179–82. doi: 10.1089/gte.2006.0531. [DOI] [PubMed] [Google Scholar]
- 31.Lu-Kuo J, Ward DC, Spritz RA. Fluorescence in situ hybridization mapping of 25 markers on distal human chromosome 2q surrounding the human Waardenburg syndrome, type I (WS1) locus (PAX3 gene). Genomics. 1993;16:173–9. doi: 10.1006/geno.1993.1155. [DOI] [PubMed] [Google Scholar]
- 32.Pasteris NG, Trask BJ, Sheldon S, Gorski JL. Discordant phenotype of two overlapping deletions involving the PAX3 gene in chromosome 2q35. Hum Mol Genet. 1993;2:953–9. doi: 10.1093/hmg/2.7.953. [DOI] [PubMed] [Google Scholar]
- 33.Soejima H, Fujimoto M, Tsukamoto K, Matsumoto N, Yoshiura KI, Fukushima Y, Jinno Y, Niikawa N. Three novel PAX3 mutations observed in patients with Waardenburg syndrome type 1. Hum Mutat. 1997;9:177–80. doi: 10.1002/(SICI)1098-1004(1997)9:2<177::AID-HUMU11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 34.Tassabehji M, Newton VE, Leverton K, Turnbull K, Seemanova E, Kunze J, Sperling K, Strachan T, Read AP. PAX3 gene structure and mutations: close analogies between Waardenburg syndrome and the Splotch mouse. Hum Mol Genet. 1994;3:1069–74. doi: 10.1093/hmg/3.7.1069. [DOI] [PubMed] [Google Scholar]
- 35.Wu BL, Milunsky A, Wyandt H, Hoth C, Baldwin C, Skare J. In situ hybridization applied to Waardenburg syndrome. Cytogenet Cell Genet. 1993;63:29–32. doi: 10.1159/000133495. [DOI] [PubMed] [Google Scholar]
- 36.Clark JG. Uses and abuses of hearing loss classification. ASHA. 1981;23:493–500. [PubMed] [Google Scholar]
- 37.Arias S, Mota M. Apparent non-penetrance for dystopia in Waardenburg syndrome type 1 with some hints on the diagnosis of dystopia canthorum. J Genet Hum. 1978;26:103–31. [PubMed] [Google Scholar]
- 38.Farrer LA, Arnos KS, Asher JH, Jr, Baldwin CT, Diehl SR, Friedman TB, Greenberg J, Grundfast KM, Hoth C, Lalwani AK, Landa B, Leverton K, Milunsky A, Morell R, Nance WE, Newton V, Ramesar R, Rao VS, Reynolds JE, San Agustin TB, Wilcox ER, Winship I, Read AP. Locus heterogeneity for Waardenburg syndrome is predictive of clinical subtypes. Am J Hum Genet. 1994;55:728–37. [PMC free article] [PubMed] [Google Scholar]
- 39.Nayak CS, Isaacson G. Worldwide distribution of Waardenburg Syndrome. The 62. Ann Otol Rhinol Laryngol. 2003;112:817–20. doi: 10.1177/000348940311200913. [DOI] [PubMed] [Google Scholar]
- 40.Oysu C, Baserer N, Tinaz M. Audiometric manifestations of Waardenburg's syndrome. Ear Nose Throat J. 2000;79:704–9. [PubMed] [Google Scholar]
- 41.Newton V. Hearing loss and Waardenburg’s syndrome: implications for genetic counselling. J Laryngol Otol. 1990;104:97–103. doi: 10.1017/s002221510011196x. [DOI] [PubMed] [Google Scholar]
- 42.Toriello HV, Reardon W, Gorlin RJ. (2004). Hereditary hearing loss and its syndromes. Oxford University Press. [Google Scholar]
- 43.Wang J, Li S, Xiao X, Wang P, Guo X, Zhang Q. PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1. Mol Vis. 2010;16:1146–53. [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh SK, Bandopadhyay D, Ghosh A, Biswas SK, Mandal RK. Waardenburg syndrome: A report of three cases. Indian J Dermatol Venereol Leprol. 2010;76:550–2. doi: 10.4103/0378-6323.69089. [DOI] [PubMed] [Google Scholar]
- 45.Eigelshoven S, Kameda G, Kortüm AK, Hübsch S, Angerstein W, Singh P, Vöhringer R, Goecke T, Mayatepek E, Ruzicka T, Wildhardt G, Meissner T, Kruse R. Waardenburg syndrome type I with heterochromia iridis and circumscribed hypopigmentation of the skin. Pediatr Dermatol. 2009;26:759–61. doi: 10.1111/j.1525-1470.2009.01033.x. [DOI] [PubMed] [Google Scholar]